

Abstract
The bones of the vertebrate face develop from transient embryonic branchial arches that are populated by cranial neural crest cells. We have characterized a mouse mutant for the Forkhead family transcription factor Foxi3, which is expressed in branchial ectoderm and endoderm. Foxi3 mutant mice are not viable and display severe branchial arch-derived facial skeleton defects, including absence of all but the most distal tip of the mandible and complete absence of the inner, middle and external ear structures. Although cranial neural crest cells of Foxi3 mutants are able to migrate, populate the branchial arches and display some elements of correct proximo distal patterning, they succumb to apoptosis from embryonic day 9.75 onwards. We show this cell death correlates with a delay in expression of Fgf8 in branchial arch ectoderm and a failure of neural crest cells in the arches to express FGF responsive genes. Zebrafish foxi1 is also expressed in branchial arch ectoderm and endoderm, and morpholino knockdown of foxi1 also causes apoptosis of neural crest in the branchial arches. We show that heat shock induction of fgf3 in zebrafish arch tissue can rescue cell death in foxi1 morphants. Our results suggest that Foxi3 may play a role in the establishment of signaling centers in the branchial arches that are required for neural crest survival, patterning and the subsequent development of branchial arch derivatives.
Keywords: Pharyngeal arch, Craniofacial development, Neural Crest, FGF
INTRODUCTION
Skeletal elements of the middle ear and jaw are derived from the most anterior of five pairs of branchial arches (BA) that develop on the ventral side of the embryo at the level of the hindbrain (reviewed in Noden and Trainor, 2005; Szabo-Rogers et al., 2010). Initially, each arch comprises an ectodermal surface, a core of mesoderm, and an endodermal lining. As arch outgrowth progresses, cranial neural crest cells migrate laterally and ventrally from the hindbrain and posterior midbrain to populate the arch mesenchyme, eventually forming the bulk of the mesenchyme (Lumsden et al., 1991; Schilling and Kimmel, 1994; Serbedzija et al., 1992). Bones and cartilage of the lower face are mainly derived from these neural crest cells, whose descendants form the mandible, maxilla, tympanic ring, and bones of the middle ear, among other structures. Perturbations in proliferation, migration, patterning, and differentiation of cranial neural crest cells can therefore lead to malformations of craniofacial skeletal structures (Passos-Bueno et al., 2009; Szabo-Rogers et al., 2010).
As the branchial arches expand ventrally, their anterior and posterior borders are defined by a series of contact points between pharyngeal ectoderm and underlying pharyngeal endoderm, which form morphological barriers called pharyngeal pouches. The pouches isolate populations of mesenchyme in each arch, and are also points of communication between ectoderm and endoderm (Couly et al., 2002). They are sources of signaling molecules that promote neural crest cell survival and contribute to the establishment of proximal distal and anterior posterior axes within each arch (Brito et al., 2006; Couly et al., 2002; David et al., 2002).
We have previously identified a transcription factor, Forkhead Box i3 (Foxi3), that is expressed in the pharyngeal region of mouse embryos in a segmented pattern between the branchial arches (Ohyama and Groves, 2004). Foxi3 is one of three Foxi transcription factors present in the mouse genome, all of which are closely related to the zebrafish foxi1 transcription factor. Mouse Foxi1 expression is limited to the dorsal otic vesicle, and Foxi1 mutant mice exhibit only balance defects (Hulander et al., 2003; Hulander et al., 1998). However, zebrafish foxi1 is expressed in the pharyngeal epithelium during arch development (Solomon et al., 2003b). A zebrafish foxi1 mutant, hearsay, lacks an otic vesicle and presents severe defects in skeletal structures of the face that develop from the anterior arches (Nissen et al., 2003; Solomon et al., 2003a). We have generated a mouse Foxi3 mutant, and found a facial skeleton phenotype that is similar to zebrafish hearsay mutants. Foxi3 mutants lack much of the lower jaw and other branchial arch derivatives, such as the entire middle and external ear apparatus. Here, we characterize the mechanism underlying the branchial arch defects of Foxi3 mutants. We show that cranial neural crest cells emigrate normally from the brain of Foxi3 mutants, but then undergo apoptosis as they populate the branchial arches. Since neural crest cells do not express Foxi3, this suggests that Foxi3 may regulate the expression of trophic or survival factors in arch ectoderm or endoderm. We show that the activity of Foxi3 in pharyngeal epithelia is required for early expression of Fgf8 in arch ectoderm. We also show a conservation of this pathway in zebrafish; here, fgf3 is expressed in branchial arch ectoderm and requires the expression of foxi1. We show that ectopic expression of Fgf3 in pharyngeal ectoderm can reduce neural crest cell death in zebrafish foxi1 morphants. We propose that Foxi1 and 3 expression is required for normal pharyngeal pouch morphology in zebrafish and mouse respectively, that it establishes signaling centers in the developing branchial arches necessary for crest survival, and that the craniofacial phenotype seen in Foxi3 mutants is due to reduced FGF8 signaling in the pharyngeal region.
MATERIALS AND METHODS
Generation of Foxi3 Mutant Mice
The targeting vector for the mouse Foxi3-floxed-neo allele was constructed using BAC recombineering (Warming et al., 2005). Briefly, an approximately 11kb genomic DNA fragment containing exon 2 of mouse Foxi3 was retrieved from a BAC clone bMQ 285H11 of 129Sv BAC genomic library obtained from the Wellcome Trust Sanger Institute (Adams et al., 2005) Using recombineering, a loxP site was inserted upstream of exon 2, and an Frt-PGKNeo-Frt-LoxP sequence as inserted downstream of exon 2 (Figure 2A) (Meyers et al., 1998). Electroporation of the targeting vector into ES cells, screening of the targeted ES cells and blastocyst injection were performed by the transgenic core facility at Norris Cancer Center of the University of Southern California. Germline Foxi3-floxed-neo founder mice were identified and confirmed by genomic Southern blotting to detect the extra EcoRV and NheI sites introduced by the Frt-PGKNeo-Frt-LoxP sequence (Figure 2B). The Foxi3-del allele used in this study was generated by crossing the Foxi3-floxed-neo allele with CMV-Cre line (JAX Mice, stock #003465).
Figure 2. Generation of Foxi3 mutant mice.

A: Diagram of the mouse Foxi3 locus, composed of two coding exons. A targeting vector was generated containing a loxP site upstream of exon 2 and an Frt-PGKNeo-Frt-LoxP sequence downstream of exon. B: The successfully targeted allele introduced an extra NheI and EcoRV site into the locus that could be detected by Southern blotting with 5’ and 3’ probes. Crossing successfully targeted offspring with CMV-Cre mice recombined the loxP sites to generate the Foxi3-del allele,
Mouse Genotyping
The Foxi3 deletion allele (Foxi3-del) was maintained by breeding heterozygous mice. Primers used to genotype embryos were f3G1 (5’-GGC CTT GTC TCA ACC AAC AG-3’), f3G2 (5’-GTT TCC TGT ATC CCT GGC TG-3’) and f3G3 (5’-CTT GGA ATG GGT TGA CTG AG-3’). f3G1 and f3G2 produce a 350bp band corresponding to the wild-type allele and f3G1 and f3G3 yield a 600bp band corresponding to the Foxi3-del allele.
Whole Mount DAPI Imaging
Embryos were fixed, washed in PBS with 1% Triton X-100, incubated for 5 minutes in DAPI solution, and washed in PBS with 0.1% Tween 20. Embryos were mounted in PBS in a depression slide and photographed on a confocal microscope using the methods described by Trainor and colleagues (Sandell et al., 2012).
Skeletal Staining
Mouse embryos were deeply anaesthetized in PBS on ice and decapitated. Heads were scalded in 70°C water and skin was removed. Skulls were stained with Alizarin Red and Alcian Blue as described (Ovchinnikov, 2009). Briefly, skin was removed from the embryos and fixed in 95% ethanol, the heads were defatted in overnight in acetone, cartilage was stained overnight in 0.015% Alcian Blue in 80% ethanol:20% acetic acid. The heads were rinsed in 70% ethanol, cleared in 2% potassium hydroxide and counter-stained with 0.005% Alizarin Red in 1% potassium hydroxide. After staining, the embryos were cleared in 1% potassium hydroxide followed by stabilization and storage in a 1:1 solution of glycerol and ethanol.
Probe Synthesis and In situ Hybridization
Digoxygenin labeled, cRNA probes were synthesized for whole mount in situ hybridization as described(Stern, 1998) using plasmid clones from the following sources: Dlx2 ( John Rubenstein), Dlx5 (Jin Xian Liu), Sox10 (Michael Wegner), Erm (Annette Neubuser), Fgf8 and Spry2 (Gail Martin), Dlx3 (Maria Morasso), Pax9 (Rudi Balling), Gsc (Richard Behringer), Lhx7 (Maria Grigoriou), and Pitx1 (Dan Bernard).The Foxi3 probe was generated by our lab and previously described (Ohyama and Groves, 2004). Whole mount in situ hybridization was performed as recently described (Khatri and Groves, 2013). After in situ hybridization, stained embryos were equilibrated in 15% sucrose in PBS and embedded in 7.5% gelatin with 15% sucrose in PBS for sectioning.
Immunohistochemistry
Embryos were fixed and embedded in gelatin (7.5% gelatin, 15% sucrose in PBS). 14μm thick sections were collected on Superfrost Plus slides. For cleaved caspase-3 detection, cleaved caspase-3 antibody (AF835, R&D Systems) was diluted 1:200 in blocking buffer (PBST with 0.1% Triton X-100 and 10% goat serum). Secondary antibody (AlexaFluor 488 goat anti-rabbit, Invitrogen) was diluted 1:1000 in blocking buffer. For AP2α detection, slides were boiled in 10mM citric acid for 10 minutes prior to antibody application. The 3B5 AP2α monoclonal antibody developed by Trevor Williams was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the University of Iowa, Department of Biology, Iowa City, IA 52242. AP2α antibody was diluted 1:100 in blocking buffer. Slides were incubated for 15 minutes at room temperature in 0.012% hydrogen peroxide prior to secondary antibody application. Staining was detected with biotinylated secondary antibody (Mouse Vectastain ABC kit) in conjunction with PerkinElmer TSA Plus Cyanine-3 System. All slides were mounted in Fluoromount G (Southern Biotech). For dephosphorylated ERK, whole embryos were incubated with p44/p42 MAPK rabbit polyclonal (Cell Signaling #9101) diluted 1:35 in PBS with 0.02% Tween 20 and 5% goat serum. dpERK was detected with Vectastain HRP ABC kit with nickel intensification. Embryos were then embedded in gelatin and sectioned at 14μm.
Quantification of Apoptotic Cells in Mouse
Cells located within BA1 at any point ventral to the floor plate of the neural tube that were stained with the cleaved caspase-3 antibody were counted in 14 μm sections from 6 wild type and 5 Foxi3 mutant embryos at 25 somites of development. The average number of apoptotic cells per section in each embryo was calculated and these values were used to determine the average number of apoptotic cells and the standard error in Foxi3 mutants and wild types. Statistical significance was determined with a two-tailed t-test assuming unequal variance.
Zebrafish Experiments
Wild type zebrafish embryos were derived from AB line (Eugene, OR). In this study used transgenic lines Tg(hsp70:fgf8a)x17 (Millimaki et al., 2010) and Tg(hsp70:fgf3)x27 (Sweet et al., 2011), referred to hereafter as hsfgf8 and hsfgf3. Except where noted, embryos were maintained at 28.5 °C in fish water (aqueous solution of .008% Instant Ocean salts) containing methylene blue and PTU (1-phenyl 2-thiourea, 0.3 mg/ml, Sigma) to block melanin formation. For global mis-expression of hsfgf3, embryos were incubated at 37°C for 30 minutes and then transferred to 33 °C to maintain weak transgene activity for an extended period. For global mis-expression of hsfgf8, embryos were incubated at 35 °C for 6 hours. For morpholino injections approximately 5ng foxi1 morpholino oligomer was injected per embryo at 1-cell stage. The sequence of foxi1-MO has been published previously (Solomon et al., 2003a). Whole mount in-situ hybridization and cryosectioning were performed as described previously (Phillips et al., 2001; Vemaraju et al., 2012). For cell death analysis, embryos were dechorionated and incubated in fish water containing 2ug/ml acridine orange for one hour. The embryos were then washed twice with fish water prior to observation. Laser activation of hs:fgf3 was performed using a MicroPoint system with a NL100 nitrogen laser (Stanford Research Systems). Laser power was partially attenuated to avoid cell ablation and applied with a 20x objective in a series of 15-18 pulses per region.
Quantification of Apoptotic Cells in Zebrafish
Acridine orange-positive cells were counted in the region between the midbrain-hindbrain border and the first somite, and between the pharyngeal endoderm and the roof of the otic vesicle. 11-18 embryos were analyzed for cell death quantification in most experiments, but 7 foxi1 morphants were analyzed following a 37°C 30 min heat shock and 9 embryos were quantified for focal hsfgf3 rescue experiments. Both wild type and foxi1 morphant embryos showed generalized elevation of cell death during laser activation (i.e. on both the irradiated and non-irradiated sides) due to prolonged exposure to tricaine anesthetic.
RESULTS
Foxi3 is expressed in pharyngeal endoderm and ectoderm
During branchial arch development, dorsal-ventral, proximal-distal, and oral aboral domains of ectoderm are defined by unique gene expression patterns (Danesh et al., 2009; Liu et al., 2005; Maemura et al., 1996). We extended our previous work on the expression pattern of Foxi3 expression (Ohyama and Groves, 2004) to gain further insights into its developmental function. Prior to branchial arch outgrowth, Foxi3 is expressed broadly in the pharyngeal region in both ectoderm and endoderm at the six somite stage (6ss; Figure 1). As the first branchial arch (BA1) becomes morphologically distinct, Foxi3 expression is down-regulated in expanding arch ectoderm and its expression domain bifurcates into two distinct patches of tissue anterior and posterior to the arch itself. By the 15ss, Foxi3 expression is restricted to epithelium between the two morphologically identifiable arches present at this stage, BA1 and BA2. At this point, three Foxi3 expression domains are observable: anterior to BA1 at low levels, in the cleft between BA1 and BA2, and posterior to BA2. Foxi3 is continues to be expressed in the ectoderm and in the endoderm underlying these three domains (Figure 1). As the remaining branchial arches develop, Foxi3 expression divides further and is expressed in the endoderm of each pharyngeal pouch as well as the overlying cleft ectoderm, with stronger expression around posterior, later-developing arches and the expression anterior arches gradually becoming fainter (Figure 1). It is important to note that Foxi3 is expressed neither in branchial arch mesoderm nor in the cranial neural crest cells that populate arch mesenchyme (Figure 1, section).
Figure 1. Foxi3 is expressed in pharyngeal ectoderm and endoderm.

Prior to branchial arch outgrowth, Foxi3 is expressed broadly in the pharyngeal region in both ectoderm and endoderm. As arch development progresses, Foxi3 expression is refined to the clefts between the arches (arrowheads). Foxi3 is expressed in both ectoderm and endoderm, but is not expressed in the mesoderm or neural crest cells that make up the mesenchyme of the arches. A1 – branchial arch 1, A2 – branchial arch 2, P1 – pharyngeal pouch 1, P2 – pharyngeal pouch 2, ss – somite stage of development. Scale bars are 100μm.
Foxi3 mutant mice have aberrant pharyngeal morphology and severe defects in arch derived skeletal structures
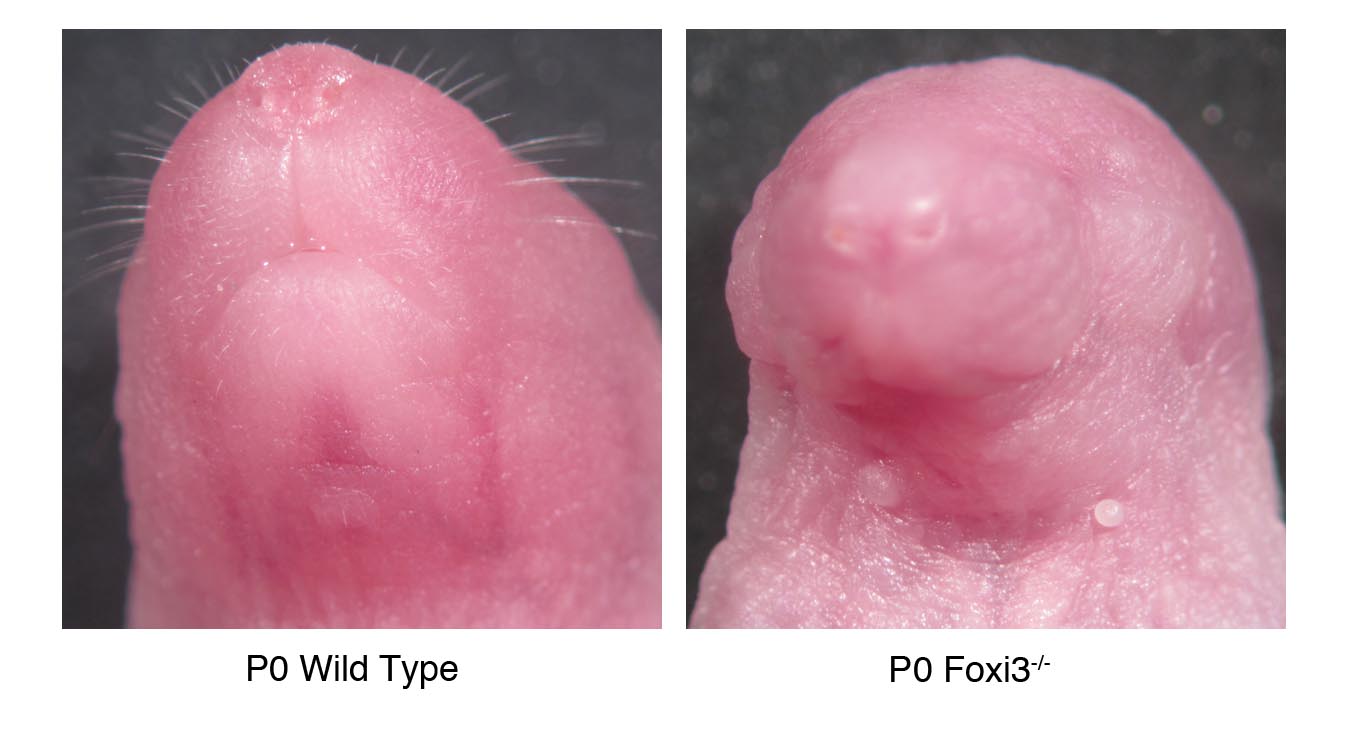
We generated Foxi3 mutant mice by targeting the second of two exons coding for Foxi3 using a conditional strategy. (Figure 2). To determine the function of Foxi3 in pharyngeal development, we examined Foxi3 mutant mice at various stages in embryonic development, beginning at embryonic day 8.0 (E8.0) through birth (P0). Foxi3 mutants are not viable, with embryos dying across a range of ages beginning around E9.5. Some Foxi3 mutant embryos are born, but die immediately after birth. These pups are readily identifiable by their lack of mouth, with a continuous ectodermal covering over the lower half of the face, and by the absence of external ear pinnae (Figure 3). Although whisker follicles are found on the snouts of Foxi3 mutant pups, whiskers are absent (Supplementary Figure 1), consistent with the expression of Foxi3 in hair follicles (Drogemuller et al., 2008). Dead pups have been noted in cages while the dam is giving birth to the remainder of the litter. As-wild type and heterozygous pups do not yet display milk spots in these litters, it appears unlikely that the mutants die due to starvation. More probably the disrupted development of their lower faces renders mutants unable to breathe.
Figure 3. Neural crest derived bones of the head are malformed or absent in Foxi3 mutants.

In Foxi3 mutants at E16.5, Meckel's cartilage (mc) is misshapen and a distinct mandible is absent. At E18.5 the full range of Foxi3 mutant skeletal defects are apparent. A truncated mandible (m*) is fused to the maxilla (mx*). The maxilla is malformed and the jugal (j) is absent (missing structures indicated by arrowheads), and the squamosal bone (sq*) and palatines (p*) are misshapen. The middle and inner ears are absent, indicated by an asterisk (*). Removed from the maxilla, the fused portion of mandible is small, truncated, and asymmetrical. Foxi3 mutant pups lack an external ear and the lower half of the face is covered in continuous ectoderm. In a magnified view of the Foxi3 mutant ear all ear structures are absent, including tympanic ring (t), malleus (ma), incus (i), and stapes (s). Basisphenoid – bs; premaxilla – pmx; vomer – v.
The apparent lack of mouths and the smaller size of their faces suggests some skeletal alterations in Foxi3 mutants. In order to carefully catalog changes in bone and cartilage structures resulting from loss of FOXI3, we stained the heads of mutant and wild type mouse embryos with Alcian Blue and Alizarin Red to reveal cartilage and bone respectively (Figure 3). Skeletal staining of the heads of E18.5 Foxi3 mutant embryos reveals a number of severe defects in bone and cartilage development. In Foxi3 mutants, only a small anterior portion of the mandible develops and is fused to the distal end of maxilla. The maxilla itself is also malformed, as is the squamosal portion of the temporal bone. The jugal is absent and the palatines are reduced in size. Additionally, there is no sign of the tympanic ring or incus, malleus, or stapes of the middle ear, and the entire temporal bone and inner ear are absent (Figure 3). A full description of the inner ear phenotype of Foxi3 mutant mice will be published separately. Although the mandible is almost entirely absent, bone and cartilage staining at E16.5 reveals that at least some elements of Meckel's cartilage are present in Foxi3 mutant embryos. However, it is misshapen, curving downward and away from the midline (Figure 3). To document any other roles of Foxi3 in skeletal development, we also examined E18.5 whole embryos. We observed no gross morphological defects in any skeletal elements below the neck in Foxi3 mutants, although more than half of embryos that develop to E18.5 displayed a wide gap between the frontal bones of the skull (data not shown).
Interestingly, the affected skeletal structures we observed in Foxi3 mutants are derivatives of the cranial neural crest cells that populate BA1 and 2, cell populations that do not express Foxi3 (Figure 1). We therefore examined embryos at earlier embryonic stages to assess whether absence of FOXI3 from ectoderm and endoderm resulted in a developmental defect that might eventually lead to loss of neural crest derived skeletal structures. Indeed, we found the anterior pharyngeal region to be morphologically disrupted early in development in Foxi3 mutants. At 15ss, when a distinct BA1 is readily apparent in wild-type mouse embryos, coronal sections through the pharyngeal region of Foxi3 mutants show a failure to individuate distinct arches. The pharyngeal pouches that segregate mesenchyme of one arch from the next are not present; there are no contact points between pharyngeal endoderm and ectoderm and all arch mesenchyme is continuous (Figure 4A). This morphological defect is particularly striking a day later in development in 25ss whole-mount embryos, where well defined arches in wild-type embryos are replaced by a smooth expanse of ectoderm in Foxi3 mutants (Figure 4A). Notably, the transcription factor Pax9, normally expressed most strongly in the pharyngeal pouches during early arch outgrowth, is expressed continuously throughout the pharyngeal endoderm of 15 somite Foxi3 mutants (Figure 4B)(Neubuser et al., 1995). In a pattern highly reminiscent of endodermal Foxi3 expression, Pax 9 expression diminishes from the pouches in a gradient, with lower expression in anterior pouches than in posterior pouches by 25 somites. In Foxi3 mutants at 25ss as in 15ss, expression is continuous throughout the endoderm and persists in the anterior pharyngeal region (Figure 4B). This altered pattern suggests a failure of endoderm to establish pouch-and arch lining-specific domains in the absence of Foxi3. However, in spite of the failure of pharyngeal pouches to form morphological barriers between arches in Foxi3 mutants, we observed that anterior Hox-negative and posterior Hoxb2 positive populations of neural crest cells in the arches do not intermingle, suggesting that initially neural crest identity may not be affected in Foxi3 mutants (data not shown).
Figure 4. Foxi3 mutant mice do not form pharyngeal pouches.

(A) Coronal sections through 12ss embryos stained with DAPI reveal absence of a distinct pharyngeal pouch 1 (p1) between the first two pharyngeal arches (a1, a2) in the Foxi3 mutant (asterisk). At 25 somites, confocal imaging of DAPI-stained embryos shows that Foxi3 mutant embryos have clearly failed to form distinct arches (asterisk). (B) Whole mount embryos at 15 somites and 25 somites show less distinct patterns of Pax9 expression around the presumptive pharyngeal pouches. Coronal sections through these embryos reveal continuous expression of Pax9 along the entire extent of pharyngeal endoderm at both ages in Foxi3 mutants, whereas Pax9 is restricted to pouches in wild type embryos.
Neural Crest Cells in Foxi3 mutants migrate to the arches and begin to differentiate, but then die
Although Foxi3 is not expressed in migrating neural crest cells, these cells must migrate past Foxi3-expressing ectoderm and endoderm in order to enter the arches. To address the possibilities that neural crest cells are not entering the branchial arches, or are unable to differentiate in the altered pharyngeal environment of Foxi3 mutants, we examined mutant and wild type embryos for expression of migratory and post-migratory neural crest cell markers. We observed Sox10-expressing cells exiting the neural tube at 15ss, suggesting that cranial neural crest cell formation and early emigration from the midbrain and hindbrain are unaffected. Although the migration pattern of Sox10-positive cells is slightly altered in Foxi3 mutants due to the absence of the otic placode and pharyngeal pouches, we saw no obvious difference in the numbers of cranial neural crest cells entering the pharyngeal region (Figure 5A).
Figure 5. Neural crest cells migrate, populate and pattern the Foxi3 mutant pharyngeal region.

A: In 15 somite staged embryos, neural crest cells expressing Sox10 migrate out of the neural tube and into the pharyngeal region (asterisks), but do not divide into separate arch populations (asterisk in wild type embryo). After entering the arches, neural crest cells establish a proximal-distal axis through nested and combinatorial expression of Dlx transcription factors (illustration in A). B: In 18 somite staged Foxi3 mutants, neural crest cells adopt the nested expression pattern (A), but the population of cells expressing the intermediate and distal factors Dlx5 and Dlx3 are smaller in the mutant first arch. Scale bars are 100μm.
During early arch development, post-migratory neural crest cells establish a proximal-distal axis in each arch via a nested pattern of expression of six Dlx transcription factors (Figure 5A) (Qiu et al., 1997). To ascertain whether neural crest cells are able to initiate a differentiation program in Foxi3 mutant arches, we examined 18ss embryos for expression of Dlx2, 3 and 5. In spite of the failure of Foxi3 mutants to form distinct arches, neural crest cells do qualitatively adopt the typical nested Dlx pattern. We saw no significant differences in the extent of Dlx2, which is expressed throughout the proximo-distal extent of the first arch (Figure 5B). However, the numbers of cells expressing the distal marker Dlx3 are noticeably reduced in the mesenchyme at the level of presumptive BA1 (Figure 5B). The size and intensity of the intermediate to distal Dlx5-expressing population is likewise reduced, but to a lesser extent than Dlx3.
Although neural crest cells migrate into the pharyngeal region of Foxi3 mutants, the structures derived from many of those cells are absent, leading to the question of what becomes of the BA1 mesenchyme. An obvious possibility is apoptosis of neural crest cells. We stained transverse sections through presumptive BA1 in Foxi3 mutants for activated caspase-3, a marker of apoptosis, and for the transcription factor AP2α, expressed in neural crest cells and pharyngeal ectoderm but not in pharyngeal mesoderm (Brewer et al., 2002). Prior to 25ss we saw no increase in apoptosis among cells in presumptive BA1 of Foxi3 mutants (data not shown); however, beginning at 25ss a population of mesenchymal cells near the ventral edge of the pharyngeal ectoderm contain activated caspase-3, indicating an abnormal increase in cell death in cells corresponding to the distal population of cells in BA1 (Figure 6A). The difference in apoptotic cells is significant, with wild type embryo sections containing an average of 5.8 dying cells per section versus 14.7 cells in Foxi3 mutants (Figure 6B). Dying cells are concentrated in the posterior region of presumptive BA1. Adjacent sections from the same Foxi3 mutant embryos stained with AP2α suggest that these dying cells are neural crest rather than mesoderm (Figure 6A).
Figure 6. Neural crest cells undergo apoptosis in Foxi3 mutant arches.

(A) Neural crest cells, stained with AP2α, undergo apoptosis, indicated by active caspase-3 in the distal tip of Foxi3 mutant arches (box 3). Some apoptosis in the proximal region of wild type and Foxi3 mutant arches (boxes 1 and 2). We excluded these cells from neural crest cell death quantification because they exist in both populations and are AP2α-negative. Higher magnification images of the regions highlighted with white boxes are shown below. Scale bars are 100μm. (B) The increase in apoptosis in Foxi3 mutant arches is statistically significant (p=0.04). Foxi3 mutants n=5; wild type n=6 embryos. Error bars represent the standard error of the mean.
Fgf8 expression in pharyngeal ectoderm is delayed in Foxi3 mutants
Foxi3 is a transcription factor expressed in epithelial cells, but the bulk of the affected structures in the arch region of Foxi3 mutants are neural crest-derived. It is therefore likely that a signaling molecule that is expressed in Foxi3-positive cells communicates from pharyngeal epithelium to neural crest mesenchyme and that expression of such a signaling factor might be altered in Foxi3 mutants. An excellent candidate for this signaling factor is Fibroblast growth factor 8 (Fgf8). Fgf8 has a well-documented role in cell survival in the pharyngeal region of amniotes (Abu-Issa et al., 2002; Tucker et al., 1999). The requirement for FGF signaling to support cranial neural crest cell survival and branchial arch development is conserved from fish to mammals, and loss of pharyngeal Fgfs leads to missing craniofacial skeletal structures in vertebrates (Crump et al., 2004; Trumpp et al., 1999). Foxi3 and Fgf8 are expressed in similar patterns in the pharyngeal region, and Foxi3 mutants and Fgf8 branchial ectoderm-conditional knockouts have similar phenotypes, in particular, a significantly reduced mandible and concomitant apoptosis of cranial neural crest cells in the first arch (Abu-Issa et al., 2002; Crossley and Martin, 1995; Trumpp et al., 1999). In Foxi3 mutants, Fgf8 expression in the pharyngeal ectoderm is delayed until well after BA1 normally develops. At 15ss, Fgf8 is absent from pharyngeal ectoderm of Foxi3 mutants while strongly expressed in wild-type embryos (Figure 7A). Fgf8 expression is present but reduced in the endoderm at these stages (Figure 7A). Fgf8 is eventually expressed in pharyngeal ectoderm hours later at about 22ss (Figure 7A).
Figure 7. Fgf8 expression is delayed in Foxi3 mutant pharyngeal ectoderm.

The expression pattern of Fgf8 at 15 somites is similar to the Foxi3 expression pattern at the same stage. In 15 somite staged Foxi3 mutants, Fgf8 expression is substantially reduced and is present only in pharyngeal endoderm but not pharyngeal ectoderm. Expression of downstream transcription factor Erm is reduced in Foxi3 mutants to a few cells adjacent to the endoderm. MAPK signaling as indicated by di-phosphorylated Erk (pErk) is detectable in many fewer mesenchymal cells in Foxi3 mutant embryo arches. The level of staining in Foxi3 mutants was similar to that seen in wild type embryos incubated for 30 minutes in the MAPK inhibitor U0126 (not shown). Expression of Fgf8 partially recovers in pharyngeal ectoderm by 22 somites, but is still not expressed as broadly as in wild type embryos of corresponding ages. Scale bars are 100μm.
Although Fgf8 is present in the pharyngeal endoderm of Foxi3 mutants, the residual FGF8 appeared insufficient to induce a typical signaling response in arch mesenchyme. At 15ss, expression of Erm, an ETS-domain containing transcription factor induced by FGF signaling is absent from the distal mesenchyme of presumptive BA1 in Foxi3 mutants but not in the brain (Figure 7B). Likewise, Foxi3 mutant mesenchyme displays reduced MAPK signaling, as assessed by reduced presence of phosphorylated ERK in mesenchymal cells (Figure 7B).
To clarify the extent to which reduced and delayed FGF8 signaling from the ectoderm influences not just neural crest cell survival, but also overall pattering in BA1, we examined expression of additional transcription factors. Both goosecoid (Gsc) and Lhx7 in the BA1 mesenchyme are expressed around E9.5 in response to FGF8 (Grigoriou et al., 1998; Trumpp et al., 1999). In Foxi3 mutants, as in wild type embryos at 25ss, Gsc is expressed in the proximal region of BA1 between the mandibular and maxillary prominences (Figure 8). Lhx7, in contrast, is expressed in two distinct domains at 25ss: at the distal tip of BA1 and at the anterior extreme of BA1 where the maxillary prominence is just beginning to expand (Figure 8). In Foxi3 mutants, only one domain is present in the truncated BA1, suggesting an absence of much of the intervening region of the arch (Figure 8). A possible explanation for this difference may be that Gsc expressing tissues are still present in the absence of Foxi3, but the severely truncated arches in the mutants have specifically lost transcriptionally unique intermediate or distal domains.
Figure 8. Patterning of truncated arches is minimally altered in Foxi3 mutants.

At both 15 somites and 25 somites, Pitx1 expression around BA1 is unaltered. In the Foxi3 mutant there is a reduction in Pitx1-positive cells anterior to BA1 (asterisk in Pitx1 25ss). Gsc expression remains unchanged in Foxi3 mutants (arrowheads in Goosecoid). At 25 somites, Lhx7 is found in two distinct domains in wild type embryos (arrowheads in Lhx7). Only one domain is found in Foxi3 mutant embryos at the same age (asterisk in Lhx7). Similarly, at 25 somites in wild type embryos, Msx1 is found in two domains, whereas only one domain is present in Foxi3 mutants. This pattern may represent a loss of the distal Lhx7 and Msx1 domains, or a fusion of the two domains for each of these factors.
That hypothesis may be supported by evidence from transcription factors regulated through other signaling pathways in the arches. Pitx1 expression and Msx1 expression have been described to be unaltered by loss of Fgf8 from the ectoderm (Liu et al., 2005; Trumpp et al., 1999). Interestingly, the pattern of Msx1 expression is similar to Lhx7, as is the pattern change in Foxi3 mutants, with two BA1 Msx1 domains in the wild type embryo reduced to one domain in the Foxi3 mutant. Although the major morphological differences between Foxi3 mutants and wild type embryos result in a noticeable decrease in Pitx1 expression area, the relative location in which it is expressed is unchanged in Foxi3 mutant arches at both 15ss and 25ss (Figure 8). The changes seen in Foxi3 mutants are consistent with a loss of intermediate and distal arch transcriptional domains. Of note, but beyond the scope of this study, Pitx1 expression anterior to BA1 is reduced in Foxi3 mutants at 25ss (asterisk, Figure 8).
Restoration of Fgf signal in zebrafish foxi mutants rescues the cell death and skeletal phenotypes
To confirm that loss of ectodermal FGF signaling was responsible for neural crest cell death in the arches of Foxi mutants, we preformed a rescue experiment in zebrafish. The functional homologues of mouse Foxi3 and pharyngeal Fgf8 in zebrafish are foxi1 and fgf3 (Nissen et al., 2003; Solomon et al., 2003a). Accordingly, zebrafish foxi1 is expressed in pharyngeal ectoderm and endoderm but is excluded from neural crest-derived pharyngeal arches(Solomon et al., 2003a). A previous study reported that nascent migratory neural crest cells expressing krox20 initially co-express foxi1(Nissen et al., 2003). However, analysis of gene expression under high magnification revealed that migratory streams of neural crest expressing krox20 remain distinct from the pharyngeal domain of foxi1, with no cells co-expressing both markers (Figure 9A, A’). We confirmed in tissue sections that zebrafish fgf3 is expressed in pharyngeal pouch endoderm during appropriate stages to affect pharyngeal arch morphogenesis (Figure 9C, C’). To test if foxi1 also regulates fgf3 expression to promote neural crest survival, we knocked down foxi1 with previously characterized translation-blocking morpholinos (Solomon et al., 2003b). Pharyngeal expression of fgf3 is down-regulated in foxi1 morphants as early as 22 hours post fertilization (hpf), followed by a wave of apoptosis in pharyngeal arch tissue peaking at around 26 hpf, with an average of 102.1 apoptotic cells in the pharyngeal arches of a morphants (Figure 10 A, B, n=7) (Nissen et al., 2003).
Figure 9. Fgf mis-expression in zebrafish rescues the cell death phenotype in foxi1 morphants.

A, A’: At 14 hpf (10 somites), krox20 (red) marks streams of nascent neural crest migrating from the hindbrain whereas foxi1 (black) marks non-neural ectoderm abutting the hindbrain. No cells co-express both markers. The boxed area in A is magnified in A’. Images show a dorsal view with anterior to the left. B-I’: Embryos at later stages are oriented with anterior to the left and dorsal up, with the otic vesicle circled with a dashed line and pharyngeal pouches marked with arrows. B-C’: Expression of fgf3 in wild-type control embryos marks pharyngeal pouch endoderm at 22 hpf (B) and 24 hpf (C, C’). The vertical line in C marks the plane of section in C’. D, D’: A hs:fgf3 transgenic embryo injected with foxi1-MO shows a reduced level of fgf3 expression on the non-activated side at 22 hpf (D) but shows dramatically elevated expression on the laser-activated side (D’). Note the horizontal axis in D is inverted to facilitate comparison. Laser-activation was performed at 20 hpf, focusing on the pharyngeal region, shown in the inset to panel D’. E-I’: Live embryos were incubated with acridine orange at 26 hpf to label cells undergoing apoptosis. A wild-type control embryo shows very few apoptotic cells (E) whereas a foxi1 morphant shows a marked increase in apoptosis (F). The cell death phenotype normally seen in foxi1 morphants was strongly suppressed by global low-level activation of hs:fgf3 (37°C for 30 minutes beginning at 20 hpf) (G) or hs:fgf8 (35°C for 6 hours beginning at 20 hpf) (H). A hs:fgf3 transgenic embryo injected with foxi1-MO showed elevated apoptosis on the non-activated side (I, horizontal axis inverted for easier comparison), but apoptosis was strongly suppressed on the laser-irradiated side (I’).
Figure 10. Over-expression of fgf8 or fgf3 in zebrafish foxi1 morphants significantly reduces numbers of apoptotic cells in the arches.

Foxi1 morphants have a significant increase in apoptosis in the arch mesenchyme over wild type zebrafish. The apoptosis phenotype can be partially rescued by global heat shock to over-express fgf8 (A) or fgf3 (B), both under the control of heat shock promoters. Fgf induction by heat shock in the absence of foxi1 knock-down does not increase cell death in the arches (A and B). The rescue effect is specific to ectodermal fgf. Induction of fgf3 in the arch ectoderm using a low energy laser significantly rescues cell death in the arches of foxi1 morphants (C). For all graphs, error bars represent the standard error of the mean, and tests of statistical significance are shown by brackets. *** = p<0.05.
To investigate the relationship between altered Fgf signaling and elevated cell death, we injected foxi1 morpholinos into transgenic zebrafish containing heat shock-inducible transgenes encoding fgf3 or fgf8. High level mis-expression of either transgene caused developmental defects precluding meaningful interpretation. However, global low-level activation of hs:fgf3 (37°C, 30 minutes) beginning at 20 hpf significantly suppressed cell death in foxi1 morphants, reducing the average number of apoptotic cells in the arches nearly by half to 52.7 cells per embryo (Figure 9G, Figure 10B, n=20). Similarly, global activation of hs:fgf8 (35°C for 6 hours) beginning at 20 hpf also significantly suppressed cell death in foxi1 morphants, resulting in an average of 45.3 apoptotic cells in the arches (Figure 9H, Figure 10A, n=20).
Because global activation of these transgenes might not suitably mimic endodermal fgf3, we used an ablative laser set on low power to locally activate hs:fgf3 in the pharyngeal region beginning at 20 hpf (see inset cartoon in Figure 9D’). Subsequent assessment of fgf3 expression confirmed the transgene remained active on the laser-irradiated side through at least at 22 hpf (Figure 9D, D’). Moreover, all specimens showed significant suppression of cell death on the laser-irradiated side, whereas cell death was not rescued on the non-irradiated side: an average of 76.6 apoptotic cells on the irradiated side, versus 165.2 on the non-irradiated side. (Figure 9I, I’, Figure 10C, n=8). In contrast, laser activation of hs:fgf3 after 22 hpf did not rescue the cell death phenotype in foxi1 morphants (n=3, data not shown), indicating that the requirement for Fgf signaling is highly stage-dependent.
DISCUSSION
The complex orchestration of cell migration, differentiation and death underlying the formation of the vertebrate face and jaw is regulated by a wide range of signaling molecules whose expression is established by many families of transcription factors (Cobourne and Sharpe, 2003; Depew et al., 2005; Medeiros and Crump, 2012) Subtle changes in the timing, location or strength of gene expression can serve as a substrate for cranial morphological diversity in vertebrates, or alternatively lead to a wide range of craniofacial abnormalities (Minoux and Rijli, 2010; Passos-Bueno et al., 2009; Szabo-Rogers et al., 2010). In the present study, we show that loss of Foxi3, a mouse Forkhead family transcription factor leads to severe craniofacial defects consistent with a loss of most anterior branchial arch derivatives. We show that although Foxi3 is expressed exclusively in branchial arch ectoderm and endoderm, the principle defect in Foxi3 mutants is a significant loss of arch neural crest cells due to apoptosis. We propose that Foxi3 indirectly promotes the survival of arch neural crest cells by establishing the expression of Fgf8 in branchial clefts. Moreover, we show that zebrafish foxi gene regulates arch survival in a similar fashion: loss of foxi1 from the zebrafish arch region leads to cell death that can be rescued by re-activating expression of fgf3 in the arch region.
Foxi3 is expressed exclusively in pharyngeal ectoderm and endoderm (Figure 1), but neural crest cells that migrate into the branchial arches are unable to develop into skeletal arch derivatives in the absence of Foxi3. In Foxi3 mutants, neural crest cells appear to be induced normally, exit the neural tube in expected numbers, migrate along roughly correct paths to the presumptive branchial arches, and populate the space between pharyngeal ectoderm and endoderm (Figure 5). In Foxi3 mutants, the normal eversion of pharyngeal endoderm and its apposition with pharyngeal ectoderm fail to occur (Figure 4). Interestingly, one marker of the pharyngeal pouches, Pax9, is expanded in Foxi3 mutants throughout the endoderm. In spite of a failure in pharyngeal pouch formation or other obvious barriers between BA1 and BA2, Hox-negative post-migratory neural crest cells from the midbrain and rhombomeres 1 and 2 that normally populate BA1 do not appear to mix with the Hoxb2-expressing neural crest from more posterior rhombomeres that populates more posterior arches. Alternatively, it is possible that mesodermal signals that have been suggested to modulate Hox gene expression in post-migratory crest are still present Foxi3 mutants (Trainor and Krumlauf, 2000).
Upon arrival in the presumptive arches, neural crest cells begin to respond to arch-specific differentiation signals and establish the nested pattern of Dlx transcription factor expression that defines the proximal-distal axis within the arch mesenchyme (Depew et al., 2005). It is at this point - when neural crest cells have completed their migration and begun differentiation - that their development goes awry in Foxi3 mutants. Since Foxi3 is expressed in the epithelium of the branchial clefts and pouches, it cannot directly contribute to survival and differentiation of neural crest cells; it must be acting through a pathway with additional components.
One good candidate for such a survival factor is Fgf8: it is expressed in pharyngeal arches as crest cells invade, and conditional or hypomorphic alleles of Fgf8 display crest cell death and severe morphological defects in arch derivatives (Abu-Issa et al., 2002; Crossley and Martin, 1995). Our data suggest that Foxi3 regulates Fgf8 expression in pharyngeal cleft ectoderm, and that the transient loss of Fgf8 expression in Foxi3 mutants is sufficient to cause crest cell death. We do not see Fgf8 expression in the developing arch ectoderm of Foxi3 mutants at the 15ss (approximately E8.75-E9.0), Neural crest cells at the most ventral region of the pharyngeal tissue in Foxi3 mutants, the presumptive distal tip of BA1, begin to undergo apoptosis around E9.75. The timing of this cell death is consistent with the death associated with reduced or absent FGF8 signaling from the pharyngeal ectoderm in Fgf8neo/+ mutants(Abu-Issa et al., 2002). Interestingly, by this stage of development Fgf8 expression has recovered in the ectoderm of Foxi3 mutants (Figure 7A). This finding suggests that the critical period for FGF8 signaling to promote neural crest cell survival is a window between E8.25-9.25 when the cells are migrating past a region of strong FGF8 signaling into the arches. In exploring this hypothesis via rescue experiments in zebrafish embryos, we found that induction of fgf3 expression in ectoderm in zebrafish foxi1 morphants can indeed rescue the cell death phenotype, bypassing the need for foxi expression. Thus the proximal cause for neural crest cell death in Foxi3 mutant mice and foxi1 mutant zebrafish appears to be a transient absence of Fgf signaling at a crucial time point during neural crest cell migration.
We propose two potential mechanisms by which Foxi3 functions in relation to FGF signaling in the course of pharyngeal development. The first possibility is a direct genetic pathway. Foxi3 may regulate a transcriptional program that promotes Fgf8 expression. The Fgf8 regulatory region is extremely large. Loci known to regulate specific Fgf8 expression domains have been identified hundreds of kilobases away from the coding region, and are found within other nearby genes (Beermann et al., 2006; Komisarczuk et al., 2009; Marinic et al., 2013). Although no experimentally validated binding site for mammalian Foxi3 has been identified, the DNA binding domain of Foxi proteins is highly conserved among homologs and across vertebrate species. Two binding site sequences for human Foxi1, one shared with zebrafish foxi1 have been identified in yeast experiment (Zeng et al., 2008). There are six regions of DNA within 500kb downstream of Fgf8 that are conserved between chick, mouse, and human, and that contain the predicted Foxi1 binding motif. In chick embryos, none of these conserved sequences fused to a minimal promoter induced reporter gene expression in the pharyngeal region (data not shown). This does not rule out the possibility that FOXI3 directly regulates Fgf8 expression, but if FOXI3 is responsible for inducing Fgf8 it may be acting through transcriptional regulation of one or more an additional intermediate factors.
The second possibility we propose for Foxi3 function is based on the unusual pharyngeal morphology of Foxi3 mutants; Foxi3 may be involved in modifying epithelial morphology in the pharyngeal region, specifically the development of pharyngeal pouches. Endoderm ablation experiments in chick embryos have demonstrated that contact with endoderm is required for the induction of Fgf8 in pharyngeal ectoderm (Haworth et al., 2004; Haworth et al., 2007). Thus, the failure in pouch formation seen in Foxi3 mutants may lead to absence of ectodermal Fgf8 and may be the underlying cause of the craniofacial defects in these embryos. In this case, the transcriptional role of FOXI3 could be in modulating expression of genes involved with tissue remodeling or cell adhesion. Certainly the refinement of Foxi3 expression to areas of contact between ectoderm and endoderm would be consistent with a function in promoting expression of adhesion factors required to maintain pharyngeal pouch integrity under the onslaught of neural crest cell migration. A transcriptional profiling experiment may be illuminating in determining the precise transcriptional role of Foxi3 in epithelial cells.
Whether Foxi3 acts through genetic regulation of Fgf8, through regulation of tissue remodeling and adhesion factors, or through some other mechanism, it is clearly of critical importance to pharyngeal development. Further exploration of the genetic pathways in which FOXI3 is integrated could open the way to better understanding of signaling pathways in the pharyngeal region and the conserved morphogenetic processes responsible for generating one defining factor of the vertebrate morphology – the branchial arches.
Supplementary Material
{kind=link}
ACKNOWLEDGEMENTS
We thank Gail Martin, John Rubenstein, Maria Morasso, Jin Xian Liu, Michael Wegner and Annette Neubüser for probes, Trevor Williams for advice on the AP2α antibody, Tegy Vadakkan for assistance with confocal imaging and Alyssa Faught, Hongyuan Zhang and Huiling Li for excellent technical assistance. The 3B5 monoclonal antibody to AP-2α developed by Trevor Williams was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242. This work was supported by RO1 DC013072 and RO1 DC004675 (A.K.G.), RO3 DC007349 (T.O.) and RO1 DC003806 (B.B.R.). R.K.E. was supported in part by T32 HD055200.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Foxi3 mutant mice were designed and generated by Takahiro Ohyama, who also provided the data in Figure 2. All experiments except those in Figures 2, 9 and 10 were designed by Renée Edlund and Andy Groves and performed by Renée Edlund. The zebrafish experiments in Figure 9 and 10 were designed by Renée Edlund, Husniye Kantarci, Bruce Riley and Andy Groves and performed by Husniye Kantarci and Bruce Riley. The manuscript was written by Renée Edlund and edited by Andy Groves, Takahiro Ohyama Husniye Kantarci and Bruce Riley.
REFERENCES
- Abu-Issa R, Smyth G, Smoak I, Yamamura K, Meyers EN. Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development. 2002;129:4613–4625. doi: 10.1242/dev.129.19.4613. [DOI] [PubMed] [Google Scholar]
- Adams DJ, Quail MA, Cox T, van der Weyden L, Gorick BD, Su Q, Chan WI, Davies R, Bonfield JK, Law F, Humphray S, Plumb B, Liu P, Rogers J, Bradley A. A genome-wide, end-sequenced 129Sv BAC library resource for targeting vector construction. Genomics. 2005;86:753–758. doi: 10.1016/j.ygeno.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Beermann F, Kaloulis K, Hofmann D, Murisier F, Bucher P, Trumpp A. Identification of evolutionarily conserved regulatory elements in the mouse Fgf8 locus. Genesis. 2006;44:1–6. doi: 10.1002/gene.20177. [DOI] [PubMed] [Google Scholar]
- Brewer S, Jiang X, Donaldson S, Williams T, Sucov HM. Requirement for AP-2alpha in cardiac outflow tract morphogenesis. Mechanisms of development. 2002;110:139–149. doi: 10.1016/s0925-4773(01)00579-2. [DOI] [PubMed] [Google Scholar]
- Brito JM, Teillet MA, Le Douarin NM. An early role for sonic hedgehog from foregut endoderm in jaw development: ensuring neural crest cell survival. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:11607–11612. doi: 10.1073/pnas.0604751103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobourne MT, Sharpe PT. Tooth and jaw: molecular mechanisms of patterning in the first branchial arch. Archives of oral biology. 2003;48:1–14. doi: 10.1016/s0003-9969(02)00208-x. [DOI] [PubMed] [Google Scholar]
- Couly G, Creuzet S, Bennaceur S, Vincent C, Le Douarin NM. Interactions between Hox-negative cephalic neural crest cells and the foregut endoderm in patterning the facial skeleton in the vertebrate head. Development. 2002;129:1061–1073. doi: 10.1242/dev.129.4.1061. [DOI] [PubMed] [Google Scholar]
- Crossley PH, Martin GR. The mouse Fgf8 gene encodes a family of polypeptides and is expressed in regions that direct outgrowth and patterning in the developing embryo. Development. 1995;121:439–451. doi: 10.1242/dev.121.2.439. [DOI] [PubMed] [Google Scholar]
- Crump JG, Maves L, Lawson ND, Weinstein BM, Kimmel CB. An essential role for Fgfs in endodermal pouch formation influences later craniofacial skeletal patterning. Development. 2004;131:5703–5716. doi: 10.1242/dev.01444. [DOI] [PubMed] [Google Scholar]
- Danesh SM, Villasenor A, Chong D, Soukup C, Cleaver O. BMP and BMP receptor expression during murine organogenesis. Gene expression patterns : GEP. 2009;9:255–265. doi: 10.1016/j.gep.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David NB, Saint-Etienne L, Tsang M, Schilling TF, Rosa FM. Requirement for endoderm and FGF3 in ventral head skeleton formation. Development. 2002;129:4457–4468. doi: 10.1242/dev.129.19.4457. [DOI] [PubMed] [Google Scholar]
- Depew MJ, Simpson CA, Morasso M, Rubenstein JL. Reassessing the Dlx code: the genetic regulation of branchial arch skeletal pattern and development. Journal of anatomy. 2005;207:501–561. doi: 10.1111/j.1469-7580.2005.00487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drogemuller C, Karlsson EK, Hytonen MK, Perloski M, Dolf G, Sainio K, Lohi H, Lindblad-Toh K, Leeb T. A mutation in hairless dogs implicates FOXI3 in ectodermal development. Science. 2008;321:1462. doi: 10.1126/science.1162525. [DOI] [PubMed] [Google Scholar]
- Grigoriou M, Tucker AS, Sharpe PT, Pachnis V. Expression and regulation of Lhx6 and Lhx7, a novel subfamily of LIM homeodomain encoding genes, suggests a role in mammalian head development. Development. 1998;125:2063–2074. doi: 10.1242/dev.125.11.2063. [DOI] [PubMed] [Google Scholar]
- Haworth KE, Healy C, Morgan P, Sharpe PT. Regionalisation of early head ectoderm is regulated by endoderm and prepatterns the orofacial epithelium. Development. 2004;131:4797–4806. doi: 10.1242/dev.01337. [DOI] [PubMed] [Google Scholar]
- Haworth KE, Wilson JM, Grevellec A, Cobourne MT, Healy C, Helms JA, Sharpe PT, Tucker AS. Sonic hedgehog in the pharyngeal endoderm controls arch pattern via regulation of Fgf8 in head ectoderm. Developmental biology. 2007;303:244–258. doi: 10.1016/j.ydbio.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Hulander M, Kiernan AE, Blomqvist SR, Carlsson P, Samuelsson EJ, Johansson BR, Steel KP, Enerback S. Lack of pendrin expression leads to deafness and expansion of the endolymphatic compartment in inner ears of Foxi1 null mutant mice. Development. 2003;130:2013–2025. doi: 10.1242/dev.00376. [DOI] [PubMed] [Google Scholar]
- Hulander M, Wurst W, Carlsson P, Enerback S. The winged helix transcription factor Fkh10 is required for normal development of the inner ear. Nature genetics. 1998;20:374–376. doi: 10.1038/3850. [DOI] [PubMed] [Google Scholar]
- Khatri SB, Groves AK. Expression of the Foxi2 and Foxi3 transcription factors during development of chicken sensory placodes and pharyngeal arches. Gene expression patterns : GEP. 2013;13:38–42. doi: 10.1016/j.gep.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komisarczuk AZ, Kawakami K, Becker TS. Cis-regulation and chromosomal rearrangement of the fgf8 locus after the teleost/tetrapod split. Developmental biology. 2009;336:301–312. doi: 10.1016/j.ydbio.2009.09.029. [DOI] [PubMed] [Google Scholar]
- Liu W, Selever J, Murali D, Sun X, Brugger SM, Ma L, Schwartz RJ, Maxson R, Furuta Y, Martin JF. Threshold-specific requirements for Bmp4 in mandibular development. Developmental biology. 2005;283:282–293. doi: 10.1016/j.ydbio.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Lumsden A, Sprawson N, Graham A. Segmental origin and migration of neural crest cells in the hindbrain region of the chick embryo. Development. 1991;113:1281–1291. doi: 10.1242/dev.113.4.1281. [DOI] [PubMed] [Google Scholar]
- Maemura K, Kurihara H, Kurihara Y, Oda H, Ishikawa T, Copeland NG, Gilbert DJ, Jenkins NA, Yazaki Y. Sequence analysis, chromosomal location, and developmental expression of the mouse preproendothelin-1 gene. Genomics. 1996;31:177–184. doi: 10.1006/geno.1996.0029. [DOI] [PubMed] [Google Scholar]
- Marinic M, Aktas T, Ruf S, Spitz F. An integrated holo-enhancer unit defines tissue and gene specificity of the Fgf8 regulatory landscape. Developmental cell. 2013;24:530–542. doi: 10.1016/j.devcel.2013.01.025. [DOI] [PubMed] [Google Scholar]
- Medeiros DM, Crump JG. New perspectives on pharyngeal dorsoventral patterning in development and evolution of the vertebrate jaw. Developmental biology. 2012;371:121–135. doi: 10.1016/j.ydbio.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers EN, Lewandoski M, Martin GR. An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nature genetics. 1998;18:136–141. doi: 10.1038/ng0298-136. [DOI] [PubMed] [Google Scholar]
- Millimaki BB, Sweet EM, Riley BB. Sox2 is required for maintenance and regeneration, but not initial development, of hair cells in the zebrafish inner ear. Developmental biology. 2010;338:262–269. doi: 10.1016/j.ydbio.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minoux M, Rijli FM. Molecular mechanisms of cranial neural crest cell migration and patterning in craniofacial development. Development. 2010;137:2605–2621. doi: 10.1242/dev.040048. [DOI] [PubMed] [Google Scholar]
- Neubuser A, Koseki H, Balling R. Characterization and developmental expression of Pax9, a paired-box containing gene related to Pax1. Developmental biology. 1995;170:701–716. doi: 10.1006/dbio.1995.1248. [DOI] [PubMed] [Google Scholar]
- Nissen RM, Yan J, Amsterdam A, Hopkins N, Burgess SM. Zebrafish foxi one modulates cellular responses to Fgf signaling required for the integrity of ear and jaw patterning. Development. 2003;130:2543–2554. doi: 10.1242/dev.00455. [DOI] [PubMed] [Google Scholar]
- Noden DM, Trainor PA. Relations and interactions between cranial mesoderm and neural crest populations. Journal of anatomy. 2005;207:575–601. doi: 10.1111/j.1469-7580.2005.00473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyama T, Groves AK. Expression of mouse Foxi class genes in early craniofacial development. Developmental dynamics : an official publication of the American Association of Anatomists. 2004;231:640–646. doi: 10.1002/dvdy.20160. [DOI] [PubMed] [Google Scholar]
- Ovchinnikov D. Alcian blue/alizarin red staining of cartilage and bone in mouse. Cold Spring Harbor protocols. 20092009 doi: 10.1101/pdb.prot5170. pdb prot5170. [DOI] [PubMed] [Google Scholar]
- Passos-Bueno MR, Ornelas CC, Fanganiello RD. Syndromes of the first and second pharyngeal arches: A review. American journal of medical genetics. Part A. 2009;149A:1853–1859. doi: 10.1002/ajmg.a.32950. [DOI] [PubMed] [Google Scholar]
- Phillips BT, Bolding K, Riley BB. Zebrafish fgf3 and fgf8 encode redundant functions required for otic placode induction. Developmental biology. 2001;235:351–365. doi: 10.1006/dbio.2001.0297. [DOI] [PubMed] [Google Scholar]
- Qiu M, Bulfone A, Ghattas I, Meneses JJ, Christensen L, Sharpe PT, Presley R, Pedersen RA, Rubenstein JL. Role of the Dlx homeobox genes in proximodistal patterning of the branchial arches: mutations of Dlx-1, Dlx-2, and Dlx-1 and -2 alter morphogenesis of proximal skeletal and soft tissue structures derived from the first and second arches. Developmental biology. 1997;185:165–184. doi: 10.1006/dbio.1997.8556. [DOI] [PubMed] [Google Scholar]
- Sandell LL, Kurosaka H, Trainor PA. Whole mount nuclear fluorescent imaging: convenient documentation of embryo morphology. Genesis. 2012;50:844–850. doi: 10.1002/dvg.22344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling TF, Kimmel CB. Segment and cell type lineage restrictions during pharyngeal arch development in the zebrafish embryo. Development. 1994;120:483–494. doi: 10.1242/dev.120.3.483. [DOI] [PubMed] [Google Scholar]
- Serbedzija GN, Bronner Fraser M, Fraser SE. Vital dye analysis of cranial neural crest cell migration in the mouse embryo. Development. 1992;116:297–307. doi: 10.1242/dev.116.2.297. [DOI] [PubMed] [Google Scholar]
- Solomon KS, Kudoh T, Dawid IB, Fritz A. Zebrafish foxi1 mediates otic placode formation and jaw development. Development. 2003a;130:929–940. doi: 10.1242/dev.00308. [DOI] [PubMed] [Google Scholar]
- Solomon KS, Logsdon JM, Jr., Fritz A. Expression and phylogenetic analyses of three zebrafish FoxI class genes. Developmental dynamics : an official publication of the American Association of Anatomists. 2003b;228:301–307. doi: 10.1002/dvdy.10373. [DOI] [PubMed] [Google Scholar]
- Stern CD. Detection of multiple gene products simultaneously by in situ hybridization and immunohistochemistry in whole mounts of avian embryos. Current topics in developmental biology. 1998;36:223–243. doi: 10.1016/s0070-2153(08)60505-0. [DOI] [PubMed] [Google Scholar]
- Sweet EM, Vemaraju S, Riley BB. Sox2 and Fgf interact with Atoh1 to promote sensory competence throughout the zebrafish inner ear. Developmental biology. 2011;358:113–121. doi: 10.1016/j.ydbio.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo-Rogers HL, Smithers LE, Yakob W, Liu KJ. New directions in craniofacial morphogenesis. Developmental biology. 2010;341:84–94. doi: 10.1016/j.ydbio.2009.11.021. [DOI] [PubMed] [Google Scholar]
- Trainor PA, Krumlauf R. Patterning the cranial neural crest: hindbrain segmentation and Hox gene plasticity. Nature reviews. Neuroscience. 2000;1:116–124. doi: 10.1038/35039056. [DOI] [PubMed] [Google Scholar]
- Trumpp A, Depew MJ, Rubenstein JL, Bishop JM, Martin GR. Cre-mediated gene inactivation demonstrates that FGF8 is required for cell survival and patterning of the first branchial arch. Genes Dev. 1999;13:3136–3148. doi: 10.1101/gad.13.23.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker AS, Yamada G, Grigoriou M, Pachnis V, Sharpe PT. Fgf-8 determines rostral-caudal polarity in the first branchial arch. Development. 1999;126:51–61. doi: 10.1242/dev.126.1.51. [DOI] [PubMed] [Google Scholar]
- Vemaraju S, Kantarci H, Padanad MS, Riley BB. A spatial and temporal gradient of Fgf differentially regulates distinct stages of neural development in the zebrafish inner ear. PLoS genetics. 2012;8:e1003068. doi: 10.1371/journal.pgen.1003068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. Simple and highly efficient BAC recombineering using galK selection. Nucleic acids research. 2005;33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng J, Yan J, Wang T, Mosbrook-Davis D, Dolan KT, Christensen R, Stormo GD, Haussler D, Lathrop RH, Brachmann RK, Burgess SM. Genome wide screens in yeast to identify potential binding sites and target genes of DNA-binding proteins. Nucleic acids research. 2008;36:e8. doi: 10.1093/nar/gkm1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.