

Abstract
The condensin complex is required for chromosome condensation during mitosis; however, the role of this complex during interphase is unclear. Neuroblastoma is the most common extracranial solid tumor of childhood, and it is often lethal. In human neuroblastoma, MYCN gene amplification is correlated with poor prognosis. This study demonstrates that the gene encoding the condensin complex subunit SMC2 is transcriptionally regulated by MYCN. SMC2 also transcriptionally regulates DNA damage response genes in cooperation with MYCN. Downregulation of SMC2 induced DNA damage and showed a synergistic lethal response in MYCN-amplified/overexpression cells, leading to apoptosis in human neuroblastoma cells. Finally, this study found that patients bearing MYCN-amplified tumors showed improved survival when SMC2 expression was low. These results identify novel functions of SMC2 in DNA damage response, and we propose that SMC2 (or the condensin complex) is a novel molecular target for the treatment of MYCN-amplified neuroblastoma.
Keywords: condensin complex, DNA damage response, MYCN, neuroblastoma, synergistic lethal response
Introduction
The condensin complex, which is highly conserved from bacteria to humans, is essential for proper chromosome condensation and segregation during mitosis and meiosis. There are 2 condensin complexes in human, condensin I and condensin II. These complexes share 2 common core subunits, SMC2/CAP-E and SMC4/CAP-C, and have 3 unique non-SMC subunits. Condensin I contains CAP-D2, CAP-H, and CAP-G, while condensin II contains CAP-D3, CAP-H2, and CAP-G2.1
It has recently been reported that the condensin complex also has a number of functions during interphase, including DNA repair and transcriptional regulation. Human condensin I is required for the recruitment of PARP-1 to single-strand DNA breaks in HeLa and 293T cells.2 In fission yeast, Cnd2 (human CAP-H), which is the non-SMC subunit of condensin, is synthetic lethal with the DNA replication protein RecQ helicase (human WRN, BLM and RecQL proteins) and is required for Cds1 (human CHK2) activation.3 Furthermore, the fission yeast SMC2 homolog Cut14 is required for DNA annealing.4 Human CAP-G2 is involved in the transcriptional regulation of c-kit in erythroid cells via an interaction with an erythroid-lineage-specific bHLH transcription factor.5 In addition, a genome-wide analysis of budding yeast revealed that the chromosomal condensin pattern does not alter during the cell cycle, and that the minimal condensin-binding consensus comprises a B-box element recognized by RNA polymerase III transcription factor.6
Although a condensin knockout mouse model is currently unavailable,1 knockdown of Smc2 induces cell death in mouse embryonic stem cells but not in immortalized mouse embryonic fibroblasts.7 These findings suggest that the condensin complex is not essential for viability and may be differentially regulated across tissues or during development.
The MYC family of proteins comprises MYC (c-myc), MYCN, and MYCL. MYCN encodes a transcription factor with a β-helix-loop-helix domain that is specifically expressed in neuronal tissues. Multiple target genes are regulated by MYC, including DNA damage response (DDR) genes.8-12 Cancer cells undergo many stresses, including oxidative and replicative stress.13 According to the oncogene-induced DNA damage model of cancer development,14 genomic instability is induced by oncogenes themselves. In fact, MYC induces DNA damage through reactive oxygen species (ROS) production15 and replicative stress.16
The DDR is a network of signaling pathways involved in DNA damage repair, cell cycle checkpoints, and apoptosis.17 The MRN complex has been implicated in all aspects of DNA double-strand break (DSB) processing, including initial detection, triggering signaling pathways, and facilitating repair. The MRN complex also activates ataxia-telangiectasia mutated (ATM) and related kinases that promote rapid phosphorylation of multiple proteins and of chromatin structure around the break sites. The 2 major DSB repair pathways are homologous recombination and non-homologous end-joining (NHEJ).18 BRCA1 is a versatile protein that links DNA damage sensing and DDR effectors. This protein is directly involved in homologous recombination-mediated repair of DSBs and may also function in other DNA repair pathways, including NHEJ and single-strand annealing.
Inhibiting genes that are synthetic lethal with cancer-associated mutations should exclusively kill cancer cells; therefore, identification of such genes is important for identifying new therapeutic targets.19 One of the most well-characterized therapeutic combinations comprises a BRCA1 mutation and a poly-ADP-ribose polymerase inhibitor.20,21 To date, multiple specific combinations of genes have been found to show synergistic lethal responses with MYC or MYCN.22-31
Neuroblastoma originates from precursor cells of the sympathetic nervous system. MYCN oncogene amplification and mutations in the gene encoding anaplastic lymphoma kinase (ALK) are both critically involved in the development of a high-risk clinical phenotype and poor survival probabilities.32-36 There are several animal models of neuroblastoma, including MYCN and mutated ALK transgenic mice.37 MYCN transgenic (Tg) mice, in which MYCN expression is targeted to the sympathetic neuron lineage by rat tyrosine hydroxylase,38 serve as a model of neuroblastoma. These mice develop aggressive neuroblastomas and tumorigenesis, positively correlated with the MYCN transgene dosage or the development of additional genetic mutations.39
Here, we show that SMC2 regulates several DDR genes in cooperation with MYCN, and that knockdown of SMC2 has a synergistic lethal effect with MYCN amplification. SMC2 controls several DDR genes simultaneously; therefore, it may be an effective molecular target for the treatment of MYCN-amplified cancers. In addition, we show that patients bearing MYCN-amplified tumors tend to benefit from low SMC2 expression. The results presented here suggest that SMC2 (or the condensin complex) is a molecular target of MYCN-amplified cancers.
Results
Smc2 expression in neuroblastoma model mice and human neuroblastoma cell lines
To gain insights into the molecular pathways governing neuroblastoma development, the expression profiles of superior mesenteric ganglia of 2-wk-old wild-type (wt) mice, precancerous lesions of 2-wk-old homozygote MYCN Tg mice, and terminal tumors of 6-wk-old homozygote MYCN Tg mice were examined (GSE43419). The expression levels of 79 genes were higher in precancerous lesions and tumors of MYCN Tg mice than in ganglia of wt mice. Among these genes, Smc2 was selected and characterized further. The level of Smc2 expression gradually increased as the severity of the disease progressed (Fig. 1A). To confirm this finding, semi-quantitative and quantitative RT-PCR (RT-qPCR) analyses of precancerous lesions of 2-wk-old MYCN hemizygous mice were performed (Fig. 1B); these lesions are reportedly similar to human MYCN-amplified neuroblastoma.39 Consistent with the microarray data, mouse Smc2 was highly expressed in the precancerous lesion samples (Fig. 1B).
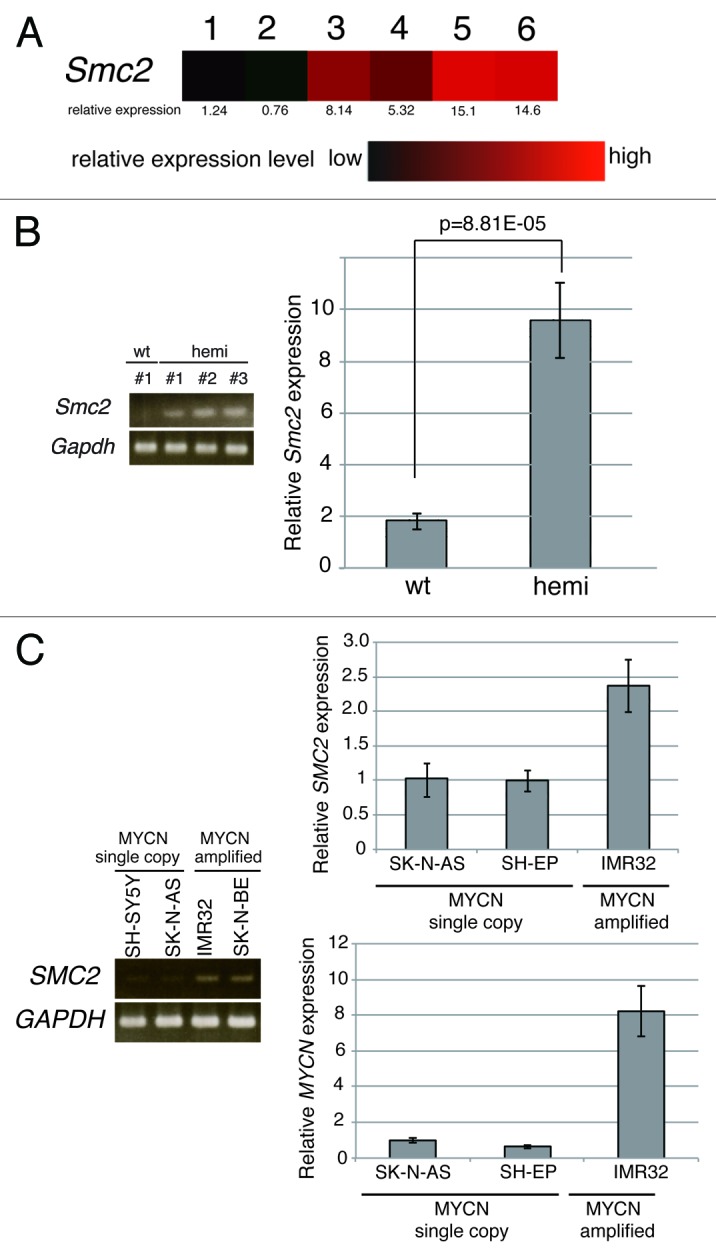
Figure 1.Smc2 expression in neuroblastoma model mice and SMC2 expression in human neuroblastoma cell lines. (A) Results of a microarray analysis of the relative expression levels of Smc2 in ganglia of wt mice (lanes 1 and 2), and precancerous (lanes 3 and 4) and tumor lesions (lanes 5 and 6) of homozygous MYCN Tg mice. (B) Semi-quantitative (left) and quantitative (right) RT-PCR analyses of Smc2 and Gapdh (control) expression levels in 3 precancerous lesion samples from hemizygous MYCN Tg mice (hemi) and ganglia of wt mouse. (C) Semi-quantitative (left) and quantitative (right) RT-PCR analyses of human SMC2 expression levels in various human neuroblastoma cell lines. SH-SY5Y, SK-N-AS, and SH-EP cells have a single copy of MYCN, and IMR32 and SK-N-BE(2) have amplified MYCN. The expression levels of Smc2 and SMC2 detected by RT-qPCR were normalized to those of Gapdh and GAPDH respectively.
The expression of SMC2 was then examined in human neuroblastoma cell lines. Human SMC2 expression was higher in MYCN-amplified cell lines (IMR32 and SK-N-BE[2]) than in MYCN single copy cell lines (SH-SY5Y, SK-N-AS and SH-EP) (Fig. 1C). These results indicate that SMC2 expression is elevated in MYCN Tg mice and MYCN-amplified human neuroblastoma cells and is correlated with MYCN expression.
Overexpression of MYCN induces SMC2 expression in human neuroblastoma cells
To determine whether overexpression of MYCN induces SMC2 expression, a CMV-driven MYCN plasmid or a CMV-Venus plasmid (as a control) were introduced into SH-EP MYCN single copy cells using a lentivirus. After live cell sorting, RT-PCR and immunoblotting analyses were performed. The levels of SMC2 mRNA and protein were higher in SH-EP cells constitutively expressing MYCN than in control cells (Fig. 2A and B).

Figure 2. Overexpression of MYCN induces SMC2 expression in human neuroblastoma cells. (A and B) A CMV-driven plasmid containing MYCN was introduced into the SH-EP MYCN single copy cell line. After live cell sorting, the levels of human SMC2 mRNA and protein were measured by semi-quantitative (A, left) and quantitative (A, right) RT-PCR, as well as by immunoblotting (B). The arrowhead in (B) indicates a non-specific band. The expression levels of SMC2 detected by RT-qPCR were normalized to those of GAPDH. (C) Schematic representation of the E-boxes identified in the human SMC2 gene. The light gray boxes indicate the 5′ and 3′ UTRs; the dark gray boxes indicate the exons; and the black circles represent putative E-boxes (MYCN-binding sites). The sequences of the E-boxes are shown. (D) ChIP followed by qPCR analysis of the E-boxes in the SMC2 gene was performed using control IgG or an anti-MYCN antibody in SH-EP cells expressing Venus (control) or MYCN. The 5-kb sequence upstream of the SMC2 gene was examined as a negative control. The data show the percentage of the target DNA precipitated with the control IgG or MYCN antibody and are represented as the mean ± SE of at least n = 3 independent experiments.
MYC binds to a canonical consensus DNA sequence (CACGTG) named the E-box. MYC also binds to several other non-canonical DNA motifs in vitro, including CATGTG, CATGCG, CACGCG, CACGAG, and CAACGTG.40 Three canonical E-boxes (E-box1, E-box2, and E-box3) were detected within the introns of SMC2 (Fig. 2C). A chromatin immunoprecipitation (ChIP) assay was used to examine whether MYCN is able to bind to the identified E-boxes (Fig. 2D). When MYCN was overexpressed, the amount of MYCN that bound to E-box2 increased (Fig. 2D). In the control cells, the amount of MYCN bound to E-box2 was also higher than the amount of IgG antibody bound to E-box2 (Fig. 2D); however, this difference is likely due to non-specific binding, because the amount of MYCN bound to the 5-kb upstream region also increased (Fig. 2D). These results indicate that MYCN regulates SMC2 expression by binding to its E-box2 motif.
Knockdown of SMC2 induces DNA damage and apoptosis
A short hairpin RNA (shRNA) targeting SMC2 inhibited proliferation of MYCN-overexpressing SH-EP cells but not of MYCN single copy control SH-EP cells (Fig. 3A). By contrast, the proliferation of MYCN-overexpressing SH-EP cells and control SH-EP cells infected with a non-target shRNA was similar (Fig. 3A), indicating that overexpression MYCN and a reduction in SMC2 expression has a synergistic lethal response. The knockdown efficiency of each cell is shown in Figure 3B.

Figure 3A–C. Knockdown of SMC2 induces DNA damage and apoptosis. (A) Growth of MYCN-overexpressed SH-EP cells and control SH-EP cells infected with non-target shRNA or SMC2-specific shRNAs. Counting started 3 d after infection. On each day, 3 plates were counted and averaged. Data are shown as a ratio of the number of cells at 3 d after transfection and are represented as the mean ± SD of n = 3 independent repeats. (B) SMC2 knockdown efficiency of (A). (C) TUNEL staining of apoptotic IMR32 cells at 6 d after infection (upper panel) and quantification of apoptotic IMR32 cells at the indicated time-points (lower panel). On each day, 3 plates were counted and averaged. (D) Immunofluorescence of γ-H2AX and DAPI staining of IMR32 (MYCN-amplified), SK-N-BE(2) (MYCN-amplified), SK-N-AS (MYCN-single copy), and SH-EP (MYCN-single copy) cells infected with non-target or SMC2-specific shRNA. Images were captured 3 d after infection. (E) Quantification of the γ-H2AX-positive cells shown in (D). Data are represented as the mean ± SD of n = 3 independent repeats. Their homoscedasticities were checked by f test. Statistical significance was evaluated with a 2-tailed, unpaired t test. (F) The percentage viability of SH-EP or MYCN overexpressed SH-EP cells infected with non-target or SMC2-specific shRNA and treated with cisplatin (left) or campthotecin (right). Data are represented as the mean ± SE of n = 3 independent experiments. Their homoscedasticities were checked by f test. Statistical significance was evaluated with a 2-tailed, unpaired t test.
Next, a TUNEL assay was used to determine whether knockdown of SMC2 and amplification of MYCN causes apoptosis. Very few FITC-positive cells were detected in non-infected or non-target shRNA-infected IMR32 cells, but almost all cells infected with SMC2-specific shRNA were FITC-positive (Fig. 3C). Most of the SMC2-knockdown IMR32 cells were TUNEL-positive at 6 d after virus infection. DNA damage induces apoptosis;41 therefore, the level of DNA damage in these cells was also examined. Histone H2A phosphorylation at a serine residue (γ-H2AX) is a sensitive marker of DSBs.42 The number of γ-H2AX-positive cells was markedly higher in SMC2-knockdown IMR32 and SK-N-BE(2) cells than in IMR32 and SK-N-BE(2) cells infected with a non-specific shRNA (Fig. 3D and E); however, the number of γ-H2AX-positive cells was not increased by knockdown of SMC2 in the SK-N-AS and SH-EP cell lines. Most of the SMC2-knockdown IMR32 cells, and approximately 40% of the non-target shRNA-infected IMR32 cells, displayed γ-H2AX foci. The mechanism that induced DNA damage in the non-target lentivirus-infected MYCN-amplified cells is unknown. Despite this outcome, the results suggest that knockdown of SMC2 induces additional DNA damage in MYCN-amplified cells.

Figure 3D. See Figure 3A–Clegend.
The induction of ROS production or replication stress by MYC causes DNA damage.15,16 We hypothesized that overexpression of MYCN also induces DNA damage, most of which is repaired in the normal condition, thereby allowing MYCN-amplified cells to survive. However, once SMC2 expression is reduced, cells are unable to repair the DNA damage, resulting in subsequent cell death. If these hypotheses are correct, SMC2-knockdown cells would be expected to be sensitive to DNA damage even in MYCN single copy neuroblastoma cell lines. This hypothesis was tested by treating SH-EP cells, which contain a single copy of MYCN, with cisplatin and camptothecin to cause DNA damage. Cisplatin mainly causes intrastrand cross-linking,43 whereas camptothecin, a topoisomerase I inhibitor, causes replication-dependent DSBs.44 Treatment of SMC2-knockdown SH-EP cells with cisplatin or camptothecin decreased cell viability (Fig. 3F, upper panels), and treatment of SMC2-knockdown MYCN-overexpressed SH-EP cells with cisplation or camptothecin was more sensitive (Fig. 3F, lower panels). These results suggest that MYCN-induced DNA damage is required for the synergistic lethal response with SMC2 knockdown.
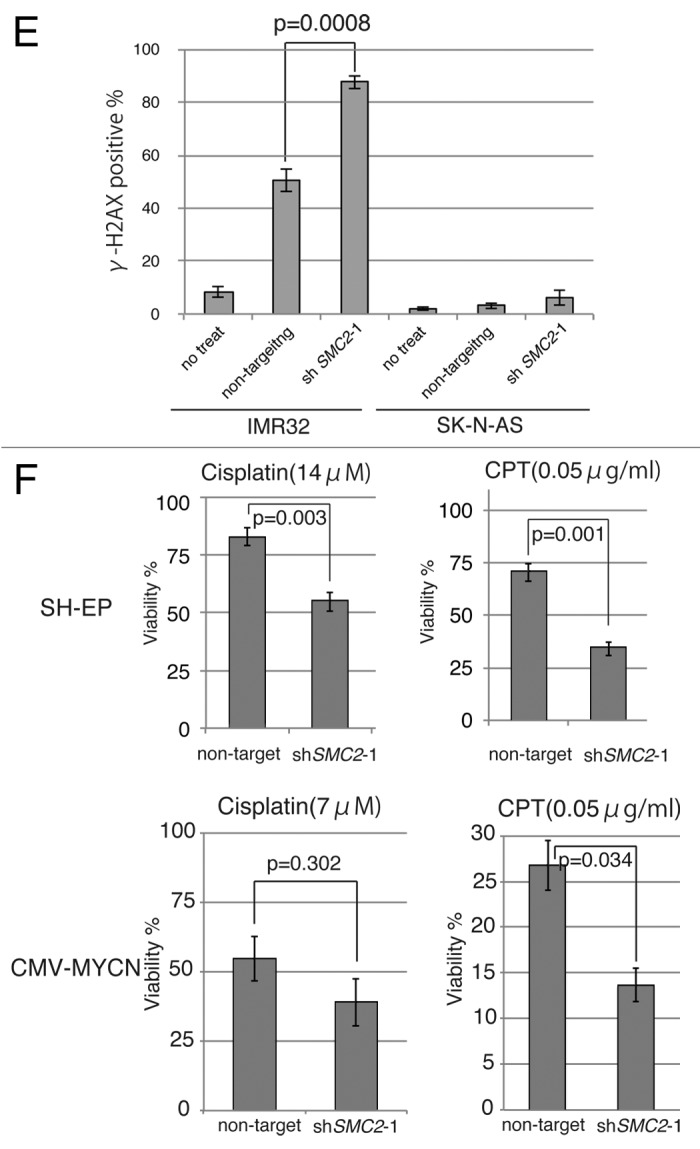
Figure 3E and F. See Figure 3A–Clegend.
SMC2 regulates the expression of DDR genes
We next investigated how SMC2 is involved in DNA damage repair. Table 1 shows mini-ontology data of 3 cohorts, which are available at the R2 microarray analysis and visualization platform (http://r2.amc.nl). The first cohort consisted of 88 neuroblastoma patients from the Academic Medical Center in Amsterdam,45 the second cohort consisted of 101 neuroblastoma patients from the Children’s Hospital of Philadelphia,46 and the third cohort consisted of 30 neuroblastoma patients from the Leeds and Newcastle NHS Trusts.47
Table 1. Mini-ontology of genes correlated with SMC2 expression from published human neuroblastoma expression array data sets (GSE16476, GSE3960, GSE13136) created using the R2 bioinformatic platform (http://r2.amc.nl).
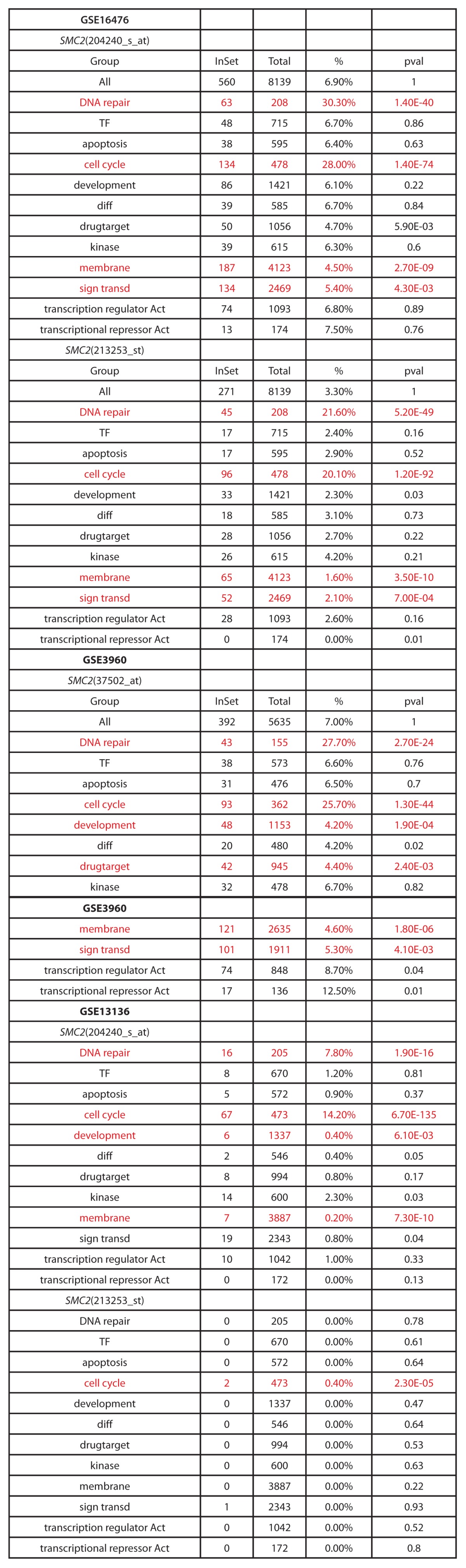
DDR genes are involved in DNA repair. DDR gene expression increased in parallel with SMC2 expression (Table 1), suggesting a relationship between SMC2 expression and DDR gene expression. Some DDR genes, including BRCA1, MRE11, NBS1, RAD50, and ATM, are transcriptionally regulated by MYC.8-12 To determine whether SMC2 is involved in controlling the transcription of DDR genes, qPCR analyses were used to measure the expression levels of some DDR genes regulated by MYC when SMC2 was knocked down in single copy or in MYCN-amplified neuroblastoma cell lines. In IMR32 cells, knockdown of SMC2 did not affect β-ACTIN transcription, but the levels of BRCA1, MRE11, NBS1, RAD50, and ATM mRNAs decreased (Fig. 4A, left panel). By contrast, knockdown of SMC2 in SK-N-AS cells had a smaller effect on the expression levels of these genes (Fig. 4A, right panel). Similar results were obtained when experiments were performed with MYCN-overexpressed SH-EP cells and control SH-EP cells (Fig. 4B). These results suggest that SMC2 regulates DDR gene expression in cooperation with MYCN.

Figure 4A and B. SMC2 interacts with MYCN and transcriptionally regulates DDR genes. (A) RT-qPCR analysis of the relative expression levels of DDR genes in IMR32 cells (MYCN-amplified, top-left panel) and SK-N-AS cells (MYCN single copy, top-right panel) infected with non-targeting or SMC2-specific shRNA. Expression levels in SMC2-specific shRNA-infected cells were normalized to those in the control cells. Bottom panels are shown SMC2 knockdown efficiency in each cells. (B) RT-qPCR analysis of the relative expression levels of DDR genes in MYCN-overexpressed SH-EP cells and control SH-EP cells as similar in (A). (C) Pull-down assay showing that MYCN interacts with SMC2 and SMC4. CMV-driven MYCN, along with CMV-driven Halo-control, Halo-SMC2 or Halo-SMC4 and Halo-MYCN proteins were expressed in 293T cells. A pull-down assay was performed using a Halo-tag. The proteins were detected with the indicated antibodies. WCE (TMR), TMR Direct ligand stained Halo-tag proteins in whole cell extract. (D) SMC2 and MYCN bind to the E-box motif in the NBS1 gene. The left panel shows the results of a ChIP assay of the E-box in the NBS1 gene performed using an anti-MYCN antibody in cells expressing Venus (control) or MYCN. The right panel shows the results of a ChIP assay of the E-box in the NBS1 gene performed using an anti-SMC2 antibody in MYCN-amplified IMR32 cells. A region situated 14.5 kb upstream of the E-box was used as a negative control. Data show the percentages of the target DNA precipitated with the antibodies and are represented as the mean ± SE of at least n = 3 independent qPCR experiments. (E) The protein level of SMC4 and CAP-D2 when SMC2 is knockdown in MYCN expressing SH-EP (MYCN single copy) cells and control Venus expressing SH-EP cells. β-ACTIN is used as loading control.
MYCN interacts with SMC2 and SMC4
The possibility of an interaction between SMC2 and MYCN was investigated using a pull-down assay. The results indicated that SMC2, SMC4, and MYCN all interacted with each other (Fig. 4C). Choi and colleagues reported that MYCN associates with SMC4 and SMC2 in FLAG-tagged MYCN-expressing HeLa cells,48 suggesting that this interaction occurs in multiple cell types.

Figure 4C–E. See Figure 4A and Blegend.
The MYC binding site in the NBS1 gene is the E-box within intron 1.11 To test whether MYCN also binds to this motif, a ChIP analysis was performed using an anti-MYCN antibody. MYCN bound to the E-box motif in the NBS1 gene (Fig. 4D, left panel). SMC2 bound to the same motif (Fig. 4D, right panel). These results suggest that SMC2 regulates NBS1 transcription in cooperation with MYCN.
In addition, we found that the protein level of condensin subunit decreased when SMC2 knockdown (Fig. 4E). This suggests that condensin complex becomes unstable when SMC2 is lost.
Genome-wide analysis of genes regulated by SMC2
To identify the genes regulated by SMC2, RNA-seq of SMC2-knockdown SH-EP cells constitutively expressing MYCN or Venus as a control was performed (SRA081723). A remarkable number of the genes that were induced or repressed in these cells were related to the DDR, DNA repair, or the cell cycle (Table 2; Table S1). The results indicated that knockdown of SMC2 had a larger effect on ATM and RAD50 transcription in SH-EP cells overexpressing MYCN than in the control cells, which agrees with the results of the RT-qPCR analyses shown in Figure 4A. These results suggest that SMC2 effects the expression of a number of genes involved in DNA repair, the DDR, and the cell cycle.
Table 2. List of induced or repressed GO categolies belonging to DDR, DNA repair, and cell cycle-related classes in SMC2-knockdown Venus- or MYCN-expressing SH-EP cells. Non-target shRNA-infected cells were used as a control.
| Gene ontology classes enriched in SMC2-knockdown Venus-expressing SH-EP cells | ||||
|---|---|---|---|---|
| GO term | Total No. genes in set | No. genes regulated | P value | |
| cell cycle | 828 | 225 | 2.55E-12 | |
| cell cycle process | 647 | 185 | 5.77E-12 | |
| cell cycle phase | 548 | 158 | 2.52E-10 | |
| mitotic cell cycle | 470 | 139 | 5.97E-10 | |
| DNA metabolic process | 594 | 157 | 1.58E-07 | |
| regulation of cell cycle | 526 | 139 | 2.00E-06 | |
| M phase | 344 | 97 | 1.95E-05 | |
| chromosome | 471 | 124 | 1.72E-05 | |
| interphase of mitotic cell cycle | 257 | 76 | 7.92E-05 | |
| M phase of mitotic cell cycle | 246 | 73 | 1.19E-04 | |
| interphase | 261 | 76 | 1.37E-04 | |
| G1/S transition of mitotic cell cycle | 126 | 45 | 1.21E-04 | |
| mitosis | 239 | 71 | 1.44E-04 | |
| chromosome, centromeric region | 124 | 44 | 1.72E-04 | |
| chromosome segregation | 98 | 37 | 2.82E-04 | |
| cell division | 295 | 82 | 3.30E-04 | |
| condensed chromosome, centromeric region | 69 | 29 | 3.80E-04 | |
| chromosome organization | 518 | 125 | 0.001 | |
| mitotic prometaphase | 73 | 29 | 0.001 | |
| regulation of cell cycle process | 292 | 78 | 0.003 | |
| condensed chromosome | 124 | 41 | 0.003 | |
| condensed chromosome kinetochore | 64 | 25 | 0.008 | |
| DNA replication | 147 | 45 | 0.008 | |
| response to DNA damage stimulus | 427 | 103 | 0.009 | |
| DNA conformation change | 159 | 47 | 0.013 | |
| chromatin | 206 | 56 | 0.028 | |
| negative regulation of cell cycle | 194 | 53 | 0.037 | |
| cell cycle checkpoint | 180 | 50 | 0.037 | |
| DNA repair | 287 | 72 | 0.041 | |
| kinetochore | 75 | 26 | 0.047 | |
| chromatin remodeling | 67 | 24 | 0.049 | |
| Gene ontology classes enriched in SMC2-knockdown MYCN-expressing SH-EP cells | ||||
| GO term | Total No. genes in set | No. genes regulated | P value | |
| gene expression | 1680 | 581 | 3.86E-15 | |
| response to DNA damage stimulus | 427 | 179 | 1.29E-10 | |
| DNA metabolic process | 594 | 223 | 1.36E-10 | |
| cell cycle process | 647 | 244 | 4.60E-09 | |
| mitotic cell cycle | 470 | 188 | 4.64E-09 | |
| cell cycle | 828 | 299 | 5.90E-09 | |
| chromatin modification | 328 | 139 | 2.72E-08 | |
| DNA repair | 287 | 124 | 7.03E-08 | |
| covalent chromatin modification | 194 | 90 | 3.67E-07 | |
| histone modification | 192 | 89 | 4.78E-07 | |
| cell cycle phase | 548 | 204 | 8.24E-07 | |
| regulation of gene expression | 2625 | 796 | 1.02E-06 | |
| chromosome organization | 518 | 194 | 1.18E-06 | |
| chromatin organization | 396 | 154 | 3.28E-06 | |
| transcription factor binding | 284 | 116 | 1.18E-05 | |
| regulation of cell cycle | 526 | 191 | 2.23E-05 | |
| cell division | 295 | 118 | 3.28E-05 | |
| mitosis | 239 | 98 | 1.05E-04 | |
| chromosome | 471 | 171 | 1.05E-04 | |
| M phase of mitotic cell cycle | 246 | 100 | 1.25E-04 | |
| chromosome, centromeric region | 124 | 58 | 2.26E-04 | |
| regulation of transcription, DNA-dependent | 2324 | 692 | 3.00E-04 | |
| transcription cofactor activity | 345 | 130 | 3.10E-04 | |
| protein binding transcription factor activity | 352 | 132 | 3.42E-04 | |
| transcription factor binding transcription factor activity | 349 | 131 | 3.52E-04 | |
| condensed chromosome, centromeric region | 69 | 37 | 4.79E-04 | |
| interphase of mitotic cell cycle | 257 | 101 | 6.15E-04 | |
| interphase | 261 | 102 | 7.27E-04 | |
| M phase | 344 | 127 | 0.001 | |
| regulation of gene expression, epigenetic | 96 | 46 | 0.001 | |
| chromatin remodeling | 67 | 35 | 0.002 | |
| chromosomal part | 389 | 140 | 0.002 | |
| negative regulation of gene expression | 580 | 197 | 0.002 | |
| mitotic prometaphase | 73 | 37 | 0.002 | |
| DNA-dependent transcription, initiation | 76 | 38 | 0.002 | |
| positive regulation of transcription, DNA-dependent | 664 | 220 | 0.003 | |
| condensed chromosome kinetochore | 64 | 33 | 0.004 | |
| cell cycle checkpoint | 180 | 72 | 0.008 | |
| posttranscriptional regulation of gene expression | 243 | 92 | 0.008 | |
| transcription, DNA-dependent | 794 | 254 | 0.01 | |
| condensed chromosome | 124 | 53 | 0.011 | |
| regulation of transcription from RNA polymerase II promoter | 792 | 253 | 0.011 | |
| positive regulation of gene expression | 712 | 230 | 0.011 | |
| chromosome segregation | 98 | 44 | 0.012 | |
| transcription initiation from RNA polymerase II promoter | 51 | 27 | 0.012 | |
| regulation of cell cycle process | 292 | 106 | 0.013 | |
| transcription coactivator activity | 197 | 76 | 0.016 | |
| regulation of cell cycle arrest | 188 | 73 | 0.017 | |
| spindle | 160 | 64 | 0.018 | |
| DNA recombination | 130 | 54 | 0.02 | |
| kinetochore | 75 | 35 | 0.025 | |
| microtubule organizing center | 326 | 114 | 0.035 | |
| negative regulation of transcription, DNA-dependent | 521 | 171 | 0.039 | |
| protein serine/threonine kinase activity | 364 | 125 | 0.039 | |
| DNA damage response, signal transduction by p53 class mediator | 71 | 33 | 0.04 | |
| G2/M transition of mitotic cell cycle | 101 | 43 | 0.049 | |
Analysis of the potential involvement of SMC2 in neurogenesis
High-risk neuroblastomas may have defects in neuritogenesis genes.49 During zebrafish development, smc2 and smc4 are expressed in some regions of the central nervous system,50 and a mutation in the microcephalin gene disregulates condensin II and causes autosomal recessive primary microcephaly.51 Therefore, we re-analyzed mouse expression array data published in ArrayExpress (E-GEOD-11356, http://www.ebi.ac.uk/microarray-as/ae/). Mycn was highly expressed in E8.5 (neural tube closure) samples and gradually decreased in E13.5 (dorsal root ganglion) and P90 (adrenal medulla) samples. The expression pattern of Smc2 was similar to that of Mycn (Fig. S1). These data suggest that, along with MYCN, SMC2 (or the condensin complex) may have a role in sympathetic neurogenesis.
Clinical data
We hypothesized that patients bearing MYCN-amplified tumors would benefit from low SMC2 expression. Therefore, we investigated whether SMC2 expression correlated with patient prognosis in a previous cohort used by Wang et al.46 In MYCN-amplified patients, low levels of SMC2 were associated with good prognosis (overall survival, P = 0.051; event-free survival, P = 0.053) (Fig. 5A). These results indicate that low SMC2 expression in MYCN-amplified patients tends to correlate with good prognosis.

Figure 5. Clinical data showing the relationship between SMC2 expression and patient prognosis in the Wang cohort.48 (A) The effects of SMC2 expression on the overall survival (OS) and event-free survival (EFS) rates of patients bearing MYCN-amplified and non-amplified tumors. Within each of the 2 tumor subsets considered, those with expression levels of SMC2 greater than the median (blue or green line) were compared with the remainder of the tumors in the subset (red or purple line) using a Kaplan–Meier analysis. (B) Expression levels of condensin I- and condensin II-specific subunits and their relationship to MYCN amplification or expression. The data were obtained from a published data set (GSE3960). The red line indicates low MYCN expression or no MYCN amplification, and the blue line indicates high MYCN expression or MYCN amplification.
The expression levels of other condensin I and II subunits were higher in precancerous and tumor lesions from MYCN Tg mice than in wt mouse ganglion samples; however, the difference was less pronounced than that observed for Smc2 (Fig. S2). Table S2 shows mini-ontology data of three cohorts available at the R2 bioinformatic platform (http://r2.amc.nl). The expression levels of most of the condensin I and II subunits correlated with DDR gene expression. We also examined the expression levels of the condensin subunits in the Wang cohort.46 Expression of the condensin I-specific subunits was related to the level of MYCN amplification or expression, whereas the expression of condensin II-specific subunits was not (Fig. 5B). Figure S3 shows the relationship among MYCN expression, condensin subunit expression, and prognosis of 101 neuroblastoma patients included in the Wang cohort.46 We separated the samples into 2 groups, namely MYCN high expression and MYCN low expression. If patients bearing MYCN-high tumors benefit from low condensin subunit expression, only the subunit-high group of MYCN-high patients would show poor prognosis. That means only MYCN-high group would show the significant difference (P < 0.05). Figure S3 shows that patients expressing high levels of MYCN and condensin I subunit (CAP-D2) or SMC4 had poor prognosis, although prognosis of those expressing condensin I subunit (CAP-H) was not always related to MYCN expression. However, prognosis of patients expressing condensin II subunit was not related to MYCN expression. These results suggest that SMC2 may function as a condensin complex (probably as condensin I), rather than alone, in this phenotype.
Discussion
This study demonstrates that (1) SMC2 is regulated by MYCN; (2) SMC2, in cooperation with MYCN, regulates DDR genes; and (3) downregulation of SMC2 and concomitant MYCN amplification induces DNA damage and has a synergistic lethal effect, resulting in apoptosis of human neuroblastoma cells. This study also demonstrates that patients bearing MYCN-amplified tumors tend to benefit from low SMC2 expression. A model summarizing these findings is shown in Figure 6. In MYCN-amplified cells, MYCN induces DNA damage, most likely via the production of ROS and replicative stress. On the other hand, since SMC2 is overexpressed in these cells, the DDR may also be highly activated. As a consequence, cells are viable, but some mutations would still remain (Fig. 6A, left). In this situation, knockdown of SMC2 would impair the DDR, resulting in cell death (Fig. 6A, right). By contrast, in MYCN single copy cells, the extent of DNA damage is lower, and knockdown of SMC2 does not affect cell viability (Fig. 6B).
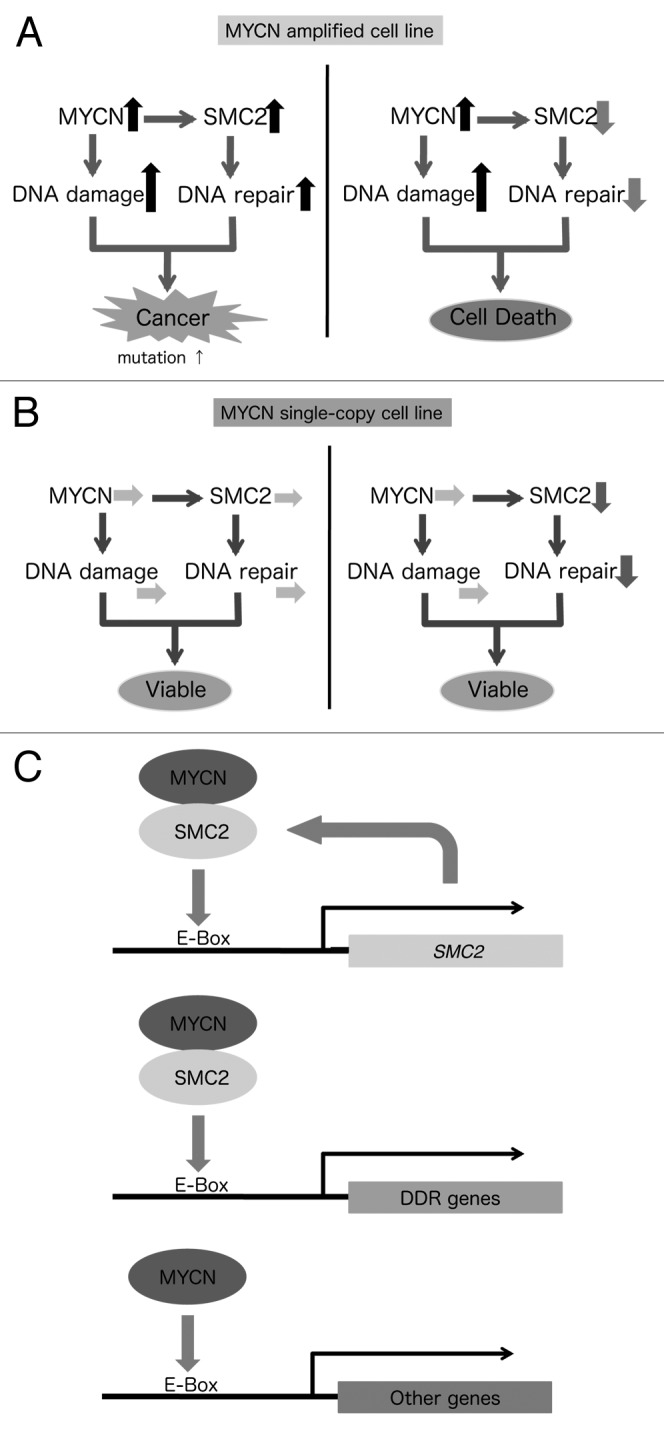
Figure 6. Schematic model showing the synergistic effects of SMC2 and MYCN in MYCN-amplified (A) and MYCN single copy (B) cells. This model is based on the proposed transcriptional regulation by MYCN and SMC2 (C).
Several reports have suggested that the condensin complex is required for the recruitment of DNA repair proteins to damage sites.2,52,53 In this study, we found that SMC2 interacts with MYCN and is involved in transcriptional regulation of DDR genes, and that SMC2 expression is regulated by MYCN as well as by Wnt54 (Fig. 6C). There are several advantages of using SMC2 as a molecular target for the treatment of MYCN-amplified neuroblastoma. First, knockdown of SMC2 has similar effects to knockdown of several DDR genes. Second, if MYC functions similarly to MYCN, inhibition of SMC2 would be expected to have growth-inhibitory activity in MYC-overexpressed/amplified cancers. If so, SMC2 knockdown is a powerful tool for both MYCN- and MYC-overexpressed/amplified cancers.
There are still some unanswered questions. First, other than MYCN, which other transcription factors interact with SMC2 or the condensin complex? CAP-G associates with bHLH transcription factors in erythroid cells;5 therefore, SMC2 (or the condensin complex) may co-operate with various transcription factors in different tissues or developmental stages. The second question relates to the mechanism by which SMC2 is involved in transcriptional regulation. SMC2 associates with MYCN, but its specific role in the MYCN complex remains unknown. SMC2 may act as a co-factor or alter the chromosomal conformation. Third, what is the functional difference between MYC and MYCN? MYCN-amplified neuroblastoma cells express low levels of MYC, while MYCN single copy neuroblastoma cells express higher levels.24 As described above, knockdown of SMC2 had a synergistic lethal effect with amplification or overexpression of MYCN, but this effect was not seen in MYCN single copy cells. This result implies that knockdown of SMC2 is synergistic lethal with MYCN but not with MYC. It might also suggest that the gene set that shows a synergistic lethal effect with MYCN is different from that associated with MYC amplification or overexpression. CDK2,26 Aurora A,27 and CHK122 kinases show specific synergistic lethal responses with MYCN, but not with MYC in MYCN single copy cells. However, another report demonstrated that MYC and MYCN are functionally redundant to some extent,55 and the gene(s) that are synthetic lethal with MYC overexpression in human foreskin fibroblasts are also synthetic lethal with MYCN, but not with MYC in MYCN single copy cells.24,56 Further studies are required to elucidate the relationship between MYC and MYCN.
Materials and Methods
Mice
MYCN Tg mice38 were maintained in the animal facility at Nagoya University Graduate School of Medicine where they were housed in a controlled environment and provided with standard nourishment and water. Normal ganglia and precancerous and tumor tissues from wt, hemizygous, and homozygous MYCN Tg mice were dissected and minced, and then total RNA was extracted. This study was approved by the Animal Care and Use Committee of Nagoya University Graduate School of Medicine, Nagoya, Japan.
Gene expression profiling
Total RNA was isolated using ISOGEN reagent (Nippon Gene), according to the manufacturer's instructions. Samples were analyzed with a GeneChip Mouse Genome 430 2.0 array (Affymetrix). Preparation of target cDNA from total RNA, hybridization to the microarray, washing, staining with an antibody amplification procedure, and scanning were all performed according to the manufacturer's instructions. The expression value (Signal) of each probe set was calculated using GeneChip Operating Software (GCOS) version 1.3 (Affymetrix). The data described in this manuscript have been deposited in the Gene Expression Omnibus database of NCBI (http://www.ncbi.nlm.nih.gov/geo) under the accession number GSE43419.
Cell lines, virus infection, and transfection
IMR32 cells were obtained from JCRB (JCRB9050); 293T (RCB2202) cells were obtained from the RIKEN Cell Bank; SH-SY5Y, SK-N-AS, and SK-N-BE(2) cells were purchased from ATCC (CRL-2266, CRL-2137, and CRL-2271); SH-EP cells were a gift from Dr Schwab (Division of Tumor Genetics, German Cancer Research Center), and NB39 cells were a gift from Dr Chiba (Fukushima Medical University).
IMR32 cells were grown in minimum essential medium (Sigma) supplemented with 10% fetal bovine serum (HyClone, Thermo Scientific) and 1% non-essential amino acids (GIBCO-Life Technologies). SK-N-AS cells were grown in Dulbecco modified Eagle medium (MP Biomedicals, LLC) supplemented with 10% fetal bovine serum and 1% non-essential amino acids. SK-N-BE(2) and SH-SY5Y cells were grown in a 1:1 ratio of minimum essential medium and Ham-F12 (GIBCO-Life Technologies) supplemented with 10% fetal bovine serum (HyClone, Thermo scientific), 1% sodium pyruvate (Sigma), 1% GlutaMAX (GIBCO-Life Technologies), and 1% non-essential amino acids. 293T cells were grown in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum. SH-EP cells were grown in RPMI1640 (Sigma) supplemented with 10% fetal bovine serum.
Replication-defective, self-inactivating lentiviral vectors were used.57,58 HEK293T cells were co-transfected with shRNA plasmids in addition to psPAX2 (Addgene) and pMD2.G (Addgene) or cDNA expression plasmids . Neuroblastoma cell lines were infected in the presence of 15 μg/ml polybrene (Sigma). 293T cells were transfected with various plasmids using FuGENE HD reagent (Promega), according to the manufacturer’s instructions.
Plasmids
Constructs were generated by PCR using specific primers and a gateway cassette containing a full-length clone of human MYCN. This fragment was initially cloned into pDONR221 (Invitrogen) and then into the CSII-CMV-RfA-IRES-Venus plasmid. The successful introduction of this fragment into each construct was confirmed by DNA sequencing. The CSII-CMV-RfA-IRES-Venus plasmid and the CSII-CMV-Venus control plasmid were kindly provided by H Miyoshi (RIKEN). Commercially available non-targeting (Sigma) and SMC2-specific shRNA vectors (TRCN0000062540, TRCN0000062542, TRCN0000062538; Thermo Scientific Open Biosystems) were used. The shRNA sequences are listed in Table S3.
For pull-down and ChIP assays, the following Halo-tag Flexi ORF clones (Promega) were used: Halo-control (G6591), Halo-SMC2 (pFN21AE9255), Halo-SMC4 (pFN21AB7320), and Halo-MYCN (pFN21AB8396). A list of the plasmids used in the study is provided in Table S4. Further details are available upon request.
DNA damaging agents
Cells were treated with cisplatin (NIPPON-KAYAKU) or camptothecin (Sigma). Treatments were performed at 37 °C for overnight and at 14 μM (for SH-EP cells) or 7 μM (for MYCN-overexpressed SH-EP cells) cisplatin and 0.05 μg/ml camptothecin.
Primers
The sequences of the primers used in the study are listed in Table S5.
Isolation of RNA, RT-PCR, and qPCR
Total RNA was isolated from cells or tissues using ISOGEN II reagent (Nippongene, Japan). For RT-PCR and RT-qPCR analyses, total RNA (1 μg) was incubated with DNase I (Invitrogen) to eliminate contaminating genomic DNA, and then reverse transcribed with oligo(dT) and random hexamer primers and the ThermoScript RT-PCR system (Invitrogen). Quantitative PCR analyses were performed using an Mx3000p or Mx3005p instrument (Agilent Technologies) and the KAPA SYBR Fast qPCR kit (KAPA Biosystems). The expression levels of mouse Smc2 and Gapdh (control), and human SMC2, MYCN, GAPDH (control), BRCA1, MRE11, NBS1, RAD50, ATM, and β-ACTIN were determined using the ΔΔCT method.
TUNEL assay
The TUNEL assay was performed using the APO-DIRECT kit (556381, BD Pharmingen). The manufacturer’s protocol for cell fixation and staining was followed and then the APO-DIRECT samples were analyzed using a FACSCanto flow cytometer (BD Pharmingen) and Diva software.
Antibodies
The following antibodies were used: anti-MYCN monoclonal antibodies (OP13, Calbiochem and NB200-109, Novus Biologicals); anti-MYCN monoclonal antibody (B8.4.B) (sc-53993, Santa Cruz); anti-SMC2 rabbit polyclonal antibodies (GTX10411, GeneTex; and ab10412, Abcam); anti-β-ACTIN monoclonal antibody (AC-15) (A5441, Sigma); anti-SMC4 rabbit polyclonal antibody (ab17958, Abcam); anti-CAP-D2 rabbit polyclonal antibody (A300-601A, Bethyl Laboratories); and anti-phospho-histone H2A.X (Ser139) mouse monoclonal antibody (JBW301, Millipore).
Immunofluorescence
For immunofluorescence, cells were grown on glass coverslips in 4- or 8-well plates. The coverslips were washed twice with phosphate-buffered saline (PBS) and then fixed with 4% paraformaldehyde for 1 h at room temperature. Fixed cells were washed with PBS a further 3 times and permeabilized with PBS containing 1% Triton X-100 for 30 min at room temperature. The cells were incubated with an anti γ-H2AX antibody (1:1000 dilution) for 1 h at room temperature and then with Alexa Fluor 488-conjugated anti-mouse IgG (Molecular Probes) (1:1000 dilution) for 30 min at room temperature. After incubation for 5 min with 0.1 μg/ml 4'6-diamidino-2-phenylindole, cells were mounted in FluorSave Reagent (Millipore). All images were subsequently processed using MetaMorph and Adobe Photoshop.
Protein preparation and immunoblotting
Cells were washed with ice-cold PBS and then lysed with RIPA buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% NP-40, 1% deoxycholic acid sodium salt monohydrate, 0.1% sodium dodecyl sulfate, and 1% protease inhibitor cocktail). Cell lysates were centrifuged at 4 °C for 10 min at 22,140 g and then analyzed by 10% PAGE. Proteins were transferred to PVDF membranes, which were then blocked for 1 h with PBS containing 5% non-fat dried milk and 0.05% Tween-20. The membranes were washed, probed with primary antibodies, and then exposed to horseradish peroxidase-conjugated secondary antibodies. Blots were visualized using ECL reagent (GE Healthcare) and autoradiography film.
Pull-down assay
The HaloTag Mammalian Pull-Down System (Promega) was used according to the manufacturer's instructions. CMV-driven MYCN with CMV-driven Halo-control, Halo-SMC2, or Halo-SMC4, and Halo-MYCN proteins were expressed in 293T cells. Cells were harvested 64 h after transfection, according to the manufacturer’s protocol. Briefly, HaloTag fusion proteins, along with their interacting proteins, were captured using the HaloLink resin and washed gently. Interacting proteins were eluted from the resin with SDS elution buffer and subjected to SDS-PAGE followed by electroblotting.
ChIP
Sub-confluent SH-EP cells expressing Venus or MYCN and IMR32 were cultured in 10-cm dishes and treated with 1% (v/v) formaldehyde for 10 min at room temperature. Cross-linking was stopped by adding glycine to a final concentration of 125 mM. The cells were washed with cold PBS and then harvested. The cells were pelleted, frozen at −70 °C, and then lysed by mechanical disruption. The Halo–ChIP system (Promega) was then used according to the manufacturer’s instructions, except that an anti-MYCN antibody and Dynabeads-protein G (Dynal) were used. DNA was purified twice using phenol-CIAA. The purified DNA was PCR-amplified using primers spanning the MYCN-binding site in the SMC2 and NBS1 genes. Cells immunoprecipitated with control IgGs were used as a negative control.
RNA-seq analysis
Cells were harvested 72 h after infection with non-target or SMC2-specific lentiviruses. Total RNA was prepared from the cells using the RNeasy mini kit (Qiagen), according to the manufacturer's instruction. The TruSeq RNA Sample Preparation Kit v2 (Illumina, Inc) was used to prepare RNA-Seq libraries following the manufacturer’s instructions. Libraries were sequenced with the Illumina HiSeq 2500 sequencer for 50-bp single read. The data described in this manuscript have been deposited in the Sequence Read Archive database of NCBI (http://www.ncbi.nlm.nih.gov/sra) under the accession number SRA081723.
Analysis of human neuroblastoma tumor profiles
Three clinical neuroblastoma gene expression data sets were used for the analysis. As described by Molenaar et al.49 (accession number: GSE16476, www.ncbi.nih.gov/geo), Wang et al.59 (accession number: GSE3960, www.ncbi.nih.gov/geo), and Lastowska et al.47 (accession number: GSE13136, www.ncbi.nih.gov/geo), the basis of these data sets differs markedly, both with respect to the selection of patient cohorts and to the microarray technologies used. The Molenaar data included 88 patients and microarrays were performed using the Affymetrix Human Genome U133 Plus 2.0 array. The Wang data included 101 cases, and microarrays were performed using the Affymetrix HgU95Av2 array. The Lastowska data included 30 neuroblastomas and microarrays were performed using the Affymetrix Human Genome U133 Plus 2.0 array. For the Molenaar, Wang, and Lastowska studies, the normalized microarray data deposited in the Gene Expression Omnibus database were used. For the Wang data, quality filtration was applied as described previously.46
In Figure 5A, we analyzed the gene expression and survival data in Wang cohort study.48 We divided the entire sample into four groups: (red) those with non-MYCN amplification and the SMC2 expression levels lower than the median value; (blue) those with non-MYCN amplification and the SMC2 expression levels higher than the median value; (green) those with MYCN amplification and the SMC2 expression levels higher than the median value; (purple) those with MYCN amplification and the SMC2 expression levels lower than the median value. Then, the significance of the difference in survival between red vs. blue and green vs. purple are examined by using Kaplan–Meyer plots and Log-rank test. The top 2 panels are plots with overall survival, whereas the bottom 2 panels are with event-free survival. In Figure 5B, we divided the entire sample of published data set (GSE3960) into those with higher and lower MYCN expressions than the median value. The plotted are the density estimates (density esimate is considered as a smoothed histogram which can be computed by using kernel density estimation algorithm kde in R) of condensin I- and condensin II-specific subunits. The given P values dictate the significance of the difference between 2 expression densities (by using U test).
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Y Watanabe (University of Tokyo) for critical reading of the manuscript. We also thank K Yokomori (UC Irvine) for providing the SMC2 plasmid; D Trono (Swiss Federal Institute of Technology, Switzerland) for providing the lentiviral packaging and production plasmids psPAX2 and pMD2.G; H Miyoshi (Subteam for Manipulation of Cell Fate, RIKEN BioResource Center, Japan) and A Miyawaki (RIKEN Brain Science Institute, Japan) for providing the CSII-CMV-RfA-IRES2-Venus and CSII-CMV-Venus vectors; KJ Wu (National Yang Ming University) and M Fujii (Aichi Cancer Center) for providing plasmids; the Cell Bank of RIKEN BioResource Center (Japan), the Health Science Research Resource Bank (Japan), JCRB (Japan), ATCC, M Schwab (German Cancer Research Center [DKFZ]), and H Chiba (Fukushima Medical University) for providing cells; F Ohka and K Katsushima (Aichi Cancer Center Research Institute) for technical support for the GO analysis; MS Jackson and M Lastowska (Institute of Human Genetics, New Castle University) for providing the survival data of GSE13136; and the Children’s Oncology Group, Children's Hospital of Philadelphia, University of Pennsylvania for clinical and outcome data. We thank M Foiani, F d’Adda di Fagagna, S Minucchi, and C-F Cheok (IFOM, Italy) for communicating unpublished results, and all members of our laboratory for their support and discussions. This work was funded in part by a grant from the Global COE Program (Integrated functional molecular medicine for neuronal and neoplastic disorders) to Nagoya University Graduate School of Medicine, a grant-in-aid from the National Cancer Center Research and Development Fund (22-4 to K.K.) and a Grant-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan (No. 24590376 to Y.M-T. and No. 23110002 and 20390092 to K.K.).
Author Contributions
Y.M-T., H.M., and K.K. conceived the project. Y.M-T. performed most of the experimental procedures and analyzed the data. I.T. performed the statistical analysis. Y.K. and K.S. performed RNA-seq. J-M.M. provided the prognosis data of the neuroblastoma patients. S.K. and H.I. performed the expression array of MYCN-Tg mice. Y.K. helped the data analysis of RNA-seq. Y.S. analyzed the data. Y.M-T. and H.M. wrote the manuscript. K.K. supervised the project.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/27983
References
- 1.Hirano T. Condensins: universal organizers of chromosomes with diverse functions. Genes Dev. 2012;26:1659–78. doi: 10.1101/gad.194746.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heale JT, Ball AR, Jr., Schmiesing JA, Kim JS, Kong X, Zhou S, Hudson DF, Earnshaw WC, Yokomori K. Condensin I interacts with the PARP-1-XRCC1 complex and functions in DNA single-strand break repair. Mol Cell. 2006;21:837–48. doi: 10.1016/j.molcel.2006.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aono N, Sutani T, Tomonaga T, Mochida S, Yanagida M. Cnd2 has dual roles in mitotic condensation and interphase. Nature. 2002;417:197–202. doi: 10.1038/417197a. [DOI] [PubMed] [Google Scholar]
- 4.Akai Y, Kurokawa Y, Nakazawa N, Tonami-Murakami Y, Suzuki Y, Yoshimura SH, Iwasaki H, Shiroiwa Y, Nakamura T, Shibata E, et al. Opposing role of condensin hinge against replication protein A in mitosis and interphase through promoting DNA annealing. Open Biol. 2011;1:110023. doi: 10.1098/rsob.110023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu Y, Leung CG, Lee DC, Kennedy BK, Crispino JD. MTB, the murine homolog of condensin II subunit CAP-G2, represses transcription and promotes erythroid cell differentiation. Leukemia: official journal of the Leukemia Society of America. Leukemia Research Fund, UK. 2006;20:1261–9. doi: 10.1038/sj.leu.2404252. [DOI] [PubMed] [Google Scholar]
- 6.D’Ambrosio C, Schmidt CK, Katou Y, Kelly G, Itoh T, Shirahige K, Uhlmann F. Identification of cis-acting sites for condensin loading onto budding yeast chromosomes. Genes Dev. 2008;22:2215–27. doi: 10.1101/gad.1675708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fazzio TG, Panning B. Condensin complexes regulate mitotic progression and interphase chromatin structure in embryonic stem cells. J Cell Biol. 2010;188:491–503. doi: 10.1083/jcb.200908026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Menssen A, Hermeking H. Characterization of the c-MYC-regulated transcriptome by SAGE: identification and analysis of c-MYC target genes. Proc Natl Acad Sci U S A. 2002;99:6274–9. doi: 10.1073/pnas.082005599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B. Genomic targets of the human c-Myc protein. Genes Dev. 2003;17:1115–29. doi: 10.1101/gad.1067003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Z, Van Calcar S, Qu C, Cavenee WK, Zhang MQ, Ren B. A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc Natl Acad Sci U S A. 2003;100:8164–9. doi: 10.1073/pnas.1332764100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiang YC, Teng SC, Su YN, Hsieh FJ, Wu KJ. c-Myc directly regulates the transcription of the NBS1 gene involved in DNA double-strand break repair. J Biol Chem. 2003;278:19286–91. doi: 10.1074/jbc.M212043200. [DOI] [PubMed] [Google Scholar]
- 12.Perini G, Diolaiti D, Porro A, Della Valle G. In vivo transcriptional regulation of N-Myc target genes is controlled by E-box methylation. Proc Natl Acad Sci U S A. 2005;102:12117–22. doi: 10.1073/pnas.0409097102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–5. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 15.Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, Wahl GM. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell. 2002;9:1031–44. doi: 10.1016/S1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
- 16.Herold S, Herkert B, Eilers M. Facilitating replication under stress: an oncogenic function of MYC? Nat Rev Cancer. 2009;9:441–4. doi: 10.1038/nrc2640. [DOI] [PubMed] [Google Scholar]
- 17.Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–33. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 19.Kaelin WG., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–98. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 20.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 21.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 22.Cole KA, Huggins J, Laquaglia M, Hulderman CE, Russell MR, Bosse K, Diskin SJ, Attiyeh EF, Sennett R, Norris G, et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc Natl Acad Sci U S A. 2011;108:3336–41. doi: 10.1073/pnas.1012351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Höglund A, Strömvall K, Li Y, Forshell LP, Nilsson JA. Chk2 deficiency in Myc overexpressing lymphoma cells elicits a synergistic lethal response in combination with PARP inhibition. Cell Cycle. 2011;10:3598–607. doi: 10.4161/cc.10.20.17887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Toyoshima M, Howie HL, Imakura M, Walsh RM, Annis JE, Chang AN, Frazier J, Chau BN, Loboda A, Linsley PS, et al. Functional genomics identifies therapeutic targets for MYC-driven cancer. Proc Natl Acad Sci U S A. 2012;109:9545–50. doi: 10.1073/pnas.1121119109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murga M, Campaner S, Lopez-Contreras AJ, Toledo LI, Soria R, Montaña MF, D’Artista L, Schleker T, Guerra C, Garcia E, et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat Struct Mol Biol. 2011;18:1331–5. doi: 10.1038/nsmb.2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Molenaar JJ, Ebus ME, Geerts D, Koster J, Lamers F, Valentijn LJ, Westerhout EM, Versteeg R, Caron HN. Inactivation of CDK2 is synthetically lethal to MYCN over-expressing cancer cells. Proc Natl Acad Sci U S A. 2009;106:12968–73. doi: 10.1073/pnas.0901418106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otto T, Horn S, Brockmann M, Eilers U, Schüttrumpf L, Popov N, Kenney AM, Schulte JH, Beijersbergen R, Christiansen H, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell. 2009;15:67–78. doi: 10.1016/j.ccr.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 28.Beierle EA, Trujillo A, Nagaram A, Kurenova EV, Finch R, Ma X, Vella J, Cance WG, Golubovskaya VM. N-MYC regulates focal adhesion kinase expression in human neuroblastoma. J Biol Chem. 2007;282:12503–16. doi: 10.1074/jbc.M701450200. [DOI] [PubMed] [Google Scholar]
- 29.Rottmann S, Wang Y, Nasoff M, Deveraux QL, Quon KC. A TRAIL receptor-dependent synthetic lethal relationship between MYC activation and GSK3beta/FBW7 loss of function. Proc Natl Acad Sci U S A. 2005;102:15195–200. doi: 10.1073/pnas.0505114102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kessler JD, Kahle KT, Sun T, Meerbrey KL, Schlabach MR, Schmitt EM, Skinner SO, Xu Q, Li MZ, Hartman ZC, et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science. 2012;335:348–53. doi: 10.1126/science.1212728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Engels IH, Knee DA, Nasoff M, Deveraux QL, Quon KC. Synthetic lethal targeting of MYC by activation of the DR5 death receptor pathway. Cancer Cell. 2004;5:501–12. doi: 10.1016/S1535-6108(04)00113-8. [DOI] [PubMed] [Google Scholar]
- 32.George RE, Sanda T, Hanna M, Fröhling S, Luther W, 2nd, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455:975–8. doi: 10.1038/nature07397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Janoueix-Lerosey I, Lequin D, Brugières L, Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455:967–70. doi: 10.1038/nature07398. [DOI] [PubMed] [Google Scholar]
- 34.Mossé YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455:930–5. doi: 10.1038/nature07261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455:971–4. doi: 10.1038/nature07399. [DOI] [PubMed] [Google Scholar]
- 36.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369:2106–20. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 37.Cheung N-KV, Dyer MA. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer. 2013;13:397–411. doi: 10.1038/nrc3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997;16:2985–95. doi: 10.1093/emboj/16.11.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hansford LM, Thomas WD, Keating JM, Burkhart CA, Peaston AE, Norris MD, Haber M, Armati PJ, Weiss WA, Marshall GM. Mechanisms of embryonal tumor initiation: distinct roles for MycN expression and MYCN amplification. Proc Natl Acad Sci U S A. 2004;101:12664–9. doi: 10.1073/pnas.0401083101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blackwell TK, Huang J, Ma A, Kretzner L, Alt FW, Eisenman RN, Weintraub H. Binding of myc proteins to canonical and noncanonical DNA sequences. Mol Cell Biol. 1993;13:5216–24. doi: 10.1128/mcb.13.9.5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stracker TH, Petrini JH. The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol. 2011;12:90–103. doi: 10.1038/nrm3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–67. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Basu A, Krishnamurthy S. Cellular responses to Cisplatin-induced DNA damage. J Nucleic Acids. 2010;2010 doi: 10.4061/2010/201367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shao RG, Cao CX, Zhang H, Kohn KW, Wold MS, Pommier Y. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. EMBO J. 1999;18:1397–406. doi: 10.1093/emboj/18.5.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang S, Laoukili J, Epping MT, Koster J, Hölzel M, Westerman BA, Nijkamp W, Hata A, Asgharzadeh S, Seeger RC, et al. ZNF423 is critically required for retinoic acid-induced differentiation and is a marker of neuroblastoma outcome. Cancer Cell. 2009;15:328–40. doi: 10.1016/j.ccr.2009.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Q, Diskin S, Rappaport E, Attiyeh E, Mosse Y, Shue D, Seiser E, Jagannathan J, Shusterman S, Bansal M, et al. Integrative genomics identifies distinct molecular classes of neuroblastoma and shows that multiple genes are targeted by regional alterations in DNA copy number. Cancer Res. 2006;66:6050–62. doi: 10.1158/0008-5472.CAN-05-4618. [DOI] [PubMed] [Google Scholar]
- 47.Łastowska M, Viprey V, Santibanez-Koref M, Wappler I, Peters H, Cullinane C, Roberts P, Hall AG, Tweddle DA, Pearson AD, et al. Identification of candidate genes involved in neuroblastoma progression by combining genomic and expression microarrays with survival data. Oncogene. 2007;26:7432–44. doi: 10.1038/sj.onc.1210552. [DOI] [PubMed] [Google Scholar]
- 48.Choi SH, Wright JB, Gerber SA, Cole MD. Myc protein is stabilized by suppression of a novel E3 ligase complex in cancer cells. Genes Dev. 2010;24:1236–41. doi: 10.1101/gad.1920310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J, Westerman BA, van Arkel J, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012;483:589–93. doi: 10.1038/nature10910. [DOI] [PubMed] [Google Scholar]
- 50.Mönnich M, Banks S, Eccles M, Dickinson E, Horsfield J. Expression of cohesin and condensin genes during zebrafish development supports a non-proliferative role for cohesin. Gene Expr Patterns. 2009;9:586–94. doi: 10.1016/j.gep.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 51.Marchal JA, Ghani M, Schindler D, Gavvovidis I, Winkler T, Esquitino V, Sternberg N, Busche A, Krawitz P, Hecht J, et al. Misregulation of mitotic chromosome segregation in a new type of autosomal recessive primary microcephaly. Cell Cycle. 2011;10:2967–77. doi: 10.4161/cc.10.17.16871. [DOI] [PubMed] [Google Scholar]
- 52.Chen ES, Sutani T, Yanagida M. Cti1/C1D interacts with condensin SMC hinge and supports the DNA repair function of condensin. Proc Natl Acad Sci U S A. 2004;101:8078–83. doi: 10.1073/pnas.0307976101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Przewloka MR, Pardington PE, Yannone SM, Chen DJ, Cary RB. In vitro and in vivo interactions of DNA ligase IV with a subunit of the condensin complex. Mol Biol Cell. 2003;14:685–97. doi: 10.1091/mbc.E01-11-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dávalos V, Súarez-López L, Castaño J, Messent A, Abasolo I, Fernandez Y, Guerra-Moreno A, Espín E, Armengol M, Musulen E, et al. Human SMC2 protein, a core subunit of human condensin complex, is a novel transcriptional target of the WNT signaling pathway and a new therapeutic target. J Biol Chem. 2012;287:43472–81. doi: 10.1074/jbc.M112.428466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Malynn BA, de Alboran IM, O’Hagan RC, Bronson R, Davidson L, DePinho RA, Alt FW. N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev. 2000;14:1390–9. [PMC free article] [PubMed] [Google Scholar]
- 56.Westermann F, Muth D, Benner A, Bauer T, Henrich KO, Oberthuer A, Brors B, Beissbarth T, Vandesompele J, Pattyn F, et al. Distinct transcriptional MYCN/c-MYC activities are associated with spontaneous regression or malignant progression in neuroblastomas. Genome Biol. 2008;9:R150. doi: 10.1186/gb-2008-9-10-r150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miyoshi H, Takahashi M, Gage FH, Verma IM. Stable and efficient gene transfer into the retina using an HIV-based lentiviral vector. Proc Natl Acad Sci U S A. 1997;94:10319–23. doi: 10.1073/pnas.94.19.10319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miyoshi H, Blömer U, Takahashi M, Gage FH, Verma IM. Development of a self-inactivating lentivirus vector. J Virol. 1998;72:8150–7. doi: 10.1128/jvi.72.10.8150-8157.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang K, Diskin SJ, Zhang H, Attiyeh EF, Winter C, Hou C, Schnepp RW, Diamond M, Bosse K, Mayes PA, et al. Integrative genomics identifies LMO1 as a neuroblastoma oncogene. Nature. 2011;469:216–20. doi: 10.1038/nature09609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.