

Metabolic reprogramming, a prominent phenotype in cancer cells, is the adaptation to shifts in the usage of metabolites, including glucose, fatty acids, amino acids, and glutamine. The central feature of this adaptation lies in the fact that cancer cells undergo glycolysis even in the presence of ample oxygen, contrary to normal cells.1 This aerobic glycolysis, termed “the Warburg effect,” has been considered to give tumor cells selective advantages through enhanced catabolism of glucose and glutamine, providing the raw materials for the synthesis of nucleotides, amino acids, and lipids to satiate rapidly dividing cancer cells. On the other side of the coin, however, unravelling the molecular network that dictates the Warburg effect may be potentially exploited for identifying new drug targets and drug resistance mechanisms in cancer.
Among the molecular players implicated in governing the Warburg effect, one of the central components for cellular metabolic integration is the mechanistic target of rapamycin (mTOR). mTOR kinase exists in 2 multi-protein complexes and is a critical effector downstream of phosphatidylinositol 3′-kinase (PI3K), which plays a central role in integrating growth factor receptor signaling with cellular metabolism.2 mTORC1 is a well-established cancer target, linking PI3K signaling through Akt to protein translation, glycolysis, and lipogenesis.2 In contrast, the upstream role of mTORC2, which phosphorylates Akt on serine 473, thereby maximizing its activity,2 is less well understood than that of mTORC1 in cancer. While some Akt-independent effects of mTORC2 in carcinogenesis have been elucidated,3 the impact of mTORC2 in cancer metabolism remains unclear.
Therefore, we recently set out to determine the role of mTORC2 in metabolic reprogramming of glioblastoma (GBM), the most common form of adult primary brain cancer and one of the most lethal of all human malignancies. Surprisingly, an unexpected Akt-independent role for mTORC2 in inducing metabolic reprogramming in GBM was found.4 mTORC2 renders GBM cells strongly addicted to glucose, and this is mediated by regulating the intracellular level of c-Myc, a crucial regulator of the Warburg effect.5 mTORC2 is shown to execute an Akt-independent phosphorylation of class IIa histone deacetylases, which leads to the inactivating acetylation of FoxO, a negative regulator of c-Myc. As a result, the microRNA-dependent blockade of c-Myc is relieved, potently promoting glycolytic tumor growth. Importantly, mTORC2/acetylated FoxO/c-Myc expression confers an adverse prognostic impact to GBM patients, and it can be abrogated by dual PI3K/mTOR kinase inhibition, resulting in tumor cell death of the mouse xenograft tumor models with patient-derived GBM neurosphere cells. These results provide new insight into the role of mTORC2 signaling in cancer, identifying metabolic reprogramming through a c-Myc-dependent pathway as a critical consequence.
These results have an intriguing implication; that is to say, GBM is addicted to c-Myc. c-Myc plays a central role in cancer cell metabolism,5 but the mechanisms by which activated growth factor receptor signaling pathways harness c-Myc remain to be clarified. Our recent studies demonstrate that GBM with an activated mutant form of EGFR engages c-Myc signaling at least by 2 complementary steps: (1) promotion of alternative splicing of Delta Max to modulate c-Myc function6 and (2) upregulation of cellular levels of c-Myc through mTORC2.4 This new, multistep scheme highlights the heavy reliance of GBM on c-Myc activity to promote glycolytic metabolism, which might be exploitable as a potential therapeutic opportunity.
Another therapeutic spin-off derives from the findings that, as a consequence of dual regulation of FoxO through Akt-dependent phosphorylation and mTORC2-dependent acetylation,4 GBM can evade PI3K/Akt inhibition via mTORC2-dependent FoxO acetylation and sustained c-Myc expression. FoxO and its downstream regulation of c-Myc are tightly controlled through 2 independent and highly specific pathways of post-translational modification and microRNA suppression. The net consequence of this series of events is the conferral of resistance to PI3K and Akt inhibitors in GBM. Patients with GBM have a median survival time of 12–15 months from the time of initial diagnosis, despite surgery, radiation, and chemotherapy, and new approaches are desperately needed.7 A new era of targeted cancer therapies has been heralded by progress in genomics and epigenomics, and discovery of the distinct molecular characteristics of cancer cells has led to the development of therapies that selectively zero in on these cancer-specific targets. However, drug resistance occurs even to this category of therapeutics, regardless of drug target and mechanism of action.8 Our study further sheds new light on the resistance mechanisms of GBM to targeted therapies, providing compelling rationale for the combined inhibition of PI3K/Akt and mTORC2 as a promising “combinatorial targeted therapy” for this deadly brain cancer (Fig. 1).
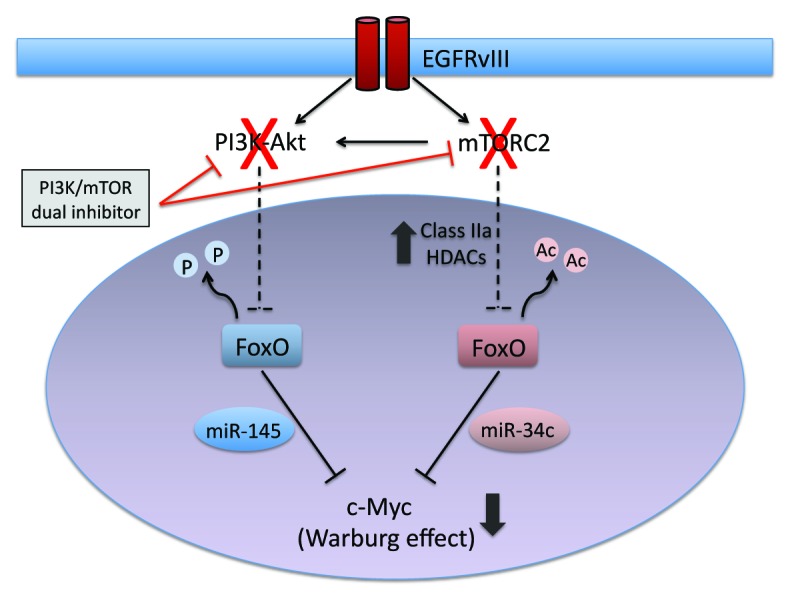
Figure 1. mTORC2 is a kingpin of the Warburg effect and drug resistance. mTORC2 inhibits FoxO via acetylation, whereas PI3K/Akt via phosphorylation, leading to the upregulation of c-Myc, a key effector of the Warburg effect. PI3K/Akt-targeted therapies alone can be bypassed by mTORC2-mediated pathways, whereas targeting both PI3K/Akt and mTORC2 potently suppresses c-Myc and the Warburg effect.
Masui K, et al. Cell Metab. 2013;18:726–39. doi: 10.1016/j.cmet.2013.09.013.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/28377
References
- 1.Warburg O. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 2.Laplante M, et al. Cell. 2012;149:274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanaka K, et al. Cancer Discov. 2011;1:524–38. doi: 10.1158/2159-8290.CD-11-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Masui K, et al. Cell Metab. 2013;18:726–39. doi: 10.1016/j.cmet.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dang CV, et al. Clin Cancer Res. 2009;15:6479–83. doi: 10.1158/1078-0432.CCR-09-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Babic I, et al. Cell Metab. 2013;17:1000–8. doi: 10.1016/j.cmet.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cloughesy TF, et al. Annu Rev Pathol. 2014;9:1–25. doi: 10.1146/annurev-pathol-011110-130324. [DOI] [PubMed] [Google Scholar]
- 8.Masui K, et al. Carcinogenesis. 2013;34:725–38. doi: 10.1093/carcin/bgt086. [DOI] [PMC free article] [PubMed] [Google Scholar]