

Human cells must faithfully duplicate billions of base pairs of DNA every cell division cycle. Insufficient precursors, DNA damage, and difficult-to-replicate sequences cause replication fork stalling. Even in the best of circumstances, hundreds of forks may stall during each S phase in a human cell, and the frequency increases in cells exposed to genotoxic or oncogenic stresses. Cells must resolve these stalled replication forks to complete DNA synthesis, as failure to do so leads to mutations or cell death. Given its importance, multiple pathways exist to recover stalled replication forks, including stabilization and restart, repriming and post-replicative repair, template switching, and double-strand break (DSB)-mediated recovery. However, much remains to be learned about these mechanisms and how they are coordinated to successfully replicate the genome trillions of times during a human lifetime.
The most important regulator of replication stress responses is the ATR kinase, which phosphorylates hundreds of substrates and is essential for every round of cell division. Many cancer cells have an elevated need for ATR due to oncogene-induced replication stress, making ATR a potential drug target for cancer therapy.1 In addition, ATR inhibition hypersensitizes cells to many chemotherapeutic agents that work by damaging DNA or interfering with DNA replication.
ATR activity is critical to prevent replication fork collapse, a situation in which a fork is no longer competent to support DNA synthesis. But what is fork collapse at the molecular level, and how does fork collapse contribute to cell death in ATR-deficient cells? These questions have been difficult to answer, especially in mammalian systems, since deletion of the ATR gene is cell lethal. The development of selective ATR kinase inhibitors provides a tool to investigate these ATR functions.
As expected, stalled replication forks in ATR inhibited cells rapidly undergo collapse into DSBs.2,3 In addition to DSBs, ATR inhibition results in the formation of excess single-stranded DNA (ssDNA) at the fork, including both the template and newly synthesized strands.2 The timing of replication origin usage is deregulated, which exacerbates the problems as new forks stall and collapse. Importantly, fork collapse into DSBs and excess ssDNA is an active process mediated by SLX4- and CtIP-dependent nucleases.2,3
The DNA structure at the stalled replication fork that is cleaved to create breaks is not known; however, one clue comes from our observation that the SMARCAL1 protein is involved in the aberrant fork processing that happens in ATR-deficient cells.2 SMARCAL1 is a SNF2 family, DNA-dependent ATPase that catalyzes branch migration of fork junctions.4 In particular, SMARCAL1 is recruited to and active at stalled replication forks that contain ssDNA on the leading template strand.5 On these substrates, SMARCAL1 catalyzes reversal of the replication fork into a chicken-foot structure, which may be the substrate for SLX4-dependent nucleases. SMARCAL1 is also able to catalyze the reverse reaction (fork restoration) when the nascent leading strand is longer than the nascent lagging strand,5 so other models for how aberrant SMARCAL1 activity leads to fork collapse could be envisioned.
Importantly, fork reversal is a common event in human cells and is an evolutionarily conserved mechanism of fork stabilization and repair at least in some circumstances.6 Furthermore, SMARCAL1 normally promotes fork restart cells that have an intact ATR pathway.7 Thus, in ATR-deficient cells, SMARCAL1-catalyzed fork remodeling leads to fork collapse, but in ATR-proficient cells SMARCAL1 maintains fork stability. This paradox is further illustrated by the observation that either too little or too much SMARCAL1 activity in cells leads to replication-associated DSBs.8
The solution to this conundrum is that ATR directly regulates SMARCAL1 to maintain the balance between too much and too little SMARCAL1 activity (Fig. 1). Specifically, ATR phosphorylates SMARCAL1 on S652 in a linker region between the 2 lobes of its ATPase domain. S652 phosphorylation happens after SMARCAL1 binds to DNA at the replication fork and inhibits its fork remodeling activities. Thus, ATR ensures the right level of SMARCAL1 activity at the damaged replication fork. Treating cells with an ATR inhibitor causes fork collapse, in part because it interferes with SMARCAL1 regulation, sending stalled forks through a pathway that includes SLX4-dependent cleavage. Were this infrequent, recombination could repair the break and restart replication, but the deregulation of origin timing when ATR is inhibited multiplies the number of collapsed forks. This leads to genome-wide problems, evidenced by pan-nuclear γH2AX staining. Furthermore, addition of replication stress to the system in the presence of an ATR inhibitor for more than 30–45 min ensures the cells will be unable to complete replication and are destined to die.2
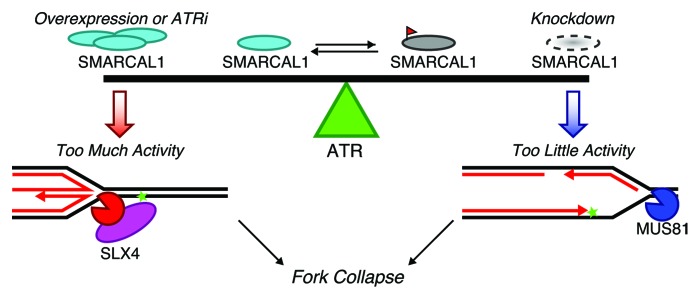
Figure 1. ATR maintains a balance between too much and too little SMARCAL1 activity. Tipping the balance toward too much SMARCAL1 activity by either inhibiting ATR or overexpressing SMARCAL1 results in fork collapse through an SLX4-dependent cleavage. Conversely, fork collapse is MUS81-dependent when there is too little SMARCAL1. The actual DNA structures that these nucleases cleave in these conditions remain to be experimentally verified.
The ATR–SMARCAL1 pathway is certainly not the only mechanism by which ATR prevents fork collapse and cell death. For example, ATR signaling likely regulates the integrity of the replisome proteins themselves, and both RNF4 and PLK1 have been implicated in this pathway.3 Given the large number of ATR substrates and mechanisms of fork repair, ATR inactivation likely disrupts multiple pathways of fork repair and maintenance, and much remains to be understood about this critical genome-maintenance activity.
Couch FB, et al. Genes Dev. 2013;27:1610–23. doi: 10.1101/gad.214080.113.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/28212
References
- 1.Toledo LI, et al. Mol Oncol. 2011;5:368–73. doi: 10.1016/j.molonc.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Couch FB, et al. Genes Dev. 2013;27:1610–23. doi: 10.1101/gad.214080.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ragland RL, et al. Genes Dev. 2013;27:2259–73. doi: 10.1101/gad.223180.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bétous R, et al. Genes Dev. 2012;26:151–62. doi: 10.1101/gad.178459.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bétous R, et al. Cell Rep. 2013;3:1958–69. doi: 10.1016/j.celrep.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Atkinson J, et al. Nucleic Acids Res. 2009;37:3475–92. doi: 10.1093/nar/gkp244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ciccia A, et al. Genes Dev. 2009;23:2415–25. doi: 10.1101/gad.1832309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bansbach CE, et al. Genes Dev. 2009;23:2405–14. doi: 10.1101/gad.1839909. [DOI] [PMC free article] [PubMed] [Google Scholar]