

Abstract
Metastatic prostate cancer (PC) is lethal and lacks effective strategies for prevention or treatment, requiring novel therapeutic approaches. Interleukin-6 (IL-6) is a cytokine which has been linked with PC pathogenesis by multiple studies. However, the direct functional role(s) of IL-6 in PC growth and progression have been unclear. In the present study, we show that IL-6 is produced in distant metastases of clinical PCs. IL-6 activated signaling pathways in PC cells induced a robust 7-fold increase in metastases formation in nude mice. We further show that IL-6 promoted migratory PC cell phenotype, including increased PC cell migration, microtubule re-organization and heterotypic adhesion of PC cells to endothelial cells. IL-6-driven metastasis was predominantly mediated by Stat3 and to lesser extent by ERK1/2. Most importantly, pharmacological inhibition of Jak1/2 by AZD1480 suppressed IL-6-induced signaling, migratory PC cell phenotypes and metastatic dissemination of PC in vivo in nude mice. In conclusion, we demonstrate that the cytokine IL-6 directly promotes PC metastasis in vitro and in vivo via Jak-Stat3 signaling pathway, and that IL-6-driven metastasis can be effectively suppressed by pharmacological targeting of Jak1/2 using Jak1/2 inhibitor AZD1480. Our results therefore provide a strong rationale for further development of Jak1/2 inhibitors as therapy for metastatic PC.
Keywords: Prostate cancer metastases, Jak1/2-inhibition, IL-6
INTRODUCTION
Development of metastatic disease is one of the key challenges in clinical management of prostate cancer (PC) (1). While localized PC can be effectively managed by surgery and radiation, the single greatest cause of PC morbidity and mortality is due to formation of distant metastases (2-6). Identification of the molecular changes leading to metastatic dissemination of PC is critical for development of therapeutic interventions to prevent PC progression to the lethal metastatic form of the disease.
Interleukin-6 (IL-6) is a multifunctional cytokine implicated in PC pathogenesis (7). IL-6 and IL-6 receptors are expressed in PC, with elevated expression in high Gleason score PCs (8, 9). Concomitantly, IL-6 has been found to be elevated in the sera of patients with castrate-resistant prostate cancer (CRPC), and IL-6 serum levels correlate with markers of PC progression (10-16). Furthermore, IL-6 is expressed at high levels in the androgen-independent PC cell lines PC3 and DU145 (15), while the androgen-dependent PC cell line LNCaP expresses lower IL-6 levels (17). IL-6 binds to the IL-6 receptor, a complex which is composed of two subunits: IL-6-R alpha (IL-6–specific) and gp130 (shared by IL-6 and related cytokines) (5, 18, 19). IL-6 first binds to the IL-6R alpha subunit (20), which then leads to recruitment of the signal-transducing gp130 subunit. Association of gp130 with IL-6 and IL-6R alpha leads to formation of the high-affinity IL-6 receptor complex and homodimerization of two gp130 subunits, resulting in activation of downstream Jak1/2-Stat3, ERK1/2-MAPK and/or PI3K-Akt signaling in PC cells (5, 19, 21-24). In androgen receptor (AR)-positive PC cell lines, it has been suggested that some of the biological effects of IL-6 are mediated via IL-6 induction of AR signaling (25-27).
The existing literature on IL-6 regulation of PC cell growth and viability have been controversial, including reports documenting both growth-stimulatory and inhibitory effects (28-34). Our previous work demonstrated that Stat3 activation predominantly affects PC progression by inducing metastatic behavior of PC cells in vitro and massive metastasis formation in vivo, rather than promoting PC cell growth (35, 36). Given that Stat3 is one of the key IL-6-activated signaling molecules in PC, we hypothesized that IL-6 promotes metastatic progression of PC.
Here, we show that the IL-6 protein is expressed in the majority of distant metastases of clinical PCs. IL-6 induced a robust increase in PC metastasis formation in nude mice in an experimental metastasis assay, which was accompanied by IL-6-driven PC cell migration and heterotypic adhesion in vitro. We further demonstrate that IL-6 stimulation leads to activation of Jak1/2 and predominantly Stat3 signaling pathway in AR-negative and AR-positive PC cells. Most important from a therapeutic perspective, pharmacological targeting of Jak1/2 by a potent adenosine triphosphate (ATP)-competitive small-molecule inhibitor of Jak1/2 kinase, AZD1480 (37) prevented IL-6-driven development of a migratory PC cell phenotype in vitro and suppressed IL-6-driven metastatic dissemination of PC in nude mice. In summary, Jak1/2 may represent a therapeutic target proteins for PC, and prevention of metastatic progression of primary PC in patients could potentially be achieved by small-molecule Jak1/2 inhibitors currently in clinical development (38, 39).
MATERIALS AND METHODS
Clinical samples of distant metastases of PCs
Archival and de-identified, formalin-fixed, paraffin-embedded specimens of distant metastases of clinical PCs (n=87) (regional lymph nodes, n=36; bone, n=3; other organs, n=48) (Table 1) were obtained from University of Turku in Finland. The immunohistochemical analysis of the de-identified archival tissues was granted an exemption from full IRB review by Thomas Jefferson University.
Table 1.
IL-6 expression in distant metastases of clinical prostate cancers.
| No. of patients | % | |
|---|---|---|
| Prostate cancer metastases (lymph node metastases, bone metastases and metastases to other organs): | ||
| IL-6 expression status; | 87 | 100 |
| Negative | 19 | 22 |
| Positive | 68 | 78 |
| Prostate cancer metastases to regional lymph nodes: | ||
| IL-6 expression status; | 36 | 100 |
| Negative | 4 | 11 |
| Positive | 32 | 89 |
| Prostate cancer metastases to bone: | ||
| IL-6 expression status; | 3 | 100 |
| Negative | 1 | 33 |
| Positive | 2 | 67 |
| Prostate cancer metastases to other organs: | ||
| IL-6 expression status; | 48 | 100 |
| Negative | 14 | 29 |
| Positive | 34 | 71 |
Cell culture and reagents
Human prostate cancer cell lines CWR22Rv1, DU145 and LNCaP (ATCC, Manassas, VA) were cultured in RPMI 1640 (Mediatech, Herndon, VA, #15-041-CV) containing 10% fetal bovine serum (FBS; Quality Biological, Gaithersburg, MD, #100106) and penicillin/streptomycin (Mediatech, Inc., 50 IU/ml and 50 μg/ml, respectively, #30-002-CL). LNCaP cells were cultured in the presence of 0.5 nM dihydrotestosterone (DHT; Sigma, St. Louis, MO, #512-18-6). CWR22Rv1 cells were obtained in 2005 from Dr. Thomas Pretlow (Casewestern Reserve Univ.) and LNCaP and DU145 cells in 2009 from ATCC. AZD1480 (37) was provided by AstraZeneca. IL-6 producing lentivirus (LV-II-human-IL-6 virus (3.8 × 108 IU/ml) was purchased from Capital Biosciences (Rockville, MD) and recombinant IL-6 was purchased from ProSpec Protein Specialists (Brunswick, NJ, #CYT-213).
Immunostaining of paraffin-embedded tissue sections
Immunohistochemistry of distant PC metastases was performed as described previously (35, 36, 40-42). The primary antibody IL-6 mAb (Santa Cruz Biotechnology; sc-130326) was used at a concentration of 5μg/ml.
Enzyme-linked immunosorbent assay (ELISA)
DU145 were mock-infected or infected with lentivirus expressing the IL-6 gene (MOI=5), followed by collection of cell culture media at 4, 10 and 20 days and analysis for IL-6 protein levels by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN, USA, #Q6000B).
Homotypic cell adhesion assay
DU145 and CWR22Rv1 cells were seeded on Matrigel (BD Biosciences, #354234), serum-starved for 18 h and treated with recombinant IL-6 (5 nM) for 24, 48, 72 or 96 h.
Heterotypic cell adhesion assay
DU145 cells, cultured in the presence of 1% charcoal-stripped fetal bovine serum (CS-FBS), were infected with IL-6 and Stat3-shRNA and/or ERK1/2-shRNA lentiviruses at (MOI=5) and/or treated with recombinant IL-6 (5 nM) with or without AZD1480 (800 nM). DU145 and CWR22Rv1 cell adhesion to the endothelial cells was determined as described previously (43) and in the Suppl. Methods.
Immunofluorescence cytochemistry of tubulin
DU145 cells grown on cover glasses were serum-starved for 18 h, treated with recombinant IL-6 (5 nM) and fixed 48 h after IL-6 treatment with 4% paraformaldehyde. Tubulin immunocytochemistry was performed as described previously and in the Suppl. Methods.
Boyden chamber migration assay
DU145 and CWR22Rv1 cells were cultured in 1% CS-FBS, phenol-red free RPMI medium in the presence or absence of IL-6 (5 nM) for 72 h. In the indicated treatment groups, the cells had been infected with lentiviruses expressing control-shRNA, Stat3-shRNA and/or ERK1/2-shRNA 24 h prior to the start of the IL-6 treatment. Also, in the indicated treatment groups, AZD1480 (800 nM) had been added to the culture medium 1 h prior to the start of the IL-6 treatment. The cells were counted and suspended to the upper chambers (2.5 × 104 cells/chamber) of the motility chamber system (8.0 μm pore size; BD BioSciences, #353097) using FBS (10%) as the chemoattractant, as described previously (35, 43). After 16 h, the cells that had traversed the membrane pores were fixed, stained and counted. Each experiment was repeated four times.
Protein solubilization, immunoprecipitation and immunoblotting
CWR22Rv1, DU145 and LNCaP cells were solubilized and immunoprecipitations and immunoblottings were performed as described previously (35, 36, 43-46). Antibodies used for immunoprecipitation and immunoblotting are described in Suppl. Methods.
Cell viability assay
DU145 cells were treated with AZD1480 or DMSO (as vehicle control) or infected with IL-6 expressing lentivirus (MOI=5) for 72 h. Cell viability was analyzed by the CellTiter 96® AQueous Assay kit (Promega, #G3580) according to the manufacturer’s protocol.
Tail-vein injections of human prostate cancer cells
Male athymic nude mice (for DU145 cells) were purchased from Taconic (Germantown, NY) and SCID mice (for CWR22Rv1 cells) were purchased from the Jackson Laboratory (Bar Harbor, ME), and cared for according to institutional guidelines. DU145 and CWR22Rv1 cells were infected with IL-6 lentivirus (MOI=5) alone or in combination with Stat3-shRNA lentivirus (MOI=5) and/or ERK1/2-shRNA (MOI=5) lentivirus. After 24 h, 1 × 106 cells were suspended in 0.2 ml of PBS and injected into the lateral tail veins of nude mice using a 27-gauge needle, as described previously (35, 36, 43). In some of the experiments, the mice were treated daily by oral gavage of AZD1480 at 50 mg/kg body weight or vehicle (0.5% HPMC/0.1% Tween-80) starting on day 3 until the mice were sacrificed (8 weeks after cell inoculation). The lungs were perfused with 1.5 ml of 15% India Ink dye in 3.7% formalin, removed and bleached in Fekete’s solution (70% ethanol, 3.7% formaldehyde, 0.75M glacial acetic acid). The livers were fixed in 10% formalin. Lung surfaces were photographed and number of surface lung metastases was scored.
Statistics
Statistical analyses are provided in Suppl. Methods.
RESULTS
IL-6 is expressed in the majority of distant metastases of clinical PCs
To test the hypothesis that IL-6 promotes metastatic behavior of human PC cells, we first determined the frequency of IL-6 expression in clinical PC metastases (n=87) using immunohistochemical detection of IL-6 in paraffin-embedded tissue sections. Representative prostate cancer metastases positive or negative for IL-6 are presented in Figure 1A. A positive immunoreaction for IL-6 was detected in 78% (68/87) of epithelial cells in PC metastases (Table 1). In PC metastases to regional lymph nodes, an intense immunoreaction for PC was detected in 89% (32/36) of the specimens, while IL-6 was expressed in 2 out of 3 bone metastases. Additionally, IL-6 was expressed in 71% (34/48) of PC metastases to distant organs other than bone. In summary, our results indicate that the majority of PC metastases robustly express IL-6 protein.
Figure 1. IL-6 is produced in distant metastases of clinical PCs, and autocrine IL-6 induces metastatic colonization of human PC cells in the lungs of nude mice in an experimental metastases assay.
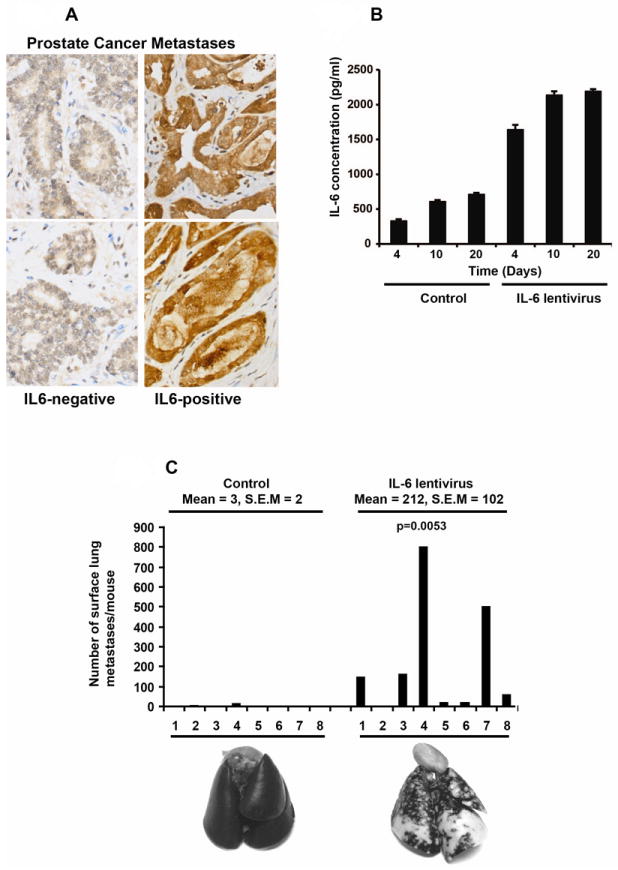
(A) IL-6 protein is expressed in distant metastases of human PCs. IL-6 in PC metastases was analyzed by immunohistochemical staining. Distant PC metastases negative (left) or positive (right) for immunostaining of autocrine IL-6 are shown. (B) ELISA demonstrating increased IL-6 protein expression in culture media of DU145 cells infected with lentivirus (MOI=5) expressing the IL-6 gene. (C) IL-6 increases metastases formation of PC cells to the lungs of athymic nude mice. Athymic nude mice were injected with DU145 cells infected with lentivirus expressing IL-6 gene at MOI=5 through the tail veins. Increased autocrine IL-6 expression in DU145 cells resulted in a significant increase (71-fold) (mean 212, S.E.M. 102) compared to control cells (mean 3, S.E.M. 2) (top). Representative photographs of India Ink-stained lungs derived from athymic nude mice (bottom).
IL-6 induces experimental metastases formation in nude mice
To determine if IL-6 affects metastatic dissemination of human PC cells in vivo, we performed in vivo experimental metastases assays in nude mice. We generated lentivirus expressing the IL-6 gene, and evaluated the efficacy of this lentiviral construct in producing IL-6 in PC cells by enzyme-linked immunosorbent assay. Lentiviral expression of the IL-6 gene in DU145 cells resulted in a 3-fold increase in IL-6 production into the culture medium (2.2 ng/ml; 0.11 nM) at 20 days compared to mock-infected cells (0.7 ng/ml; 0.04 nM) (Fig. 1B). Next, we infected DU145 cells with the IL-6 lentivirus and 24 h after the lentiviral gene delivery (MOI=5), DU145 cells were inoculated in athymic nude mice through tail vein injection (1 × 106 cells per mouse). After 8 weeks, the lungs were harvested, stained with India Ink, bleached with Fekete’s solution and scored for surface lung metastases. As demonstrated in Figure 1C, the number of metastases in mice injected with DU145 cells infected with IL-6 lentivirus was robustly increased when compared to control mice. Quantitatively, injection of DU145 cells infected with IL-6 lentivirus resulted in an average of 212 (S.E.M. = 102) metastases per lung, as compared with 3 metastases per lung using mock-infected DU145 cells (S.E.M. = 2.0). As depicted visually in Figure 1C (lower panel), IL-6 robustly promoted development of metastases in the lungs of nude mice (p=0.0053), evidenced by the density and large number of white metastatic nodules present throughout the lungs. To verify the concept that IL-6 promotes PC metastases formation in vivo, a second cell line CWR22Rv1, which is AR-positive, was infected with lentivirus expressing the IL-6 gene (MOI=5) and inoculated in mice through tail vein injections (1 × 106 cells per mouse). IL-6 increased liver metastases formation of CWR22Rv1 cells significantly (Suppl. Fig. 1A) as evidenced by increased liver weight (p=0.0079) (Suppl. Fig. 1B) and decreased overall survival (Suppl. Fig. 1C). Collectively, these data are the first demonstration that IL-6 increases the intrinsic ability of PC cells to metastasize in vivo.
IL-6 promotes PC cell migration, decreases homotypic adhesion of PC cells and increases heterotypic adhesion of PC cells to endothelial cells
Given that IL-6 was capable of inducing extensive metastases formation in vivo, we next investigated if IL-6 is involved in the regulation of metastatic behavior of prostate cancer cells in vitro. Development of metastases in the lungs and the livers of mice following tail-vein injection of PC cells requires a cascade of well-characterized biological events, including decreased homotypic adhesion, increased heterotypic adhesion, extravasation, increased migration, increased invasion of cells into the extracellular matrix and increased cell viability during migration (47). In Boyden chamber assays, migration of DU145 and CWR22Rv1 cells was increased by 46% (p=0.045) and 200% (p<0.0001), respectively, by IL-6 (5 nM) after 16 h incubation using 10% FBS as the chemoattractant (Fig. 2A and Suppl. Fig. 2A). Furthermore, IL-6 induced morphological changes characteristic of motile cells (Fig. 2B). Specifically, microtubules are known to be important for intrinsic cell polarization and directional cell migration, and can be linked to actin polymers directly or indirectly through intermediate proteins or signaling molecules (48-50). DU145 cells were stained for the presence of α-tubulin using FITC-conjugated secondary antibodies (Fig. 2B). IL-6 (5 nM) induced outward polarization of microtubules from the centrosomes, forming a dense meshwork facing the plasma membrane. In contrast, the microtubule network in control DU145 cells remained disrupted, which is characteristic of non-migratory cells (Fig. 2B).
Figure 2. IL-6 induces a migratory phenotype in PC cells.
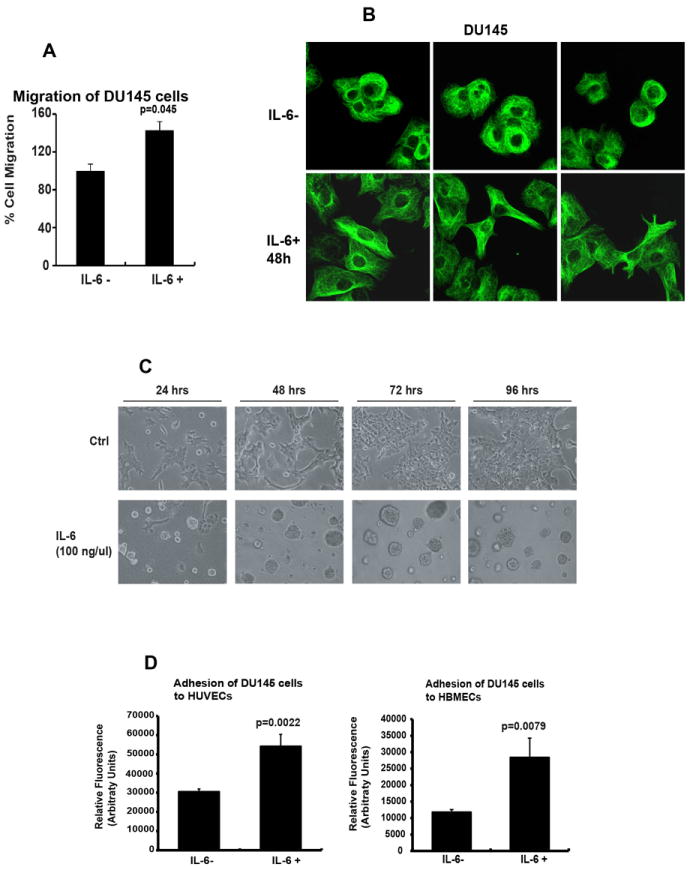
(A) IL-6 induces migration of PC cells in Boyden chamber assays. DU145 cells were cultured in the presence or absence of IL-6 (5 nM) for 72 h, counted and equal number of cells in different treatment groups were suspended within the motility chamber system using 10% FBS as a chemoattractant. The cells which traversed the membrane pores were counted after 16 h. (B) IL-6 induces polarization of the microtubule network in PC cells. DU145 cells cultured in the presence or absence of IL-6 (5 nM) for 48 h were fixed and incubated with anti-tubulin mAb followed by FITC-conjugated secondary antibody. Representative images from one of three independent experiments are shown. (C) IL-6 disrupts homotypic adhesion of PC cells. DU145 cells were seeded on Matrigel, treated with IL-6 (5 nM) for the indicated periods of time and morphological alterations were photographed. Note the dispersed and scattered cells in the IL-6 treatment group. (D) IL-6 induces heterotypic adhesion of PC cells to endothelial cells. DU145 cells were cultured in the presence or absence of IL-6 (5 nM) for 72 h, stained with a fluorescent dye and allowed to adhere to HUVECs (left) or HBMECs (right) for 60 min, after which the adherent cells were quantitated. Averages of three independent experiments are shown.
To investigate if IL-6 disrupts homotypic adhesion of PC cells, we examined the effects of IL-6 on DU145 and CWR22Rv1 cells cultured on Matrigel, a collagen-rich extracellular matrix that provides a more physiological growth environment than plastic. DU145 cells (Fig. 2C) and CWR22Rv1 cells (Suppl. Fig. 2B) were treated with recombinant IL-6 (5 nM) and cultured on Matrigel for 4 days. In the control group, DU145 cells grew as adherent sheets of cells attached to Matrigel. In contrast, cells treated with IL-6 (5 nM) were partly dispersed as single cells or small, scattered cell clusters (Fig. 2C and Suppl. Fig. 2B). These data provided evidence that IL-6 disrupts homotypic adhesion of DU145 and CWR22Rv1 cells, reducing epithelial cell-to-cell contact and suggesting increased migratory and invasive potential.
Finally, the initial arrest and attachment of cancer cells to vascular endothelium precedes extravasation from the bloodstream, and is a crucial step in the metastatic cascade. To determine if IL-6 promotes adhesion of PC cells to vascular endothelial cells, IL-6 was expressed in DU145 and CWR22Rv1 cells using IL-6 lentivirus for 72 h and tested for adhesion to two types of human endothelial cells: human umbilical vein endothelial cells (HUVEC) and human bone marrow endothelial cells (HBMEC). Notably, IL-6 increased binding of DU145 cells to HUVEC cells by 67% (p=0.0022) and to HBMEC cells by 125% (p=0.0079) (Fig. 2D). In addition, IL-6 increased binding of CWR22Ev1 cells to HBMEC cells by 51% (p<0.001) (Suppl. Fig. 2C). Collectively, the results presented here indicate that IL-6 induces a migratory phenotype in PC cells and increases heterotypic adhesion of PC cells to endothelial cells.
Stat3 is the key mediator of IL-6-driven development of a migratory PC cell phenotype in vitro
To identify signaling pathways that mediate the biological effects of IL-6 in DU145 cells, cells were starved overnight, stimulated with IL-6 (5 nM) for 30 min and immunoblotted for active phosphorylated forms of Stat3, ERK1/2 and Stat5a/b. As shown in Fig. 3A (i and ii), both Stat3 and ERK1/2, but not AKT or Stat5a/b, were activated by IL-6 stimulation in the absence of serum in the culture medium in DU145 cells. Moreover, IL-6 activated Jak2 in CWR22Rv1, DU145 and LNCaP cells and Jak1 in DU145 cells, while Jak1 was not expressed in LNCap and CWR22Rv1 cells (Fig. 3A (iii)). In CWR22Rv1 cells, IL-6 activated only Stat3 but not ERK1/2 or Akt (Suppl. Figs. 3A and 3B). Next, we generated lentiviruses expressing shRNAs targeting Stat3, ERK1 and ERK2, and verified protein knockdown efficiency in DU145 cells by Western blotting (Fig. 3B). To evaluate the contributions of Stat3 and ERK1/2 signaling to IL-6-induced PC cell migration, DU145 cells were infected with lentiviruses expressing Stat3 shRNA and/or ERK1/2 shRNA and treated with IL-6 (5 nM) for 72 h, after which the cells were tested for migratory potential in Boyden chamber assays for 16 h. IL-6 significantly increased migration of DU145 cells (p=0.0017) (Fig. 3C). Genetic knockdown of Stat3 (p=0.029) as well as ERK1/2 (p=0.057) inhibited IL-6-induced migration of DU145 cells cultured in the presence of FBS (Fig. 3C).
Figure 3. Stat3 and ERK1/2 signaling mediate the biological effects of IL-6 in induction of migratory PC cell phenotype.
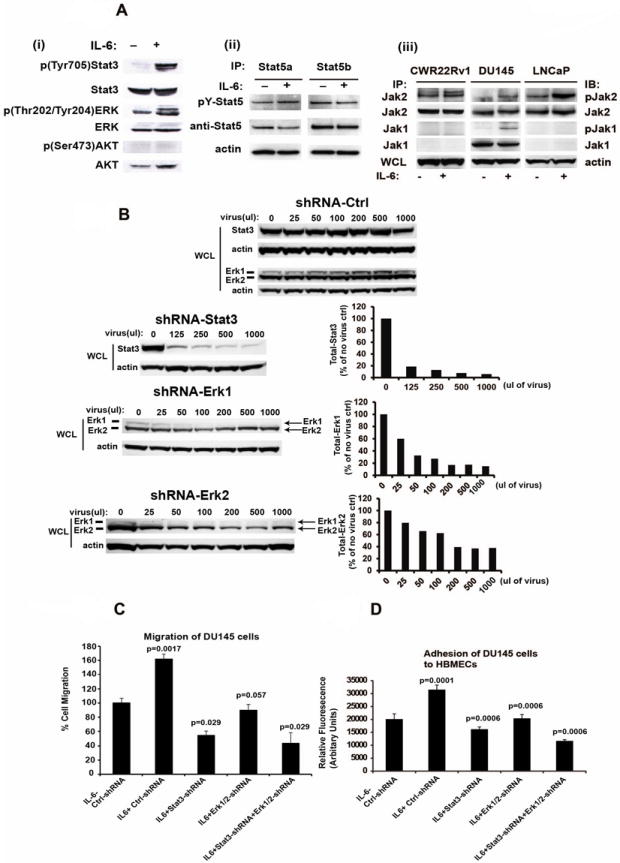
(A) IL-6 activates phosphorylation of Stat3, ERK1/2 and Jak1/2 but not Stat5 or Akt. (i) DU145 cells were serum-starved (0% FBS) overnight followed by stimulation of the cells with IL-6 (5 nM) for 20 min and immunoblotted for p(Tyr705)Stat3, Stat3, p(Thr202/Tyr204)ERK, ERK1/2 or p(Ser473)Akt or Akt. (ii) Stat5a and Stat5b were immunoprecipitated (IP) and immunoblotted for pYStat5a/b and total Stat5a/b. Whole cell lysates of parallel samples were immunoblotted for actin. (iii) CWR22Rv1, DU145 and LNCaP cells were serum-starved overnight followed by stimulation with IL-6 (5 nM) for 20 min. Jak1 and Jak2 were immunoprecipitated and blotted with anti-phosphotyrosine (4G10), anti-Jak1 or anti-Jak2 mAbs. (B) Lentivirus expressing shRNAs targeting Stat3, ERK1 and ERK2 expression in DU145 cells. DU145 cells infected with each lentiviral construct were immunoblotted with anti-Stat3, anti-actin, anti-ERK1 or anti-ERK2 antibodies. (C) Inhibition of Stat3 and/or ERK1/2 by RNA interference blocks IL-6-induced PC cell migration. DU145 cells were cultured in the presence of 1% CS-FBS and infected with lentiviruses expressing control-shRNA, Stat3-shRNA and/or ERK1/2-shRNA (MOI=5) and treated with or without IL-6 (5 nM) for 72 h. Cells were counted and equal numbers of cells in each treatment group were assayed for migration in Boyden chambers for 16 h. (D) Inhibition of Stat3 and/or ERK1/2 signaling blocks IL-6-induced increase in adhesion of PC cells to endothelial cells. DU145 cells were cultured in the presence of 1% CS-FBS and infected with lentiviruses expressing control-shRNA, Stat3-shRNA and/or ERK1/2-shRNA (MOI=5) and treated with or without IL-6 (5 nM) for 72 h. Cells were stained with a fluorescent dye and allowed to adhere to HBMEC cells for 60 min followed by quantitation of adhered cells by fluorescent reader. Averages of three independent experiments are shown.
To evaluate if Stat3 and ERK1/2 pathways mediate IL-6-induced heterotypic adhesion of PC cells to endothelial cells, DU145 cells were infected with lentiviruses expressing IL-6, Stat3 shRNA and/or ERK1/2 shRNA for 72 h and tested for adhesion to HBMEC cells. IL-6 significantly increased binding of DU145 cells to HBMEC cells (p=0.0001). Genetic knockdown of either Stat3 (p=0.0006) or ERK1/2 (p=0.0006) reduced IL-6-induced PC cell adhesion to HBMEC endothelial cells (Fig. 3D). In conclusion, the data presented here demonstrate that Stat3 signaling pathway predominantly mediates IL-6-induced PC cell migration and heterotypic adhesion in DU145 and CWR22Rv1 cells, while ERK1/2 may also contribute to mediation of IL-6 effects in DU145 cells to a lesser extent.
Pharmacological targeting of Jak2 by AZD1480 suppresses IL-6-induced Stat3 and ERK1/2 signaling and development of a migratory PC cell phenotype
Since Jak1 and Jak2 are the key tyrosine kinases activated by IL-6 in PC cells, we wanted to determine if the small-molecule Jak1/2-inhibitor AZD1480 (37) is capable of suppressing IL-6-induced metastatic behavior of human PC cells in vitro. We first tested the efficacy of AZD1480 in blocking IL-6-activated cell signaling in PC cells. AZD1480 inhibited IL-6-induced phosphorylation of Stat3 by approximately 50% (IC50) at a concentration of 9.4 nM in DU145 cells, while the IC50s for IL-6-induced ERK1 and ERK2 phosphorylation were 26 and 21 nM, respectively (Fig. 4A). Next, we evaluated if AZD1480 suppresses IL-6-driven migration of DU145 cells (Fig. 4B). DU145 cells were treated with or without recombinant IL-6 (5 nM) for 72 h in the presence or absence of AZD1480 (800 nM), after which equal numbers of cells per group were assayed for migratory ability in Boyden chambers for 16 h. IL-6 increased migration of DU145 cells by 48% (p=0.057), which was potently suppressed by AZD1480 (p=0.029) (Fig. 4B). Western blotting of cell lysates from parallel wells indicated that AZD1480 primarily inhibited Stat3 and ERK1 phosphorylation, with less inhibition of ERK2 phosphorylation, during long-term IL-6 treatment of DU145 cells (Fig 4B). Similarly, AZD1480 inhibited (p<0.0001) effectively IL-6-induced (p=0.002) migration of CWR22Rv1 cells (Suppl. Fig. 3C).
Figure 4. Pharmacological Jak1/2 inhibitor AZD1480 suppresses IL-6-activated Stat3 and ERK1/2 signaling in PC cells and IL-6-induced migratory PC cell phenotype.
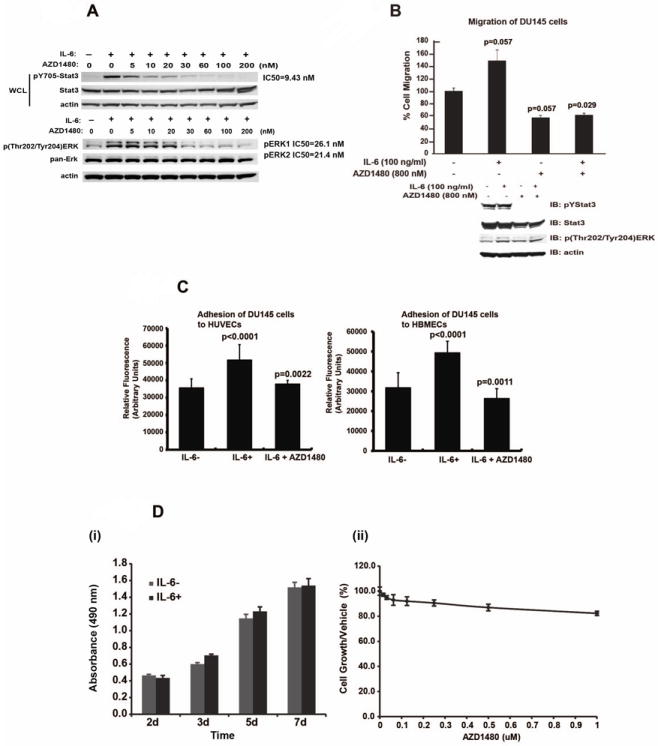
(A) AZD1480 disrupts IL-6-induced phosphorylation of Stat3 and ERK1/2 in PC cells. DU145 cells were serum-starved (0% FBS) overnight, pre-treated with AZD1480 at indicated concentrations for 1 h followed by stimulation of the cells with IL-6 (50 nM) for 20 min and immunoblotted for anti-p(Tyr705)Stat3, anti-p(Thr202/Tyr204)ERK, anti-Stat3, anti-ERK1/2 or anti-Actin, as indicated. (B) AZD1480 inhibits IL-6-induced migration of DU145 cells. DU145 cells were cultured in 1% CS-FBS in the presence or absence of IL-6 (5 nM) with or without pre-treatment of the cells with AZD1480 (800 nM). Equal numbers of cells in each treatment group were analyzed for migration in Boyden chambers for 16 h (upper panel). Cell lysates from parallel wells were immunoblotted for anti-p(Tyr705)Stat3, anti-p(Thr202/Tyr204)ERK, Stat3, or anti-actin (lower panel). (C) AZD1480 inhibits IL-6-induced heterotypic adhesion of PC cells to endothelial cells. DU145 cells were cultured in 1% CS-FBS in the presence or absence of IL-6 (5 nM) with or without pre-treatment of the cells with AZD1480 (800 nM). Averages of three independent experiments are shown. (D) IL-6 or AZD1480 did not affect the viability of DU145 cells. (i) DU145 cells were cultured in 1% CS-FBS for 7 days in the presence or absence of IL-6 (5 nM). The bars represent the numbers of viable DU145 cells in both treatment groups at indicated times. (ii) The number of DU145 cells cultured in 1% CS-FBS for 7 days in the presence or absence of AZD1480 at indicated concentration.
To investigate if AZD1480 reduces heterotypic adhesion of PC cells to endothelial cells, DU145 PC cells were treated with recombinant IL-6 (5 nM) or vehicle with or without AZD1480 (800 nM) for 72 h and assayed for binding to HUVEC and HBMEC cells (Fig. 4C). Increased heterotypic adhesion of DU145 cells to HUVEC (43%) (p<0.0001) and HBMEC cells (63%) (p<0.0001) induced by IL-6 was effectively blocked by AZD1480 (HUVEC, p=0.0022); HBMEC, p=0.0011) (Fig. 4C). Similarly, increased heterotypic adhesion of CWR22Rv1 cells to HBMEC cells by IL-6 (p=0.04) was inhibited by AZD1480 (p<0.0001).
Finally, we evaluated if the ability of AZD1480 to block IL-6-driven metastatic behavior of PC cells was due to induction of apoptosis in PC cells. Cell viability of DU145 cells cultured with or without IL-6 and/or AZD1480 was determined by MTT assay (Fig. 4D). IL-6 showed no effect on viability or growth of DU145 cells, indicating that IL-6-driven metastasis formation was not due to IL-6-stimulated PC cell proliferation. At the same time, AZD1480 failed to decrease the number of viable DU145 cells, indicating that suppression of PC cell migration and heterotypic adhesion by AZD1480 was not caused by AZD1480-induced PC cell death (Fig. 4D). In summary, the data presented here demonstrate that AZD1480 specifically blocked the stimulatory effects of IL-6 on PC cell migration and heterotypic adhesion, rather than affecting PC cell viability.
Stat3 and ERK1/2 mediate IL-6-driven metastases formation in nude mice, which can be effectively targeted by pharmacological inhibition of Jak1/2 using AZD1480
Given that IL-6 promoted metastatic behavior of PC cells in vitro, which was potently inhibited by genetic knockdown of both Stat3 and ERK1/2, we investigated if Stat3 and ERK1/2 signaling mediate IL-6-driven metastatic colonization in vivo. Most importantly, we wanted to determine if IL-6-induced migratory cell phenotype in vitro and metastatic dissemination in vivo can be suppressed pharmacologically using the Jak1/2 inhibitor AZD1480.
DU145 cells were infected with IL-6 lentivirus (MOI=5) alone or in combination with Stat3-shRNA lentivirus (MOI=5) and/or ERK1/2-shRNA lentivirus (MOI=5), after which the cells were injected into the tail veins of athymic nude mice (Fig. 5). IL-6 induced a 7-fold increase in metastases formation (787, S.E.M. = 92) compared to the control group (117, S.E.M = 28) (p<0.0001), which was substantially blocked by genetic knockdown of Stat3 alone (146, S.E.M. = 48) (p<0.0001), ERK1/2 alone (229, S.E.M. = 62) (p<0.0001) or Stat3 and ERK1/2 combined (122, S.E.M. = 44) (p<0.0001) (Fig. 5). To evaluate if AZD1480 was able to suppress IL-6-driven metastatic progression of PC, mice were treated daily by oral gavage of AZD1480, starting on day 3 until sacrifice. AZD1480 significantly suppressed IL-6-driven metastases formation in these mice (232, S.E.M = 31) (p<0.0001), compared to the IL-6 expressing group without AZD1480 treatment (787, S.E.M. = 92) (Fig. 5). Collectively, these data demonstrate that pharmacological inhibition of Jak1/2 effectively prevents IL-6-driven metastatic colonization of human PC cells in nude mice.
Figure 5. Pharmacological Jak1/2 inhibitor AZD1480 blocks IL-6-Jak-Stat3/ERK1/2-driven metastases formation in nude mice.
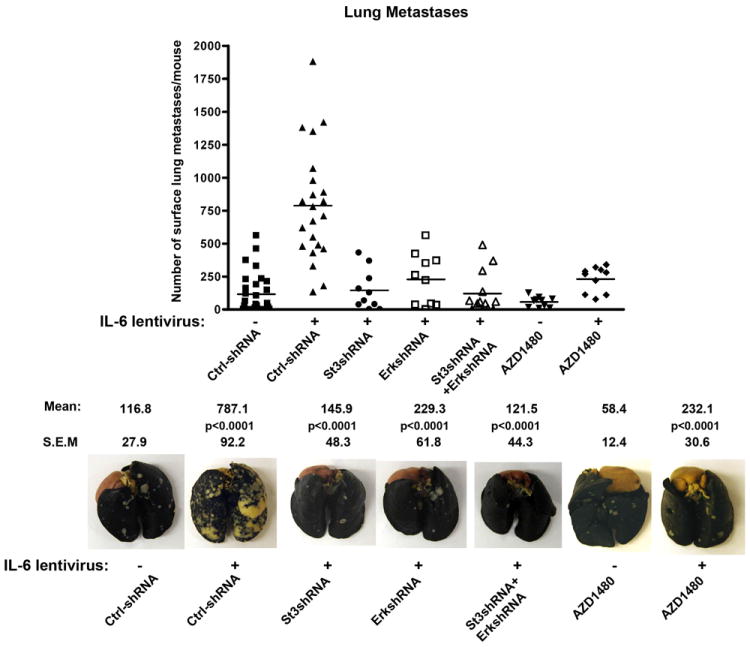
Genetic knockdown of Stat3 and/or ERK1/2, as well as pharmacological inhibition of Jak1/2 by AZD1480, suppresses PC metastases formation in nude mice. DU145 cells infected with lentivirus expressing IL-6 (MOI=5) with or without simultaneous infection of lentiviruses expressing shRNA-Stat3 (MOI=5) and/or shRNA-ERK1/2 (MOI=5) were injected in athymic nude mice through tail veins. AZD1480 was administered to indicated groups of mice daily by oral gavage (50 mg/kg). The lungs were stained with India Ink and scored for surface metastases. Representative photographs of India Ink-stained lungs (lower panel).
DISCUSSION
A significant fraction of patients with organ-confined PCs treated by radical prostatectomy develop recurrent metastatic disease (41, 42), which suggests that tumor cells with metastatic potential may have already disseminated at the time of the primary treatment. Current therapeutic strategies for metastatic PC are directed against AR signaling and eventually fail because of progression to the castrate-resistant (CR) stage of PC (2-4, 6, 23). CR PCs are treated with non-targeted cytotoxic therapeutic regimens that provide only limited additional survival benefit (2-4, 6, 23). Therefore, improved therapeutic strategies are needed both to prevent metastatic dissemination of organ-confined PC and to effectively treat existing metastatic disease. In this work, we show for the first time that IL-6-activated Jak1/2 signaling pathways induce metastatic behavior of AR-positive (CWR22Rv1) and AR-negative (DU145) PC cells in vitro and excessive metastasis formation in vivo, using experimental metastasis assays in mice. These biological pro-metastatic effects were predominantly mediated by Jak-Stat3 signaling pathway and to a lesser extent by Jak1/2-ERk1/2. Most important from a therapeutic perspective, IL-6-driven metastatic progression of PC in vivo was potently suppressed by a small-molecule pharmacological Jak1/2 inhibitor, AZD1480.
To our knowledge, this is the first evidence of IL-6-activated Jak1/2 signaling pathways promoting metastatic processes in PC. IL-6 increased PC cell migration, microtubule reorganization and heterotypic adhesion of PC cells to vascular endothelial cells, while reducing homotypic PC cell adhesion. IL-6 acts to directly upregulate metastatic processes in PC cells, since IL-6 treatment did not increase PC cell viability. Intriguingly, IL-6-activated Jak1/2 signaling resulted in a robust 7-fold increase in metastatic colonization of PC cells in the lungs of nude mice. In prior studies, IL-6 has been primarily investigated for its growth-promoting ability in PC using various model systems such as human PC cell lines, long-term exposure of PC cells to elevated IL-6 concentrations or stable PC cell clones overexpressing IL-6 (28-34). Results have been contradictory, with some studies indicating that IL-6 stimulation conferred a growth advantage on PC cells, while other studies demonstrated growth-inhibitory effects of IL-6 on PC cells (28-34). Also, IL-6 has been suggested to induce neuroendocrine differentiation of PC cells (51, 52). The current work tested the hypothesis that autocrine IL-6 activation of Jak1/2 signaling promotes metastatic progression of PC. The results presented here provide a new perspective in understanding the association of autocrine/circulating IL-6 with surrogate markers of clinical PC progression in patients, which has been previously demonstrated in numerous studies (10-16).
The biological pro-metastatic effects of IL-6 in DU145 and CWR22Rv1 PC cells were predominantly mediated by the Jak1/2-Stat3 pathway in PC cells, while ERK1/2 may also play some role in DU145 cells. The findings of the current study are in line with our previous work, which put forth a key role for Stat3 in promotion of metastatic dissemination of PC cells in vivo, induction of migratory phenotype in vitro and induction of gene expression profiles associated with metastatic processes in PC (35, 36). While IL-6 can promote AR signaling in some PC cell models (25-27), a recent study demonstrated that IL-6 secreted by endothelial cells promoted metastatic behavior of PC cells via downregulation of AR (53). Loss of AR signaling, in turn, resulted in increased TGF-beta production, followed by induction of epithelial-to-mesenchymal transition (EMT) and acquisition of an invasive PC cell phenotype (53). Our data presented here, however, show that IL-6 directly promoted metastatic processes through Jak1/2 signaling independently of AR, since DU145 cells are negative for expression of functional AR. In other words, IL-6 is capable of promoting metastatic behavior of PC cells independently of the AR-TGF-beta-axis. Future work will need to determine if IL-6 is capable of promoting metastatic processes simultaneously through multiple signaling pathways, including Jak1/2-Stat3, Jak1/2-ERK1/2 and AR-TGF-beta in PC cells positive for AR expression.
The finding of key clinical importance in the present work is that pharmacological targeting of IL-6-Jak1/2-Stat3- signaling by the Jak1/2 inhibitor AZD1480 effectively suppressed IL-6-induced migratory PC cell phenotype and metastatic dissemination in nude mice. This is critically important since numerous Jak1/2 inhibitors are currently in active clinical development for hematopoietic proliferative disorders and malignancies (38, 39). Jak1/2 inhibitors could potentially provide an adjuvant therapy for organ-confined or locally-advanced PC as a strategy to prevent metastases development after primary treatment. In addition, Jak1/2 inhibitors might provide therapeutic benefit for treatment of existing metastases in both hormone-responsive and CR PC. In the present work, we also demonstrated that IL-6 is expressed in the majority of distant metastases of clinical PCs, similar to what we have previously shown for active Stat3 expression in clinical PC metastases (35). Future work should establish if autocrine IL-6 production in PC predicts early development of recurrent metastatic PC. Moreover, it will be important to evaluate if IL-6 expression in PC metastases predicts early PC-specific death. Finally, IL-6 production/positivity of clinical PCs may serve as a biomarker to identify patients who would be most responsive to Jak2-inhibitor-based therapies. Siltuximab (CNTO328), a monoclonal antibody against IL-6, has been evaluated in several Phase I and II trials in CRPC in combination with chemotherapy or in post-chemotherapy setting (54-56). The efficacy of siltuximab was limited in these studies (54-56). However, this may be due to the fact that no patient selection based on Il-6 positivity or Stat3 activation of CRPC was conducted.
In summary, the findings presented in this work are the first to demonstrate induction of metastatic processes in PC by IL-6-Jak1/2-Stat3/ERK1/2 pathways, and the therapeutic potential of pharmacological Jak1/2 inhibition as a novel treatment strategy for advanced, metastatic PC.
Supplementary Material
Acknowledgments
This work was supported by grants from NCI (2RO1CA11358-06), AstraZeneca and Pennsylvania Department of Health to MT Nevalainen. Shared Resources of Kimmel Cancer Center at Thomas Jefferson University are partially supported by NIH Grant CA56036-08 (Cancer Center Support Grant, to KCC). We thank Geraldine Bebernitz, Shenghua Wen, Denise Hughes and Corinne Reimer at AstraZeneca for their contributions.
Footnotes
Conflict of Interest: Dennis Huszar and Michael Zinda are employees of AstraZeneca Pharmaceuticals.
References
- 1.Arya M, Bott SR, Shergill IS, Ahmed HU, Williamson M, Patel HR. The metastatic cascade in prostate cancer. Surg Oncol. 2006;15:117–28. doi: 10.1016/j.suronc.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Chen Y, Clegg NJ, Scher HI. Anti-androgens and androgen-depleting therapies in prostate cancer: new agents for an established target. Lancet Oncol. 2009;10:981–91. doi: 10.1016/S1470-2045(09)70229-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loblaw DA, Mendelson DS, Talcott JA, Virgo KS, Somerfield MR, Ben-Josef E, et al. American Society of Clinical Oncology recommendations for the initial hormonal management of androgen-sensitive metastatic, recurrent, or progressive prostate cancer. J Clin Oncol. 2004;22:2927–41. doi: 10.1200/JCO.2004.04.579. [DOI] [PubMed] [Google Scholar]
- 4.Loblaw DA, Virgo KS, Nam R, Somerfield MR, Ben-Josef E, Mendelson DS, et al. Initial hormonal management of androgen-sensitive metastatic, recurrent, or progressive prostate cancer: 2006 update of an American Society of Clinical Oncology practice guideline. J Clin Oncol. 2007;25:1596–605. doi: 10.1200/JCO.2006.10.1949. [DOI] [PubMed] [Google Scholar]
- 5.Pestell RG, Nevalainen MT, editors. Prostate Cancer: Signaling Networks, Genetics, and New Treatment Strategies. Totowa, NJ, USA: Humana Press; 2008. [Google Scholar]
- 6.Yap TA, Zivi A, Omlin A, de Bono JS. The changing therapeutic landscape of castration-resistant prostate cancer. Nat Rev Clin Oncol. 2011;8:597–610. doi: 10.1038/nrclinonc.2011.117. [DOI] [PubMed] [Google Scholar]
- 7.Culig Z, PR, Nevalainen MT. Transcription factors Stat5 and Stat3: Survival Factors for Prostate Cancer Cells. In: Nevalainen MT PR, editor. Prostate Cancer: Signaling Networks, Genetics and New Treatment Strategies. Totowa: Springer; 2008. pp. 257–190. [Google Scholar]
- 8.Hobisch A, Rogatsch H, Hittmair A, Fuchs D, Bartsch G, Jr, Klocker H, et al. Immunohistochemical localization of interleukin-6 and its receptor in benign, premalignant and malignant prostate tissue. J Pathol. 2000;191:239–44. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH633>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 9.Royuela M, Ricote M, Parsons MS, Garcia-Tunon I, Paniagua R, de Miguel MP. Immunohistochemical analysis of the IL-6 family of cytokines and their receptors in benign, hyperplastic, and malignant human prostate. J Pathol. 2004;202:41–9. doi: 10.1002/path.1476. [DOI] [PubMed] [Google Scholar]
- 10.Drachenberg DE, Elgamal AA, Rowbotham R, Peterson M, Murphy GP. Circulating levels of interleukin-6 in patients with hormone refractory prostate cancer. Prostate. 1999;41:127–33. doi: 10.1002/(sici)1097-0045(19991001)41:2<127::aid-pros7>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 11.George DJ, Halabi S, Shepard TF, Sanford B, Vogelzang NJ, Small EJ, et al. The prognostic significance of plasma interleukin-6 levels in patients with metastatic hormone-refractory prostate cancer: results from cancer and leukemia group B 9480. Clin Cancer Res. 2005;11:1815–20. doi: 10.1158/1078-0432.CCR-04-1560. [DOI] [PubMed] [Google Scholar]
- 12.Michalaki V, Syrigos K, Charles P, Waxman J. Serum levels of IL-6 and TNF-alpha correlate with clinicopathological features and patient survival in patients with prostate cancer. Br J Cancer. 2004;90:2312–6. doi: 10.1038/sj.bjc.6601814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakashima J, Tachibana M, Horiguchi Y, Oya M, Ohigashi T, asakura H, et al. Serum interleukin-6 as a prognostic factor in patients with prostate cancer. Clin Cancer Res. 2000;6:2702–6. [PubMed] [Google Scholar]
- 14.Shariat SF, Andrews B, Kattan MW, Kim J, Wheeler TM, Slawin KM. Plasma levels of interleukin-6 and its soluble receptor are associated with prostate cancer progression and metastasis. Urology. 2001;58:1008–15. doi: 10.1016/s0090-4295(01)01405-4. [DOI] [PubMed] [Google Scholar]
- 15.Twillie DA, Eisenberger MA, Carducci MA, Hseih W-S, Kim WY, Simons JW. Interleukin-6: a candidate mediator of human prostate cancer morbidity. Urology. 1995;45:542–9. doi: 10.1016/S0090-4295(99)80034-X. [DOI] [PubMed] [Google Scholar]
- 16.Wise GJ, Marella VK, Talluri G, Shirazian D. Cytokine variations in patients with hormone treated prostate cancer. J Urol. 2000;164:722–5. doi: 10.1097/00005392-200009010-00024. [DOI] [PubMed] [Google Scholar]
- 17.Hobisch A, Ramoner R, Fuchs D, Godoy-Tundidor S, Bartsch G, Klocker H, et al. Prostate cancer cells (LNCaP) generated after long-term interleukin-6 treatment express interleukin-6 and acquire an interleukin-6-partially resistant phenotype. Clin Cancer Res. 2001;7:2941–8. [PubMed] [Google Scholar]
- 18.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones SA, Scheller J, Rose-John S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest. 2011;121:3375–83. doi: 10.1172/JCI57158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamasaki K, Taga T, Hirata Y, Yawata H, Kawanishi Y, Seed B, et al. Cloning and expression of the human interleukin-6 (BSF-2/IFN beta 2) receptor. Science. 1988;241:825–8. doi: 10.1126/science.3136546. [DOI] [PubMed] [Google Scholar]
- 21.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen, Schaper F. Principles of interleukin-6 (IL-6)-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okada S, Nakamura M, Mikami Y, Shimazaki T, Mihara M, Ohsugi Y, et al. Blockade of interleukin-6 receptor suppresses reactive astrogliosis and ameliorates functional recovery in experimental spinal cord injury. J Neurosci Res. 2004;76:265–76. doi: 10.1002/jnr.20044. [DOI] [PubMed] [Google Scholar]
- 23.Pestell RG, Nevalainen MT. Prostate Cancer: Signaling Networks, Genetics and New Treatment Strategies. Totowa: Human Press; 2008. [Google Scholar]
- 24.Yang X, Qiao D, Meyer K, Friedl A. Signal transducers and activators of transcription mediate fibroblast growth factor-induced vascular endothelial morphogenesis. Cancer Res. 2009;69:1668–77. doi: 10.1158/0008-5472.CAN-07-6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen T, Wang LH, Farrar WL. Interleukin 6 activates androgen receptor-mediated gene expression through a signal transducer and activator of transcription 3-dependent pathway in LNCaP prostate cancer cells. Cancer Res. 2000;60:2132–5. [PubMed] [Google Scholar]
- 26.Hobisch A, Eder IE, Putz T, Horninger W, Bartsch G, Klocker H, et al. Interleukin-6 regulates prostate-specific protein expression in prostate carcinoma cells by activation of the androgen receptor. Cancer Res. 1998;58:4640–5. [PubMed] [Google Scholar]
- 27.Ueda T, Bruchovsky N, Sadar MD. Activation of the androgen receptor N-terminal domain by interleukin-6 via MAPK and STAT3 signal transduction pathways. J Biol Chem. 2002;277:7076–85. doi: 10.1074/jbc.M108255200. [DOI] [PubMed] [Google Scholar]
- 28.Culig Z, Steiner H, Bartsch G, Hobisch A. Interleukin-6 regulation of prostate cancer cell growth. J Cell Biochem. 2005;95:497–505. doi: 10.1002/jcb.20477. [DOI] [PubMed] [Google Scholar]
- 29.Degeorges A, Tatoud R, Fauvel Lafeve F, Podgorniak MP, Millot G, de Cremoux P, et al. Stromal cells from human benign prostate hyperplasia produce a growth-inhibitory factor for LNCaP prostate cancer cells, identified as interleukin-6. Int J Cancer. 1996;68:207–14. doi: 10.1002/(SICI)1097-0215(19961009)68:2<207::AID-IJC12>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 30.Giri D, Ozen M, Ittmann M. Interleukin-6 is an autocrine growth factor in human prostate cancer. Am J Pathol. 2001;159:2159–65. doi: 10.1016/S0002-9440(10)63067-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee SO, Lou W, Hou M, de Miguel F, Gerber L, Gao AC. Interleukin-6 promotes androgen-independent growth in LNCaP human prostate cancer cells. Clin Cancer Res. 2003;9:370–6. [PubMed] [Google Scholar]
- 32.Sanford DC, DeWille JW. C/EBPdelta is a downstream mediator of IL-6 induced growth inhibition of prostate cancer cells. Prostate. 2005;63:143–54. doi: 10.1002/pros.20159. [DOI] [PubMed] [Google Scholar]
- 33.Spiotto MT, Chung TD. STAT3 mediates IL-6-induced growth inhibition in the human prostate cancer cell line LNCaP. Prostate. 2000;42:88–98. doi: 10.1002/(sici)1097-0045(20000201)42:2<88::aid-pros2>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 34.Wallner L, Dai J, Escara-Wilke J, Zhang J, Yao Z, Lu Y, et al. Inhibition of interleukin-6 with CNTO328, an anti-interleukin-6 monoclonal antibody, inhibits conversion of androgen-dependent prostate cancer to an androgen-independent phenotype in orchiectomized mice. Cancer Res. 2006;66:3087–95. doi: 10.1158/0008-5472.CAN-05-3447. [DOI] [PubMed] [Google Scholar]
- 35.Abdulghani J, Gu L, Dagvadorj A, Lutz J, Leiby B, Bonuccelli G, et al. Stat3 promotes metastatic progression of prostate cancer. Am J Pathol. 2008;172:1717–28. doi: 10.2353/ajpath.2008.071054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gu L, Dagvadorj A, Lutz J, Leiby B, Bonuccelli G, Lisanti MP, et al. Transcription factor Stat3 stimulates metastatic behavior of human prostate cancer cells in vivo, whereas Stat5b has a preferential role in the promotion of prostate cancer cell viability and tumor growth. Am J Pathol. 2010;176:1959–72. doi: 10.2353/ajpath.2010.090653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–97. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reddy MM, Deshpande A, Sattler M. Targeting JAK2 in the therapy of myeloproliferative neoplasms. Expert Opin Ther Targets. 2012;16:313–24. doi: 10.1517/14728222.2012.662956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tibes R, Bogenberger JM, Geyer HL, Mesa RA. JAK2 inhibitors in the treatment of myeloproliferative neoplasms. Expert Opin Investig Drugs. 2012;21:1755–74. doi: 10.1517/13543784.2012.721352. [DOI] [PubMed] [Google Scholar]
- 40.Li H, Ahonen TJ, Alanen K, Xie J, LeBaron MJ, Pretlow TG, et al. Activation of signal transducer and activator of transcription 5 in human prostate cancer is associated with high histological grade. Cancer Res. 2004;64:4774–82. doi: 10.1158/0008-5472.CAN-03-3499. [DOI] [PubMed] [Google Scholar]
- 41.Li H, Zhang Y, Glass A, Zellweger T, Gehan E, Bubendorf L, et al. Activation of signal transducer and activator of transcription-5 in prostate cancer predicts early recurrence. Clin Cancer Res. 2005;11:5863–8. doi: 10.1158/1078-0432.CCR-05-0562. [DOI] [PubMed] [Google Scholar]
- 42.Mirtti T, Leiby BE, Abdulghani J, Aaltonen E, Pavela M, Mamtani A, et al. Nuclear Stat5a/b predicts early recurrence and prostate cancer-specific death in patients treated by radical prostatectomy. Hum Pathol. 2012;44:310–9. doi: 10.1016/j.humpath.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gu L, Vogiatzi P, Puhr M, Dagvadorj A, Lutz J, Ryder A, et al. Stat5 promotes metastatic behavior of human prostate cancer cells in vitro and in vivo. Endocr Relat Cancer. 2010;17:481–93. doi: 10.1677/ERC-09-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahonen TJ, Xie J, LeBaron MJ, Zhu J, Nurmi M, Alanen K, et al. Inhibition of transcription factor Stat5 induces cell death of human prostate cancer cells. J Biol Chem. 2003;278:27287–92. doi: 10.1074/jbc.M304307200. [DOI] [PubMed] [Google Scholar]
- 45.Dagvadorj A, Kirken RA, Leiby B, Karras J, Nevalainen MT. Transcription factor signal transducer and activator of transcription 5 promotes growth of human prostate cancer cells in vivo. Clin Cancer Res. 2008;14:1317–24. doi: 10.1158/1078-0432.CCR-07-2024. [DOI] [PubMed] [Google Scholar]
- 46.Dagvadorj A, Tan SH, Liao Z, Xie J, Nurmi M, Alanen K, et al. N-Terminal Truncation of Stat5a/b Circumvents PIAS3-Mediated Transcriptional Inhibition of Stat5 in Prostate Cancer Cells. Int J Biochem Cell Biol. 2011;42:2037–46. doi: 10.1016/j.biocel.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fatatis A, editor. Signaling Pathways and Molecular Mediators of Metastasis. First. Springer; 2012. [Google Scholar]
- 48.Yamazaki D, Kurisu S, Takenawa T. Regulation of cancer cell motility through actin reorganization. Cancer Sci. 2005;96:379–86. doi: 10.1111/j.1349-7006.2005.00062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Etienne-Manneville S. Actin and microtubules in cell motility: which one is in control? Traffic. 2004;5:470–7. doi: 10.1111/j.1600-0854.2004.00196.x. [DOI] [PubMed] [Google Scholar]
- 50.Ng DC, Lin BH, Lim CP, Huang G, Zhang T, Poli V, et al. Stat3 regulates microtubules by antagonizing the depolymerization activity of stathmin. J Cell Biol. 2006;172:245–57. doi: 10.1083/jcb.200503021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spiotto MT, Chung TD. STAT3 mediates IL-6-induced neuroendocrine differentiation in prostate cancer cells. Prostate. 2000;42:186–95. doi: 10.1002/(sici)1097-0045(20000215)42:3<186::aid-pros4>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 52.Deeble PD, Murphy DJ, Parsons SJ, Cox ME. Interleukin-6 and cyclic-AMP-mediated signaling potentiates neuroendocrine differentiation of LNCaP prostate tumor cells. Mol Cell Biol. 2001;21:8471–82. doi: 10.1128/MCB.21.24.8471-8482.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X, Lee SO, Xia S, Jiang Q, Luo J, Li L, et al. Endothelial cells enhance prostate cancer metastasis via IL6->androgen receptor->TGFbeta->MMP9 signals. Mol Cancer Ther. 2013;12:1026–37. doi: 10.1158/1535-7163.MCT-12-0895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dorff TB, Goldman B, Pinski JK, Mack PC, Lara PN, Jr, VanVeldhuizen PJ, Jr, et al. Clinical and correlative results of SWOG S0354: a phase II trial of CNTO328 (siltuximab), a monoclonal antibody against interleukin-6, in chemotherapy-pretreated patients with castration-resistant prostate cancer. Clin Cancer Res. 2010;16:3028–34. doi: 10.1158/1078-0432.CCR-09-3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fizazi K, De Bono JS, Flechon A, Heidenreich A, Voog E, Davis NB, et al. Randomised phase II study of siltuximab (CNTO 328), an anti-IL-6 monoclonal antibody, in combination with mitoxantrone/prednisone versus mitoxantrone/prednisone alone in metastatic castration-resistant prostate cancer. Eur J Cancer. 2012;48:85–93. doi: 10.1016/j.ejca.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 56.Hudes G, Tagawa ST, Whang YE, Qi M, Qin X, Puchalski TA, et al. A phase 1 study of a chimeric monoclonal antibody against interleukin-6, siltuximab, combined with docetaxel in patients with metastatic castration-resistant prostate cancer. Invest New Drugs. 2013;31:669–76. doi: 10.1007/s10637-012-9857-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.