

The authors performed a pooled analysis of two phase III trials comparing gemcitabine-docetaxel with capecitabine-docetaxel for the treatment of metastatic breast cancer. The analysis confirmed the lack of efficacy difference between gemcitabine-docetaxel and capecitabine-docetaxel and identified several prognostic factors associated with improved outcomes.
Keywords: Gemcitabine, Docetaxel, Capecitabine, Metastatic breast cancer, Pooled analysis
Abstract
Introduction.
In two randomized phase III trials of patients with metastatic breast cancer (MBC), gemcitabine-docetaxel (GD) and capecitabine-docetaxel (CD) had similar efficacy, but distinct safety profiles.
Methods.
Data from two GD versus CD studies were pooled; overall survival (OS), progression-free survival (PFS), and overall response rate (ORR) were determined. Cox proportional hazards models identified prognostic factors associated with improved OS and PFS. Using a multivariate prognostic model incorporating identified adverse prognostic factors, we grouped MBC patients into low-, intermediate-, and high-risk categories. Hazard ratios (HRs) of GD over CD for OS and PFS were determined for subsets of patients.
Results.
Baseline demographics of the pooled population were mostly well balanced. In the pooled population, there were no significant differences between GD versus CD for OS (HR = 1.02; p = .824), PFS (HR = 1.15; p = .079), and ORR (p = .526). In the pooled crossover population, there were trends toward improved OS (HR = 0.82; p = .171) and PFS (HR = 0.93; p = .557) with GD. Several prognostic factors (including prior adjuvant taxane) for improved OS or PFS were identified; however, there were no significant interactions between treatment arms and prognostic factors for PFS or OS, except number of metastatic sites. In the prognostic model, median OS and PFS were numerically lower in the high-risk group versus the intermediate- and low-risk groups.
Conclusion.
This analysis confirms the lack of efficacy difference between GD and CD in the pooled population, crossover population, and almost all subpopulations. Several prognostic factors were associated with improved outcomes in the pooled population.
Implications for Practice:
In two randomized phase III trials of metastatic breast cancer patients, gemcitabine-docetaxel and capecitabine-docetaxel had similar efficacy, but distinct safety profiles. This pooled analysis confirmed the lack of efficacy difference between gemcitabine-docetaxel and capecitabine-docetaxel in the pooled population, the pooled crossover population, and almost all examined subpopulations. This analysis also identified several prognostic factors (Eastern Cooperative Oncology Group performance status, estrogen receptor status, prior adjuvant taxane, and number of metastatic sites) that were associated with both improved overall survival and progression-free survival in the overall pooled population. The choice of regimen should be guided by the clinical characteristics and tolerance to toxicities of the individual patient while considering the approved indications of the drugs.
Introduction
Globally, female breast cancer accounted for 23% of total cancer cases and 14% of cancer deaths in 2008, making this the most frequently diagnosed cancer and the leading cause of cancer death among females [1]. Despite advances in treatment, the long-term prognosis for women with metastatic breast cancer (MBC) is poor [2, 3]. In these patients, systemic chemotherapy can prolong survival and improve quality of life [4, 5]. However, new and better treatment options are needed to improve outcomes. In addition, the best use of existing agents has yet to be determined.
Several agents, including gemcitabine, docetaxel, and capecitabine, have single-agent activity in advanced breast cancer [6–8]. Relative to single-agent therapy, combinations can significantly improve time to progression (TTP) and response, with a small increase in overall survival (OS) [9]. However, it is unclear whether combinations are more effective than the same agents administered sequentially [10]. In addition, combination therapy is usually associated with increased toxicity [9, 10].
In order to improve outcomes and minimize toxicity, treatments have combined drugs with distinct mechanisms of action (and sometimes synergistic activity) and partially nonoverlapping toxicities. When combined with taxanes, both gemcitabine and capecitabine have superior efficacy relative to taxane monotherapy [11, 12]. Gemcitabine-paclitaxel was associated with improved OS, TTP, and response relative to paclitaxel monotherapy and had manageable toxicity [11]. This regimen is now indicated in the U.S. for treatment of patients with MBC who have relapsed following anthracycline-based adjuvant/neoadjuvant chemotherapy unless clinically contraindicated [13] and has similar indications in the European Union and China [14, 15]. Likewise, the combination of capecitabine and docetaxel (capecitabine-docetaxel) was associated with improved OS, TTP, and response relative to docetaxel monotherapy [12] and is approved for use in the United States, European Union, and China [16–18].
Because of the synergy of gemcitabine and docetaxel in vitro [19], the combination of gemcitabine and docetaxel (gemcitabine-docetaxel) was explored in patients with MBC. This doublet was tested in nonrandomized clinical trials and demonstrated activity and tolerability [20–28].
More recently, gemcitabine-docetaxel and capecitabine-docetaxel were compared in two randomized phase III trials of patients with MBC [29, 30]. One of these trials had a planned crossover to the alternate single agent [30]. In these phase III trials, gemcitabine-docetaxel had similar efficacy to capecitabine-docetaxel, but the toxicity profile of the regimens differed. Here, we performed a pooled analysis of these phase III trials to confirm the efficacy of gemcitabine-docetaxel versus capecitabine-docetaxel in MBC patients, and to identify subsets of patients who may derive the most benefit from each regimen.
Materials and Methods
Patients
In total, 780 patients were enrolled in the two international randomized phase III trials. Both studies enrolled patients ≥18 years old with histologically or cytologically confirmed MBC [29, 30].
In the Chan et al. trial (NCT00191438), patients had measurable disease per Response Evaluation Criteria in Solid Tumors (RECIST) [31] and a Karnofsky performance status ≥70 [29]. Treatment with one prior anthracycline regimen in the neoadjuvant/adjuvant or first-line metastatic setting was required. Prior taxane treatment was permitted in the neoadjuvant/adjuvant setting if completed 6 months before enrollment.
In the Seidman et al. trial (NCT00191152), patients had an Eastern Cooperative Oncology Group performance status (ECOG PS) ≤1 and measurable or nonmeasurable disease. Patients may have completed neoadjuvant or adjuvant taxane therapy ≥6 months before enrollment [30]. Prior anthracycline, hormone, or immunotherapy, and no more than one prior line of chemotherapy for MBC were allowed. Patients who received prior taxane therapy for MBC were excluded.
In both trials, patients provided written informed consent according to local guidelines. The studies were conducted per the principles of Good Clinical Practice and the Declaration of Helsinki.
Treatment
Patients were randomized to receive either gemcitabine-docetaxel or capecitabine-docetaxel [29, 30]. In the Chan trial, patients assigned to gemcitabine-docetaxel received gemcitabine (1,000 mg/m2 30-minute i.v. infusion) on days 1 and 8 and docetaxel (75 mg/m2 60-minute i.v. infusion) on day 1 [29]. Patients assigned to capecitabine-docetaxel received oral capecitabine (1,250 mg/m2 twice daily) on days 1 through 14 and docetaxel (75 mg/m2 60-minute i.v. infusion) on day 1. The capecitabine dose was based on the label [16]. Cycles were repeated every 21 days until progressive disease or unacceptable toxicity.
In the Seidman trial, patients assigned to gemcitabine-docetaxel received gemcitabine (1,000 mg/m2 30-minute i.v. infusion) on days 1 and 8 and docetaxel (75 mg/m2 60-minute i.v. infusion) on day 1 [30]. Patients assigned to capecitabine-docetaxel received oral capecitabine (1,000 mg/m2 twice daily) on days 1 through 14 and docetaxel (75 mg/m2 60-minute i.v. infusion) on day 1. Cycles were repeated every 21 days until disease progression. The capecitabine dose was reduced because of the high incidence of diarrhea and hand-foot syndrome that was observed in the earlier Chan trial [29]. Patients who progressed on induction gemcitabine-docetaxel or capecitabine-docetaxel received single-agent capecitabine or gemcitabine, respectively (using the induction doses and schedules) within 4 weeks of documented progressive disease.
Dose reductions were described in the original reports [29, 30].
Efficacy Evaluations
In the Chan trial, the primary endpoint was progression-free survival (PFS); secondary endpoints were OS, overall response rate (ORR), time-to-treatment failure, safety, and quality of life [29]. In the Seidman trial, the primary endpoint was TTP; secondary endpoints were ORR, OS, and safety. Time-to-treatment failure was added as a post hoc analysis [30]. Tumor responses were evaluated using RECIST 1.0 criteria [31] every third cycle. Confirmatory scans were performed at least 3 weeks after the first evidence of response [29, 30].
Statistical Analyses
Patient-level data from two individual studies were pooled for analyses. OS was calculated from the date of randomization until death from any cause or censored at last known alive date. PFS was calculated from the date of randomization until first date of documented progression or death from any cause or censored last follow-up visit for patients who were still alive and progression-free. OS and PFS were estimated using the Kaplan-Meier product limit method [32]. Cox proportional hazards models [33] and log-rank tests, stratified by study, were used to calculate hazard ratios (HRs) and to compare survival curves of the two treatment arms for OS and PFS. Pooled ORR (defined as the proportion of patients with a best overall response of complete response [CR] or partial response [PR]) and disease control rate (DCR, defined as the proportion of patients with a best overall response of CR, PR, or stable disease) were also calculated in the two treatment arms and compared using the Cochran-Mantel-Haenszel test stratified by study. Potential prognostic factors were identified initially by searching available baseline variables that significantly influenced OS or PFS at a level of p < .05 in the univariate analyses, and then were included in the multivariate analyses using stepwise Cox proportional hazards modeling for OS or PFS. Factors with p values <.05 in the multivariate analyses were considered statistically significant and prognostic. All p values were two-sided and were not adjusted for multiplicity. Caution should be used when interpreting these p values.
The crossover population consisted of patients who received induction gemcitabine-docetaxel and then, upon progression, crossed over to capecitabine, and patients who received induction capecitabine-docetaxel and then, upon progression, crossed over to gemcitabine. Induction PFS was estimated for all crossover patients from the time of randomization to the date of first progressive disease or death from any cause, whichever occurred first.
Results
Patient Demographics
Table 1 shows the baseline demographics for the pooled population. From the Chan trial, 305 patients (153 gemcitabine-docetaxel; 152 capecitabine-docetaxel) were randomized [29]; from the Seidman trial, 475 patients (239 gemcitabine-docetaxel induction phase; 236 capecitabine-docetaxel induction phase) were randomized [30]. A minority of patients received prior chemotherapy for MBC (20.9% gemcitabine-docetaxel; 19.1% capecitabine-docetaxel). The arms were well balanced, with the possible exceptions of crossover status and progesterone receptor status. HER2 status was not available in the Seidman trial [30] and prior use of trastuzumab was unknown in both trials.
Table 1.
Baseline demographics of pooled population
Efficacy
Pooled Efficacy of Gemcitabine-Docetaxel Versus Capecitabine-Docetaxel
In the pooled population, OS for patients randomized to gemcitabine-docetaxel versus capecitabine-docetaxel was not statistically different (stratified log-rank p = .824, HR = 1.02, 95% CI, 0.86–1.20; median 21.5 months vs. 22.0 months) (Fig. 1A). In the pooled population, PFS for patients randomized to gemcitabine-docetaxel versus capecitabine-docetaxel was not statistically different (stratified log-rank p = .079, HR = 1.15, 95% CI, 0.98–1.35; median 8.5 months vs. 8.5 months) (Fig. 1B).
Figure 1.

Kaplan-Meier curves of the pooled population. (A): Overall survival. (B): Progression-free survival.
Abbreviations: CD, capecitabine-docetaxel; CI, confidence interval; GD, gemcitabine-docetaxel; HR, hazard ratio.
In the pooled population, the ORR was 32.1% (95% CI, 27.5–37.0) for gemcitabine-docetaxel and 34.3% (95% CI, 29.6–39.2) for capecitabine-docetaxel (Cochran-Mantel-Haenszel p = .526). The DCR (CR + PR + stable disease) was 56.6% (95% CI, 51.6–61.6) for gemcitabine-docetaxel and 57.5 (95% CI, 52.4–62.4) for capecitabine-docetaxel (Cochran-Mantel-Haenszel p = .781).
Pooled Efficacy of Crossover Population
In the pooled crossover population, although there was a trend favoring gemcitabine-docetaxel, the difference in OS among patients initially receiving gemcitabine-docetaxel versus capecitabine-docetaxel was not statistically significant (unstratified log-rank p = .171, HR = 0.82, 95% CI, 0.62–1.09; median 25.5 months vs. 23.5 months) (Fig. 2A). Likewise, there was a trend toward improved PFS of the induction phase with gemcitabine-docetaxel, but the difference in PFS in the pooled crossover population receiving gemcitabine-docetaxel versus capecitabine-docetaxel was not statistically significant (unstratified log-rank p = .557, HR = 0.93, 95% CI, 0.73–1.19; 8.3 months vs. 6.5 months) (Fig. 2B).
Figure 2.

Kaplan-Meier curves of the crossover subpopulation within the pooled population. (A): Overall survival. (B): Progression-free survival.
Abbreviations: CD-G, capecitabine-docetaxel crossed over to gemcitabine; CI, confidence interval; GD-C, gemcitabine-docetaxel crossed over to capecitabine; HR, hazard ratio.
Prognostic Factors
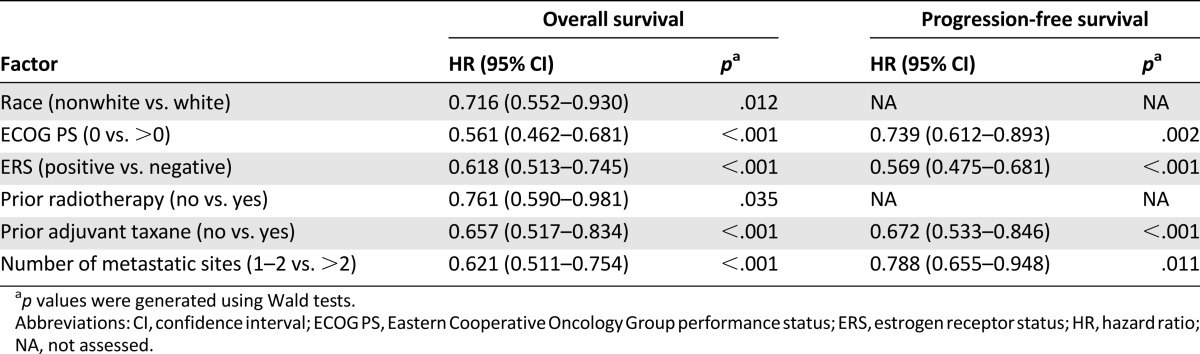
Using a univariate Cox proportional hazards model, we found that several potential prognostic factors were associated with improved OS or PFS at a significance level of p < .05 (Table 2). For OS, these were race, ECOG PS, estrogen receptor status, progesterone receptor status, prior surgery, prior radiotherapy, prior adjuvant taxane, and number of metastatic sites; for PFS, these were ECOG PS, estrogen receptor status, progesterone receptor status, prior adjuvant taxane, time since diagnosis, and number of metastatic sites. These factors were chosen for further multivariate analysis using the stepwise multivariate Cox proportional hazards modeling. As a result, race, ECOG PS, estrogen receptor status, prior radiotherapy, prior adjuvant taxane, and number of metastatic sites were significant at p < .05 for OS, and ECOG PS, estrogen receptor status, prior adjuvant taxane, and number of metastatic sites were significant at p < .05 for PFS (Table 3).
Table 2.
Prognostic factors for pooled overall survival and progression-free survival (univariate analysis)
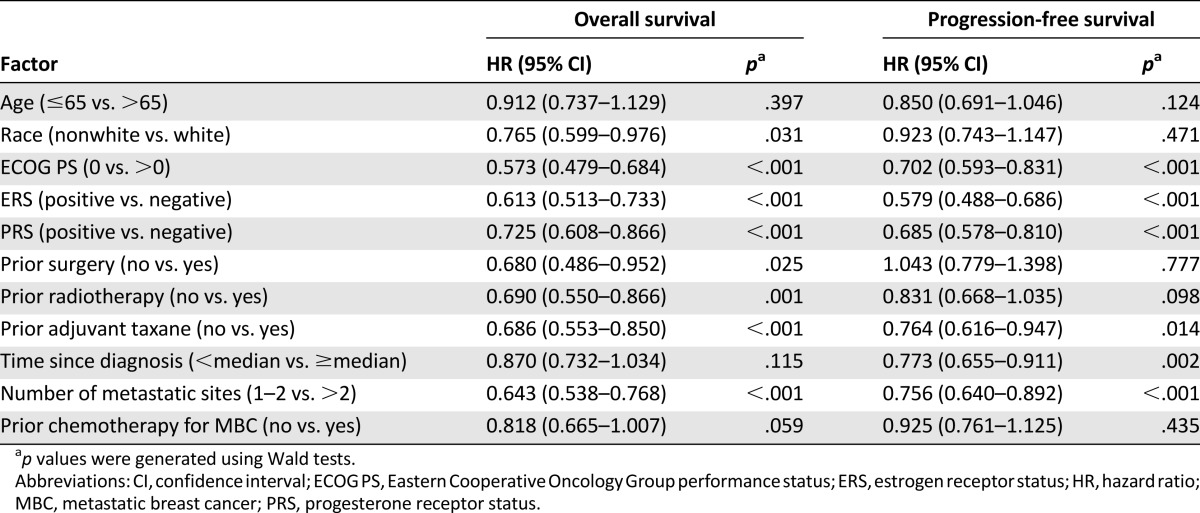
Table 3.
Prognostic factors for pooled overall survival and progression-free survival (multivariate analysis)
A multivariate prognostic model was constructed by incorporating all identified adverse prognostic factors. The prognostic factors were grouped according to the criteria shown in supplemental online Table 1. For OS, the low-risk group (n = 121) had none or one negative prognostic factor, the intermediate-risk group (n = 500) had two or three negative prognostic factors, and the high-risk group (n = 159) had four to six negative prognostic factors; for PFS, the low-risk group (n = 121) had no negative prognostic factors, the intermediate-risk group (n = 558) had one or two negative prognostic factors, and the high-risk group (n = 101) had three or four negative prognostic factors. The median OS was 11.3 (95% CI, 9.5–14.1) months in the high-risk group, 22.9 (95% CI, 20.3–24.9) months in the intermediate-risk group, and 36.5 (95% CI, 27.5–44.5) months in the low-risk group (Fig. 3A). The median PFS was 4.4 (95% CI, 3.5–5.5) months in the high-risk group, 8.6 (95% CI, 7.8–9.2) months in the intermediate-risk group, and 11.2 (95% CI, 9.3–13.4) months in the low-risk group (Fig. 3B). In all risk groups, the p value was <.001 using a stratified log-rank test.
Figure 3.

Kaplan-Meier curves of the risk groups. (A): Overall survival. (B): Progression-free survival. The stratified log-rank test for both overall survival and progression-free survival risk groups was p < .001.
Abbreviations: CI, confidence interval; Int, intermediate.
Subgroup Analyses for Prognostic Factors
Figure 4A shows the subgroup analysis for OS. Although there were trends toward a more favorable OS with capecitabine-docetaxel in some subgroups (nonwhite race, ECOG PS >0, estrogen receptor negative, prior radiotherapy, prior taxane usage, one or two metastatic sites, and high-risk group), no interaction tests were statistically significant at the p = .05 level (p values not shown), suggesting that there is no heterogeneity of treatment effects across the levels of the prognostic factors and two treatment arms. Also, in most subgroups, there were trends toward a more favorable PFS with capecitabine-docetaxel (Fig. 4B). However, with the exception of a possible interaction between metastatic sites and treatment arms (favored capecitabine-docetaxel in patients with one or two vs. more than two metastatic sites; p = .026), interaction tests for other prognostic factors were not statistically significant (p values not shown).
Figure 4.

Forest plots. Subgroups were identified using a multivariate Cox model. Squares indicate point estimates; horizontal lines indicate 95% CIs. (A): Overall survival. (B): Progression-free survival. HR >1 favors CD; HR <1 favors GD. For PFS, Wald p = .026 for treatment arm and number of metastatic sites.
Abbreviations: CD, capecitabine-docetaxel; CI, confidence interval; ECOG PS, Eastern Cooperative Oncology Group performance status; ERS, estrogen receptor status; GD, gemcitabine-docetaxel; HR, hazard ratio; OS, overall survival; PFS, progression-free survival.
Discussion
Despite improvements in outcomes for patients with MBC, the optimal use of existing chemotherapeutic agents continues to be debated. Gemcitabine and capecitabine in combination with taxanes are routinely used in first-line MBC patients who received prior anthracyclines, but the optimal use of these combinations is unknown. This pooled analysis of two international phase III trials that compared gemcitabine-docetaxel with capecitabine-docetaxel [29, 30] was performed to provide guidance for the best use of these agents.
Results obtained with the pooled population confirm the lack of efficacy difference between gemcitabine-docetaxel and capecitabine-docetaxel that had been previously reported for the individual phase III trials [29, 30]. In the pooled population, there were no between-arms differences in OS (HR = 1.02), PFS (HR = 1.15), or ORR (p = .526). The median OS (21.5 months) and median PFS (8.5 months) reported here for the pooled gemcitabine-docetaxel arm are at least equivalent to those obtained for the gemcitabine-paclitaxel combination in the registration trial (OS = 18.6 months; TTP = 6.14 months), whereas the ORR for gemcitabine-docetaxel in the pooled population (32.1%) seems lower than that in the registration trial (41.4%) [11]. Here, the capecitabine-docetaxel combination (median OS = 22.0 months; median PFS = 8.5 months) outperformed itself relative to the registration trial (median OS = 14.5 months; median TTP = 6.1 months) [12]. However, patients in the capecitabine registration trial [12] received more prior treatments than patients in the pooled population [29, 30].
Because there was no efficacy difference between gemcitabine-docetaxel and capecitabine-docetaxel in the pooled population, it was of interest to identify subsets of patients that may benefit the most from these regimens. Our data also show that although there were trends toward improved OS and PFS with gemcitabine-docetaxel in the pooled crossover population, the differences between gemcitabine-docetaxel and capecitabine-docetaxel were not statistically significant. A possible explanation is that capecitabine was better tolerated as a single agent than when combined with docetaxel. This is supported by the toxicity profiles of the Chan and Seidman trials; both trials had high rates of toxicity-related discontinuations in the capecitabine-docetaxel arms compared with the gemcitabine-docetaxel arms (Seidman = 28.4% vs. 18.0%; Chan = 27% vs. 13% in the induction phases) [29, 30]. However, this was a post hoc subset analysis, and the usual caveats regarding subset analyses should be noted [34]. It should also be noted that in the crossover population, data for secondary progression (e.g., from crossover to further progression) were not collected in the Chan trial [29], so the induction PFS in the crossover population should be viewed with caution. Given the limitations of this subset analysis, the trends toward improved OS and PFS with gemcitabine-docetaxel suggest that gemcitabine-docetaxel followed by capecitabine might be a preferred sequence option for certain MBC patients and may warrant further evaluation. However, it should be noted that gemcitabine in combination with docetaxel is currently not approved in the U.S., European Union, or China for the treatment of MBC.
Although several prognostic factors were associated with improved outcomes in the overall pooled population, there were no interactions in OS or PFS between gemcitabine-docetaxel and capecitabine-docetaxel and any of the tested prognostic factors, with the possible exception of longer PFS in the capecitabine-docetaxel arm in the subgroup of patients with one or two sites of metastases. This suggests that there is no evidence of benefiting more in either regimen within each level of prognostic factors. Using risk factors identified in a multivariate prognostic model, we categorized patients into low-, intermediate-, and high-risk groups. The high-risk group had worse outcomes (OS and PFS) than the other risk groups. However, no statistically significant interaction was found between treatment arms and risk groups.
These data show that prior adjuvant taxane was a prognostic factor for both OS and PFS. However, it should be noted that in the Seidman trial [30], a large proportion of patients had an unknown adjuvant taxane status (67.8% gemcitabine-docetaxel; 69.5% capecitabine-docetaxel). For the purpose of the pooled analysis, patients with an unknown taxane status were considered to have received no prior adjuvant taxane, but it is possible that this assumption was incorrect for some patients. Another potential issue with this analysis is that, in the Chan trial, enrolled patients were required to have received one prior anthracycline regimen [29], whereas the Seidman trial did not have this requirement, so a significant proportion of patients had not been exposed to anthracyclines (approximately 42%) [30]. Finally, it should be noted that patients with prior exposure to adjuvant taxanes may acquire drug resistance to taxanes. Thus, non–cross-resistant regimens should be evaluated in those patients toward optimization.
Finally, it should be noted that, with the exception of capecitabine, the drug doses and schedules were identical in the parent trials. The Chan trial used the approved capecitabine dose (1,250 mg/m2), whereas the Seidman trial used a reduced dose (1,000 mg/m2) [29, 30]. When used as a component of capecitabine-docetaxel or as monotherapy, the capecitabine dose can be reduced without compromising TTP or OS [35]. Therefore, the efficacy results should not have been affected by using pooled data from the previously described trials [29, 30].
Because the reduced capecitabine dose is associated with a decreased incidence of treatment-related adverse events, particularly hand-foot syndrome, diarrhea, and stomatitis [35], it would not have been appropriate to perform a safety analysis on the pooled population. Nonetheless, based on the parent trials, it is known that gemcitabine-docetaxel and capecitabine-docetaxel have different toxicity profiles; capecitabine-docetaxel is generally associated with higher incidences of grade 3-4 gastrointestinal toxicity, hand-foot syndrome, and mucositis, and gemcitabine-docetaxel is generally associated with more grade 3-4 fatigue, elevated liver enzymes, neutropenia, leukopenia, and thrombocytopenia [29, 30]. Despite these differences, toxicity-related discontinuations in the capecitabine-docetaxel arm (28.4%) were significantly greater (p = .009) than in the gemcitabine-docetaxel arm (18.0%) in the Seidman trial [30], which is consistent with toxicity-related discontinuations observed in the Chan trial (capecitabine-docetaxel = 27%; gemcitabine-docetaxel = 13%) [29].
Conclusion
Results from this analysis confirm the lack of efficacy difference between gemcitabine-docetaxel and capecitabine-docetaxel in the pooled population. In addition, there are no efficacy differences between regimens in the crossover population, as well as in almost all examined subpopulations. Several prognostic factors, such as ECOG PS, estrogen receptor status, prior adjuvant taxane, and number of metastatic sites, were associated with both improved OS and PFS in the overall pooled population. The choice of regimen should be guided by the clinical characteristics and tolerance to toxicities of the individual patient.
See http://www.TheOncologist.com for supplemental material available online.
Supplementary Material
Acknowledgments
We thank Tan Ding and Weishan Shi for assistance in statistical computation. Medical writing support was provided by Lori Kornberg (Inventiv Health Clinical). Writing support (funded by Eli Lilly and Company) included drafting of the manuscript, preparing references, and assembling figures/tables. Andrew D. Seidman and Stephen Chan contributed to this manuscript equally as co-first authors.
Author Contributions
Conception/design: Binghe Xu, Andrew D. Seidman, Stephen Chan, Jin Wang, Cong Xu
Provision of study material or patients: Binghe Xu, Stephen Chan, Cong Xu
Collection and/or assembly of data: Binghe Xu, Andrew D. Seidman, Stephen Chan
Data analysis and interpretation: Binghe Xu, Andrew D. Seidman, Stephen Chan, Jin Wang, Chao Zhu, Cong Xu
Manuscript writing: Andrew D. Seidman, Stephen Chan, Chao Zhu
Final approval of manuscript: Binghe Xu, Andrew D. Seidman, Stephen Chan, Jin Wang, Chao Zhu, Cong Xu
Disclosures
Jin Wang: Eli Lilly (E); Chao Zhu: Eli Lilly (E); Cong Xu: Eli Lilly (E). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Albain KS, de la Garza Salazar J, Pienkowski T, et al. Reducing the global breast cancer burden: The importance of patterns of care research. Clin Breast Cancer. 2005;6:412–420. doi: 10.3816/CBC.2005.n.045. [DOI] [PubMed] [Google Scholar]
- 3.Hortobagyi GN, de la Garza Salazar J, Pritchard K, et al. The global breast cancer burden: Variations in epidemiology and survival. Clin Breast Cancer. 2005;6:391–401. doi: 10.3816/cbc.2005.n.043. [DOI] [PubMed] [Google Scholar]
- 4.Mayer EL, Burstein HJ. Chemotherapy for metastatic breast cancer. Hematol Oncol Clin North Am. 2007;21:257–272. doi: 10.1016/j.hoc.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 5.Murphy CG, Seidman AD. Evolving approaches to metastatic breast cancer previously treated with anthracyclines and taxanes. Clin Breast Cancer. 2009;9(Suppl 2):S58–S65. doi: 10.3816/CBC.2009.s.006. [DOI] [PubMed] [Google Scholar]
- 6.Gradishar WJ. Clinical status of capecitabine in the treatment of breast cancer. Oncology (Williston Park) 2001;15(Suppl 2):69–71; discussion 72. [Williston Park] [PubMed] [Google Scholar]
- 7.Ravdin PM, Valero V. Review of docetaxel (Taxotere), a highly active new agent for the treatment of metastatic breast cancer. Semin Oncol. 1995;22(Suppl 4):17–21. [PubMed] [Google Scholar]
- 8.Smith IE. Overview of gemcitabine activity in advanced breast cancer. Semin Oncol. 2006;33(Suppl 9):S19–S23. doi: 10.1053/j.seminoncol.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 9.Carrick S, Parker S, Wilcken N, et al. Single agent versus combination chemotherapy for metastatic breast cancer. Cochrane Database Syst Rev. 2005;(2):CD003372. doi: 10.1002/14651858.CD003372.pub2. [DOI] [PubMed] [Google Scholar]
- 10.Carrick S, Parker S, Thornton CE, et al. Single agent versus combination chemotherapy for metastatic breast cancer. Cochrane Database Syst Rev. 2009;(2):CD003372. doi: 10.1002/14651858.CD003372.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Albain KS, Nag SM, Calderillo-Ruiz G, et al. Gemcitabine plus paclitaxel versus paclitaxel monotherapy in patients with metastatic breast cancer and prior anthracycline treatment. J Clin Oncol. 2008;26:3950–3957. doi: 10.1200/JCO.2007.11.9362. [DOI] [PubMed] [Google Scholar]
- 12.O’Shaughnessy J, Miles D, Vukelja S, et al. Superior survival with capecitabine plus docetaxel combination therapy in anthracycline-pretreated patients with advanced breast cancer: phase III trial results. J Clin Oncol. 2002;20:2812–2823. doi: 10.1200/JCO.2002.09.002. [DOI] [PubMed] [Google Scholar]
- 13.Gemzar package insert. Indianapolis, IN: Eli Lilly and Company, 2013.
- 14.European Medicines Agency. Gemzar—Article 30 referral—Annex I, II, III. London, UK: European Medicines Agency, 2008.
- 15.Gemzar package insert. Shanghai, China: Eli Lilly and Company, 2010.
- 16.Xeloda package insert. Nutley, NJ: Genentech-Hoffmann-La Roche, 2011.
- 17.European Medicines Agency. Xeloda EPAR summary for the public, 2008. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/000316/WC500058146.pdf Accessed September 11, 2013.
- 18.Xeloda package insert. Shanghai, China: Roche, 2012.
- 19.Ricotti L, Tesei A, De Paola F, et al. In vitro schedule-dependent interaction between docetaxel and gemcitabine in human gastric cancer cell lines. Clin Cancer Res. 2003;9:900–905. [PubMed] [Google Scholar]
- 20.Brandi M, Vici P, Lopez M, et al. Novel association with gemcitabine and docetaxel as salvage chemotherapy in metastatic breast cancer previously treated with anthracyclines: Results of a multicenter phase II study. Semin Oncol. 2004;31(Suppl 5):13–19. doi: 10.1053/j.seminoncol.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 21.Fountzilas G, Nicolaides C, Bafaloukos D, et al. Docetaxel and gemcitabine in anthracycline-resistant advanced breast cancer: A Hellenic Cooperative Oncology Group Phase II study. Cancer Invest. 2000;18:503–509. doi: 10.3109/07357900009012188. [DOI] [PubMed] [Google Scholar]
- 22.Kornek GV, Haider K, Kwasny W, et al. Treatment of advanced breast cancer with docetaxel and gemcitabine with and without human granulocyte colony-stimulating factor. Clin Cancer Res. 2002;8:1051–1056. [PubMed] [Google Scholar]
- 23.Laufman LR, Spiridonidis CH, Pritchard J, et al. Monthly docetaxel and weekly gemcitabine in metastatic breast cancer: a phase II trial. Ann Oncol. 2001;12:1259–1264. doi: 10.1023/a:1012247311419. [DOI] [PubMed] [Google Scholar]
- 24.Mavroudis D, Malamos N, Alexopoulos A, et al. Salvage chemotherapy in anthracycline-pretreated metastatic breast cancer patients with docetaxel and gemcitabine: A multicenter phase II trial. Ann Oncol. 1999;10:211–215. doi: 10.1023/a:1008315723253. [DOI] [PubMed] [Google Scholar]
- 25.Mavroudis D, Malamos N, Polyzos A, et al. Front-line chemotherapy with docetaxel and gemcitabine administered every two weeks in patients with metastatic breast cancer: A multicenter phase II study. Oncology. 2004;67:250–256. doi: 10.1159/000081325. [DOI] [PubMed] [Google Scholar]
- 26.O’Shaughnessy JA, Pluenneke R, Sternberg J, et al. Phase II trial of weekly docetaxel/gemcitabine as first-line chemotherapy in patients with locally recurrent or metastatic breast cancer. Clin Breast Cancer. 2006;6:505–510. doi: 10.3816/CBC.2006.n.003. [DOI] [PubMed] [Google Scholar]
- 27.Palmeri S, Vaglica M, Spada S, et al. Weekly docetaxel and gemcitabine as first-line treatment for metastatic breast cancer: Results of a multicenter phase II study. Oncology. 2005;68:438–445. doi: 10.1159/000086986. [DOI] [PubMed] [Google Scholar]
- 28.Pelegrí A, Calvo L, Antón A, et al. Docetaxel/gemcitabine administered every other week as first-line treatment for metastatic breast cancer: Final results of a phase II trial. Clin Breast Cancer. 2005;6:433–438. doi: 10.3816/cbc.2005.n.048. [DOI] [PubMed] [Google Scholar]
- 29.Chan S, Romieu G, Huober J, et al. Phase III study of gemcitabine plus docetaxel compared with capecitabine plus docetaxel for anthracycline-pretreated patients with metastatic breast cancer. J Clin Oncol. 2009;27:1753–1760. doi: 10.1200/JCO.2007.15.8485. [DOI] [PubMed] [Google Scholar]
- 30.Seidman AD, Brufsky A, Ansari RH, et al. Phase III trial of gemcitabine plus docetaxel versus capecitabine plus docetaxel with planned crossover to the alternate single agent in metastatic breast cancer. Ann Oncol. 2011;22:1094–1101. doi: 10.1093/annonc/mdq578. [DOI] [PubMed] [Google Scholar]
- 31.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 32.Kaplan EL, Meier P. Nonparametric estimation of incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 33.Cox DR, Snell EJ. Analysis of Binary Data. 2nd ed. London, UK: Chapman & Hall/CRC; 1989. [Google Scholar]
- 34.Wang R, Lagakos SW, Ware JH, et al. Statistics in medicine—Reporting of subgroup analyses in clinical trials. N Engl J Med. 2007;357:2189–2194. doi: 10.1056/NEJMsr077003. [DOI] [PubMed] [Google Scholar]
- 35.Leonard R, Hennessy BT, Blum JL, et al. Dose-adjusting capecitabine minimizes adverse effects while maintaining efficacy: A retrospective review of capecitabine for metastatic breast cancer. Clin Breast Cancer. 2011;11:349–356. doi: 10.1016/j.clbc.2011.06.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.