

Abstract
The yeast spindle pole body (SPB) component Spc110p (Nuf1p) undergoes specific serine/threonine phosphorylation as the mitotic spindle apparatus forms, and this phosphorylation persists until cells enter anaphase. We demonstrate that the dual-specificity kinase Mps1p is essential for the mitosis-specific phosphorylation of Spc110p in vivo and that Mps1p phosphorylates Spc110p in vitro. Phosphopeptides generated by proteolytic cleavage were identified and sequenced by mass spectrometry. Ser60, Thr64, and Thr68 are the major sites in Spc110p phosphorylated by Mps1p in vitro, and alanine substitution at these sites abolishes the mitosis-specific isoform in vivo. This is the first time that phosphorylation sites of an SPB component have been determined, and these are the first sites of Mps1p phosphorylation identified. Alanine substitution for any one of these phosphorylated residues, in conjunction with an alanine substitution at residue Ser36, is lethal in combination with alleles of SPC97, which encodes a component of the Tub4p complex. Consistent with a specific dysfunction for the alanine substitution mutations, simultaneous mutation of all four serine/threonine residues to aspartate does not confer any defect. Sites of Mps1p phosphorylation and Ser36 are located within the N-terminal globular domain of Spc110p, which resides at the inner plaque of the SPB and binds the Tub4p complex.
Centrosomes are microtubule-organizing centers that serve as the poles of mitotic spindles during eukaryotic cell division. In each mitotic cell cycle, the centrosome must duplicate once in preparation to form the spindle apparatus. The mitotic spindle is instrumental for proper segregation of the duplicated chromosomes into two euploid daughter cells, each receiving one of the centrosomes. Proper centrosome function is vital to cell proliferation, and errors in centrosome duplication, spindle formation, or spindle function can lead to chromosome instability, chromosome non-disjunction, and aneuploidy.
Several lines of evidence from a wide variety of organisms suggest that protein phosphorylation plays a major role in centrosome control during the cell cycle. The MPM-2 monoclonal antibody recognizes mitosis-specific centrosomal phosphoepitopes in mammalian cells, Aspergillus nidulans and Schizosaccharomyces pombe (1-3), and MPM-2 antibodies inhibit microtubule nucleation in vitro (4). Several protein kinases have been implicated in centrosome control by their mutant phenotype, including yeast Mps1p (5, 6) and Drosophila aurora (7). In vertebrates, CDK2/cyclinE is important for initiation of centrosome duplication (8-10), and Nek2p is important for centrosome separation (11). Studies involving phosphatases and phosphatase inhibitors indicate that dephosphorylation is equally important for proper centrosome regulation (12-15).
In the budding yeast S. cerevisiae, the spindle pole body (SPB)1 is functionally equivalent to the centrosome. The SPB is a multilayered cylinder embedded in the nuclear envelope. Cytoplasmic microtubules emanate from an outer plaque, and nuclear microtubules emanate from the inner plaque. The 110-kDa spindle pole component Spc110p contains a large, central coiled-coil domain, which is located in the region of the SPB between the inner and central plaques (16). The C-terminal globular domain of Spc110p is located at the central plaque of the SPB (17, 18), and has been shown to interact with the SPB components calmodulin, Spc29p and Spc42p (17, 19-22). The N-terminal globular domain of Spc110p is located at the inner plaque, where Tub4p, Spc97p, and Spc98p are found (18, 23). The N-terminal globular domain of Spc110p interacts both genetically and biochemically with these components of the Tub4p complex (19, 23-25). Spc110p is also a phosphoprotein, and phosphorylation of Spc110p at serine/threonine residues arises as cells form the mitotic spindle and disappears as cells enter anaphase (26, 27).
The dual-specificity kinase Mps1p is essential for SPB duplication and for mitotic checkpoint control in S. cerevisiae (6). Cells harboring the temperature-sensitive mps1-1 allele fail to duplicate the SPB at the restrictive temperature yet proceed through a doomed mitosis with a monopolar spindle (5). Mps1p is also involved in the mitotic checkpoint (28). High levels of Mps1p cause cells to arrest in metaphase, and these arrested cells contain hyperphosphorylated Mad1p, which is also associated with activation of the mitotic checkpoint (29). In vitro, GST-Mps1p phosphorylates Mad1p (29) as well as Spc98p, a component of the Tub4p complex (30).
Despite the recent boon in centrosome component identification (for example, see Ref. 31) and the implication that several kinases are involved in centrosome function, direct evidence of specific phosphorylation of a centrosome component by a specific kinase in vivo has been lacking. In this study, we demonstrate that the dual-specificity kinase Mps1p phosphorylates Spc110p in vitro and that these sites are important for the mitosis-specific phosphorylation of Spc110p in vivo. These phosphorylations occur within the N-terminal globular domain of Spc110p, which resides at the inner plaque of the SPB and interacts with components of the microtubule-organizing Tub4p complex. Mutating these phosphorylated residues to alanine (to prevent phosphorylation), but not to aspartate (to mimic phosphorylation), perturbs the function of Spc110p such that it can no longer support growth in the presence of mutant forms of Spc97p, a component of the Tub4p complex. This perturbation also requires another alanine substitution at Ser36 in the N-terminal globular domain, which is within an (S/T)PX(R/K) consensus sequence for phosphorylation by cdc2p/Cdc28p cyclin-dependent kinase (32, 33). Thus, proper interaction of the Tub4p complex with the yeast centrosome may require Spc110p phosphorylation at all four of these sites.
EXPERIMENTAL PROCEDURES
Media, Strains, and Genetic Manipulations
SD complete, SD-uracil (34), SD-uracil low adenine (17), YPD and YPD low adenine, and LB (35) were described previously. SD-uracil+uracil is SD-uracil supplemented with 25 μg/ml uracil. LB amp is LB medium supplemented with 100 μg/ml ampicillin. LB amp kan is LB amp medium supplemented with 6 μg/ml kanamycin. Plasmid transformations were carried out by the LiOAc method essentially as described previously (36).
Strains are listed in Table I. The spc110-4A allele was integrated into CRY1 by a two step gene replacement (37) using plasmid pJK24 cut with SnaBI, creating strain JKY1. The presence of the spc110-4A allele was confirmed by sequencing. Synthetic lethal interactions between spc110-4A and spc97-114 were tested by crossing strain JKY1 (spc110-4A) with strain TNY64-5C (spc97-114). Neither single mutant confers a temperature-sensitive phenotype. The diploid was sporulated and the tetrads dissected giving 25 tetratypes (1:3 temperature-sensitive:non-temperature-sensitive), 4 non-parental ditypes (2:2), and 3 parental ditypes (0:4). Overall, 25.8% of the spore clones from these dissections could not form colonies at 37 °C.
Table I.
Strains used in this study
| Strain name | Genotype | Source or reference |
|---|---|---|
| Wx241-2b | MATa ura3-52 his3Δ200 leu2-3,112 mps1-1 | 28 |
| Wx178-3A | MATa trp1 ura3-52 leu2-3,112 mps2-1 | |
| Wx209-8A | MATa/MATa ade2/ade2 trp1/trp1 cdc31-2/cdc31-2 | 5 |
| HSY2-12C | MATa ade2-1oc ade3Δ can1-100 his3-11,15 leu2-3,112 trp1-1 lys2Δ∷HIS3 spc110Δ∷TRP1 ura3-1 | 17 |
| JKY1 | MATa ade2-1oc can1-100 his3-11,15 leu2-3,112 trp1-1 spc110-4A ura3-1 | This study |
| TNY137 | MATa ade2-1oc ade3Δ can1-100 his3-11,15 leu2-3, 112 trp1-1 lys2Δ∷HIS3 ura3-1 spc110-221 spc97-114 (pHS26) | |
| TNY155-26A | MATa ade2-1oc ade3Δ can1-100 his3-11,15 leu2-3,112 trp1-1 lys2Δ∷HIS3 ura3-1 spc110-221 spc97-62 (pHS26) | Thu Nguyen |
| TNY113-2A | MATa ade2-1oc ade3Δ can1-100 his3-11,15 leu2-3,112 trp1-1 lys2Δ∷HIS3 ura3-1 spc110-221 spc97-113 (pHS26) | Thu Nguyen |
| TNY76-1C | MATa ade2-1oc ade3Δ can1-100 his3-11,15 leu2-3,112 trp1-1 lys2Δ∷HIS3 ura3-1 spc110-221 spc98-63 (pHS26) | 25 |
| TNY64-5C | MATa ade2-1oc ade3Δ can1-100 his3-11,15 leu2-3,112 trp1-1 lys2Δ∷HIS3 spc97-114 | Thu Nguyen |
Immunoblot Analysis, α-Factor Arrest, and Cytological Techniques
Immunoblot analysis, α-factor arrests, cell morphology characterization, and flow cytometry were performed as described (26).
Plasmids
Plasmids are listed in Table II. pCL5 expresses ArgU, an arginine-tRNAAGA/AGG (AGA and AGG are rare codons in Escherichia coli but common in yeast). pCL5 contains the pA15 origin of replication to allow for co-expression with plasmids containing the ColEI origin of replication. Plasmid pDV29 encoding GST-Spc110p-(1–183) (SPC110 GenBank™ accession number Z11582) was constructed by cloning the NcoI-EcoRI fragment from pDV17 (25) into the SmaI and EcoRI sites of pGEX-2T (Amersham Pharmacia Biotech, Piscataway, NJ). Plasmids pJK2, pJK4, and pJK7 were made by site-directed mutagenesis of plasmid pDV29 using the USE kit (Amersham Pharmacia Biotech) according to the manufacturer’s directions. All other pJK plasmids (except pJK24) were made by site-directed mutagenesis of the parent plasmid using the QuikChange kit (Stratagene, La Jolla, CA) according to the manufacturer’s directions. Plasmid pJK21 carrying URA3, CEN, and an spc110 allele was converted to integrating plasmid pJK24 by replacing the AlwNI fragment containing part of bla, CEN, and part of URA3 with the AlwNI fragment from pRS306 containing part of bla and part of URA3 but no CEN. Plasmids pDF47 and pDF48, which express 12xHIS-Spc110p fusion proteins, were constructed in several steps. First, a 6xHis tag was inserted at the initiator MET of SPC110 in plasmid pHS31 by site-directed mutagenesis as described in a previous study (26), creating pDF18. Plasmid pDF29 encodes the functional SPC110–201 allele containing an in-frame deletion of the coding sequences for residues 267–543 within the central coiled-coil (26). Plasmid pDF30 (6xHIS-SPC110–201) was constructed by swapping a 1.6-kb HindIII fragment from pDF29 into pDF18. pDF47 expresses 12xHIS-Spc110–201p(756Δ) (missing residues 267–543 within the central coiled-coil and truncated at residue 756 of Spc110p) and was constructed by cloning the SphI-SspI fragment from pDF30 into the SphI and SmaI sites of the 6xHis bacterial expression vector pQE32. pDF48 expresses 12xHIS-Spc110 P-(1–225) (truncated at residue 225 of Spc110p) and was constructed by cloning the SphI-NsiI fragment from pDF30 into the SphI and PstI sites of pQE32.
Table II.
Plasmids used in this study
| Plasmid | Parent plasmid | Relevant markers | Source or reference |
|---|---|---|---|
| pMPS1 | MPS1 | (6) | |
| pQE32 | 6×HIS tagged cloning vector | Qiagen | |
| pCL5 | arg tRNA from E. coli | This study | |
| pDF18 | pHS31 | 6×SPC110 | This study |
| pDF29 | SPC110–201 | (26) | |
| pDF30 | pDF18 | 6×SPC110–201 | (26) |
| pDF47 | pQE32 | 12×HIS-SPC110–201(756Δ) | This study |
| pDF48 | pQE32 | 12×HIS-SPC110-(1–225) | This study |
| pDV29 | pGEX-2T | GST-SPC110-(1–183) WT | This study |
| pEGKT | GAL1, -GST | ||
| pEGKTMps1Δ2 | pEGKT | GAL1, -GST-MPS1)Δ2) | (6) |
| pJK2 | pDV29 | GST-SPC110-(1–183) T68A | This study |
| pJK7 | pDV29 | GST-SPC110-(1–183) T64A,T68A | This study |
| pJK4 | pDV29 | GST-SPC110-(1–183) T64A | This study |
| pJK15 | pDV29 | GST-SPC110-(1–183) S60A | This study |
| pJK20 | pDV29 | GST-SPC110-(1–183) S60A,T64A,T68A | This study |
| pHS26 | ADE3 LYS2 SPC110 | (44) | |
| pHS31 | CEN6 ARSH4 URA3 SPC110 | (26) | |
| pJK8 | pHS31 | SPC110 S36A | This study |
| pJK11 | pHS31 | SPC110 S36A,T64A,T68A | This study |
| pJK12 | pHS31 | SPC110T64A,T68A | This study |
| pJK16 | pHS31 | SPC110 S36D,T64D,T68D | This study |
| pJK21 | pHS31 | SPC110 S36A,S60A,T64A,T68A | This study |
| pJK22 | pHS31 | SPC110 S60A,T64A,T68A | This study |
| pJK24 | pRS306 | SPC110 S36A,S60A,T64A,T68A | This study |
| pJK29 | pHS31 | SPC110 S36D,S60D,T64D,T68D | This study |
| pJK31 | pHS31 | SPC110 S36A,S60A | This study |
| pJK33 | pHS31 | SPC110 S36A,T64A | This study |
| pJK34 | pHS31 | SPC110 S36A,T68A | This study |
Unless stated otherwise, all markers from the parent plasmid are present in the new construct.
Production of Spc110p Fusion Proteins
Cultures of E. coli strain GM1 were transformed to ampicillin resistance with plasmids pDF47 or pDF48 expressing recombinant 12xHIS-Spc110–201p(756Δ) or 12xHIS-Spc110p-(1–225), respectively. Cultures of isolated transformants were diluted 1:100 into 10 ml of LB amp, grown to a density of 20 Klett units and then induced with 1–2 mm isopropyl-1-thio-β -d-galactopyranoside for 5 h at 30 °C. Cells were harvested, washed once in lysis buffer (1× phosphate-buffered saline, 10 mm β-mercaptoethanol, 1 mm phenylmethylsulfonyl fluoride) and lysed using a French Pressure cell (3/8-inch piston diameter; Aminco) at 11,000 p.s.i. to near 100% lysis. Wild-type and mutant forms of recombinant GST-Spc110p-(1–183) were co-expressed with plasmid pCL5 in strain GM1. Overnight cultures were diluted 1:200 into 200 ml of LB amp kan and grown at 37 °C to a density of 60–70 Klett units. Isopropyl-1-thio-β-d-galactopyranoside was added to a final concentration of 40 μg/ml, and cultures were incubated for an additional 1.5 h. Cells were then pelleted at 8500 × g for 10 min, and pellets were stored at −80 °C. Thawed pellets were suspended in 3 ml of cold lysis buffer (1× phosphate-buffered saline, 10 mm β-mercaptoethanol, 1 mm phenylmethylsulfonyl fluoride) and lysed in a French Pressure cell as described above. Triton X-100 was added to a final concentration of 1%, and the lysate was incubated on ice for 15 min. Cleared lysates were mixed for 1 h with 100 μl of glutathione Sepharose 4B resin (Amersham Pharmacia Biotech). The supernatant was then discarded, and the beads were washed three times with 1 ml of lysis buffer. GST-Spc110p-(1–183) was eluted in 200 μl of elution buffer (10 mm glutathione, 50 mm Tris, pH 8.0) by mixing for 10 min. The buffer was exchanged to storage buffer (50 mm Tris, pH 7.5, 10 mm MgCl2, 50 mm NaCl, 0.5 mm DTT, 5% v/v glycerol) by gel filtration on G-25 Sephadex. Samples were frozen in liquid N2 and stored at −80 °C.
Mps1p in Vitro Kinase Assay
All reactions were carried out as described previously (6) except as follows. Kinase assays used to generate phosphorylated GST-Spc110p-(1–183) for analysis of phosphorylation sites by mass spectrometry used 200 μl of GST-Mps1p bound to GSH-Sepharose in a 50% slurry, which was washed once in kinase assay buffer without ATP (KAB-ATP: 50 mm Tris, pH 7.5, 10 mm MgCl2, 2 mm DTT) and resuspended in 50 μl of KAB-ATP and 10 μl of GST-Spc110p-(1–183) at a concentration of 1 mg/ml. 40 μl of 5× KAB (final concentration, 50 mm Tris, pH 7.5, 10 mm MgCl2, 2 mm DTT, 2 mm ATP containing 200 μCi of [γ-32P]ATP) was added, and the mixture was incubated at 30 °C with shaking for 5 h. 20 μl was subjected to 12% SDS-PAGE. The gel was stained with Coomassie Brilliant Blue, dried, and quantified by PhosphorImager analysis to assess stoichiometry of phosphate incorporation. Kinase assays used to compare incorporation of phosphate into wild-type and phosphorylation site mutants of GST-Spc110p-(1–183) used 10 μl of GST-Mps1p conjugated to GSH-Sepharose beads and ~0.2 μg of GST-Spc110p-(1–183) (wild-type or mutant). The entire reaction was separated by SDS-PAGE, and the gel was stained with Coomassie Brilliant Blue prior to PhosphorImager analysis.
Proteolytic Digestion and Mass Spectrometry
Both kinase and substrate were expressed as GST fusions for these experiments, and both were bound to GSH-Sepharose resin during the reaction. Prior to proteolytic digestion the resin was washed twice and resuspended in 100 μl of 50 mm Tris, pH 7.5, 1 mm CaCl2 for digestion with trypsin, or in 100 μl of 100 mm Tris, pH 9.2, for digestion with endoproteinase Lys C (endoLys-C). Proteolytic digestion was performed at 30 °C overnight with shaking to keep the resin in suspension. The resin was removed by centrifugation, leaving the peptides in the supernatant, then washed once with 50 μl of H2O. The wash and supernatant were combined, reduced in a vacuum-concentrating microcentrifuge (Heto-Holten, Allerød, Denmark) to near dryness, and resuspended in 50 μl of 0.1–1.0% trifluoroacetic acid. Electrospray ionization, liquid chromatography mass spectrometry (ESI-LC/MS), and tandem mass spectrometry (ESI-LC/MS/MS) were carried out using either a hand-packed 500-μm i.d. HPLC column containing C18 reverse phase resin (Columbus) interfaced to an API III+ triple-quadrupole mass spectrometer (PE-Biosystems, Foster City, CA), or a 320-μm i.d. HPLC column (Micro-Tech, Sunnyvale, CA) packed with C18 reverse phase resin interfaced to an LCQ ion trap mass spectrometer (ThermoQuest, San Jose, CA). Matrix-assisted laser desorption ionization, time of flight (MALDI-TOF) mass spectrometry was carried out on 0.5 μl of analyte mixed with 0.5 μl of α-cyano-4-hydroxycinnamic acid matrix (Agilent Technologies, Hewlett-Packard), using a Voyager DE-STR mass spectrometer (Per-Septive Biosystems, Foster City, CA).
RESULTS
Mps1p Activity Is Necessary for Mitosis-specific Phosphorylation of Spc110p in Vivo
Previously we have shown that the 110-kDa SPB component Spc110p undergoes serine/threonine phosphorylation in cells containing pre-anaphase mitotic spindles, which results in a slower-migrating isoform (p120) during SDS-PAGE (26). We analyzed the Spc110p mobility shift in cells carrying the mps1-1 mutation at the restrictive temperature to determine whether the essential dual-specificity kinase Mps1p played a role in the mitosis-specific Spc110p phosphorylation. Mps1-1p is a severely crippled conditional mutant kinase, both in vivo and in vitro (38). Cells were first synchronized in G1 by the addition of the mating pheromone α-factor and then released into prewarmed 37 °C medium. At 37 °C, mps1-1 cells entered the cell cycle at the same time as the mps1-1 pMPS1 control cells (as evidenced by bud emergence at 30 min after release for both cultures), but unlike the wild-type control, the slower migrating mitosis-specific isoform of Spc110p never accumulated in mps1-1 cells (Fig. 1). Thus, MPS1 is required for the mitosis-specific phosphorylation of Spc110p in vivo.
Fig. 1. Mitosis-specific Spc110p phosphorylation is dependent upon Mps1p function.

Strain Wx241-2b (mps1-1), alone or transformed with plasmid pMPS1, was grown at room temperature in SD-uracil and SD-uracil+uracil medium, respectively (doubling time = 3.5 h). Mid-logarithmic cultures were exchanged into YPD medium. Cultures were then treated with 6 μm α-factor at room temperature for 2.5 h, after which cultures were shifted to 37 °C. After an additional 1.75 h of α-factor treatment at 37 °C (>95% shmoo morphology), cultures were released from the arrest into prewarmed YPD at 37 °C, and aliquots were taken for total cell protein (trichloroacetic acid precipitation), bud morphology, and DNA content at the indicated time points as described (26). A, protein content and bud morphology. Western blot analysis using affinity-purified anti-Spc110p antibodies was performed as described previously (26). 120- (p120) and 112-kDa (p112) Spc110p isoforms are indicated. Buds appeared 30 min after release from α-factor for both cultures. Lanes 1–7, mps1-1 cells; lanes 8–14, mps1-1 cells harboring plasmid pMPS1. B, DNA content. The time points taken just prior to release from G1 arrest (37 G1 arrest (0)) were taken at the same time as the 0-min time points shown in A. Additional aliquots were taken during asynchronous growth at room temperature (RT asynch) and G1 arrest at room temperature (RT G1 arrest) to establish the distribution of cells containing haploid DNA content (1N) and diploid DNA content (2N). Peak height reflects cell number. By the last time point (180 min after release) cells carrying the pMPS1 plasmid had returned to G1 as assessed by DNA content, whereas the mps1-1 cells exhibited a wide range of DNA content, including below 1N and above 4N as observed previously (5).
MPS1 has at least two previously identified execution points during the cell cycle, one that is essential for SPB duplication (5), and another that is involved in the mitotic spindle checkpoint (28). Because the mitosis-specific phosphorylation of Spc110p does not occur until after SPB duplication is complete (26), it is possible that the dependence for Spc110p phosphorylation on Mps1p function merely reflects a prerequisite for SPB duplication. To test this hypothesis, we performed similar analyses for Spc110p phosphorylation at the restrictive temperature in two additional mutant backgrounds that block SPB duplication, mps2-1 and cdc31-2. Both mutations cause cells to arrest with large buds and a G2 DNA content at the restrictive temperature. Cells carrying the mps2-1 mutation arrest at the restrictive temperature with a malformed duplicated SPB that fails to insert into the nuclear envelope and lacks both the inner plaque and nuclear microtubules (5). Cells carrying the cdc31-2 mutation fail completely in SPB duplication (39). The slower migrating mitosis-specific isoform of Spc110p is present at the restrictive temperature in each mutant background despite failures in SPB duplication (Fig. 2). Furthermore, ordering of execution points during the cell cycle place CDC31 function first, followed by MPS1 function and then by MPS2 function (5). Thus failure of the mitosis-specific phosphorylation of Spc110p in mps1-1 cells is not due simply to a failure in SPB duplication.
Fig. 2. Mitosis-specific Spc110p phosphorylation persists in the absence of SPB duplication.
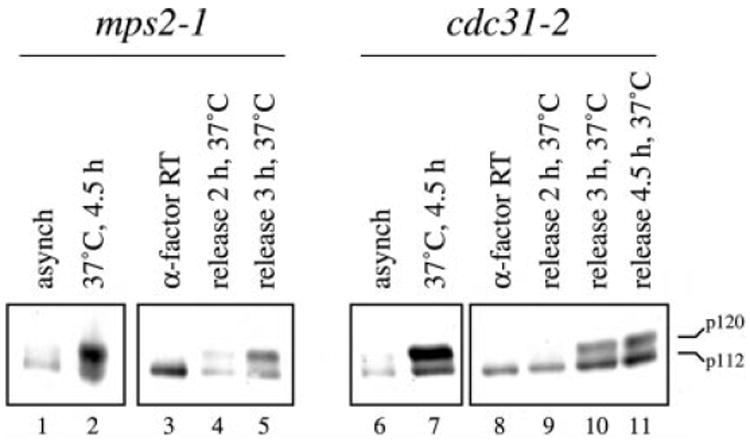
Western blot analysis using affinity-purified anti-Spc110p antibodies was performed as described previously (26). Strains Wx178-3A and Wx209-8A carrying the mutations mps2-1 or cdc31-2, respectively, were each grown at room temperature to mid-logarithmic phase (doubling times = 2.67 and 3 h, respectively). Each culture was then shifted to the restrictive temperature of 37 °C for 4.5 h, after which total cell protein extracts were obtained from 1-ml aliquots by trichloroacetic acid precipitation as described (26). Separate cultures were treated with 6 μm α-factor at room temperature, and total cell protein extracts were obtained from 1-ml aliquots after 1.5 doublings (>96% shmoo morphology). The cultures were then washed with prewarmed YPD and released from the α-factor arrest in YPD at 37 °C, and total cell extracts were prepared from 1 ml of culture after the indicated time at 37 °C. Lanes 1–5, mps2-1 cells grown at room temperature for 4.5 h (lane 1), incubated at 37 °C for 4.5 h (lane 2), arrested with α-factor at room temperature (lane 3), and released from α-factor arrest at 37 °C for 2 h or 3 h (lanes 4 or 5). Lanes 6–11: cdc31-2 cells grown at room temperature for 4.5 h (lane 6), incubated at 37 °C for 4.5 h (lane 7), arrested with α-factor at room temperature (lane 8), and released from α-factor arrest at 37 °C for 2, 3, or 4.5 h (lanes 9–11). DNA content was consistent with previously published findings for mps2-1 and cdc31-2 at the restrictive temperature (5, 39). 120- (p120) and 112-kDa (p112) Spc110p isoforms are indicated.
We also tested if MPS1 expression could promote the production of the mitosis-specific Spc110p isoform in cells blocked in G1 by the addition of α-factor, a state where the mitosis-specific isoform is normally not present (26). Cells expressing MPS1 from a galactose-inducible promoter while held at this G1 arrest clearly produce the mitosis-specific Spc110p isoform (Fig. 3, lane 9), whereas similarly arrested cells harboring control plasmids do not (Fig. 3, lane 7). A non-phosphorylatable form of Spc110p (4A, described below) is not shifted in this experiment (Fig. 3, lane 10), demonstrating that the Spc110p mobility shift promoted by the expression of MPS1 during G1 arrest does not result from phosphorylation at inappropriate sites. Because other mitosis-specific functions would be turned off during this G1 arrest, it is likely that the production of the slower-migrating Spc110p isoform, which normally results from mitosis-specific phosphorylation, here results directly from the inappropriate expression of Mps1p.
Fig. 3. Mps1p expression during G1 arrest results in the production of the mitosis-specific Spc110p isoform.

Western blot analysis using affinity-purified anti-Spc110p antibodies and preparation of total cell protein extracts was performed as in Fig. 1. CRY1 (wt) and JKY1 (4A) harboring either plasmid pEGKT (pGAL1-GST) or plasmid pEGKTMps1Δ2 (pGAL1-GST-MPS1) were grown under the following conditions. Galactose induction during the G1 arrest was performed as described previously (29) with the exception that cells were held at the arrest during the induction by the further addition of 10 μm α-factor after 2 h. Lanes 1 and 2, asynchronous cultures. Lanes 3–6, cells arrested during the G1 stage of the cell cycle by the addition of 10 μm α-factor. Lanes 7–10, galactose induction during the G1 arrest. 120-(p120) and 112-kDa (p112) Spc110p isoforms are indicated. The high mobility signal present in lanes 9 and 10 results from the galactose-induced expression of GST-Mps1p and is visualized due to cross-reactivity between the affinity-purified anti-Spc110p antibodies (raised against a GST fusion to the coiled-coil region of Spc110p (26)) and the GST-Mps1p fusion protein. The apparent molecular mass of GST-Mps1p in lanes 9 and 10 is 150 kDa, which is in agreement with previous findings (6, 38) despite a predicted molecular mass of 112 kDa. Furthermore, the 150-kDa band in lanes 9 and 10 is the only protein reacting with anti-GST antibodies in a Western blot analysis (data not shown).
Mps1p Phosphorylates Spc110p in Vitro at Sites within the N-terminal Globular Domain of Spc110p
We expressed different forms of recombinant Spc110p in E. coli, and the lysates were added to an in vitro GST-Mps1p kinase assay (6). GST-Mps1p (purified from yeast) phosphorylates 12xHIS-Spc110–201p(756Δ), which is missing residues 267–543 within the central coiled-coil region and the last 188 residues of the C-terminal globular domain (Fig. 4A, lane 2). 12xHIS-Spc110p-(1–225), which consists of the first 225 amino acids and encompasses the N-terminal globular domain of Spc110p, was also phosphorylated in vitro by GST-Mps1p (Fig. 4A, lane 3). A fusion containing a portion of the central coiled-coil (residues 265–755) was not phosphorylated by GST-Mps1p (Fig. 4A, lane 9). Phosphorylation of the various Spc110p substrates depended on the addition of GST-Mps1p and was distinct from any background phosphorylation from the E. coli extract (Fig. 4A, lane 1), or from any breakdown products resulting from Mps1p autophosphorylation (Fig. 4A, lane 4).
Fig. 4. In vitro phosphorylation of Spc110p by Mps1p.

A, GST-Mps1p in vitro kinase assays. Crude bacterial extract from cultures expressing recombinant 12xHIS-Spc110–201p(756Δ) (pDF47), 12xHIS-Spc110p-(1–225) (pDF48), or no fusion protein (pQE32) was used as substrates for phosphorylation by GST-Mps1p kinase (purified from yeast) in the presence of radiolabeled [γ-32P]ATP as described under “Experimental Procedures.” Purified 6xHIS-Spc110p-(265–755), containing residues 265–755 within the central coiled-coil (26), and myelin basic protein (MBP) were also used as substrates. Kinase assays were boiled for 4 min in SDS-PAGE buffer prior to separation on 12% SDS-polyacrylamide gels (42). The dried gels were exposed to autoradiography film or a PhosphorImager screen. Lane 1, 12xHIS-Spc110–201p(756Δ) expressed from plasmid pDF47 with no GST-Mps1p added to the reaction. Lane 2, 12xHIS-Spc110–201p(756Δ) expressed from plasmid pDF47 with GST-Mps1p added to the reaction. Lane 3, 12xHIS-Spc110p-(1–225) expressed from plasmid pDF48 with GST-Mps1p added to the reaction. Lane 4, expression plasmid pQE32 containing no fusion protein with GST-Mps1p added to the reaction. Lanes 5 and 6, Western blot analysis using anti-MRGS antibodies (Qiagen, Valencia, CA) as described previously (26) to determine the mobility of 12xHIS-Spc110–201p(756Δ) expressed from plasmid pDF47 (lane 5) and 12xHIS-Spc110-(1–225) expressed from plasmid pDF48 (lane 6). Lane 7, no substrates added to the reaction. Lane 8, MBP added to the reaction. Lane 9, 6xHIS-Spc110p-(265–755) added to the reaction. Lanes 10–12, Coomassie Brilliant Blue-stained gel shown for lanes 7–9. The relative mobilities of the kinase and substrates are indicated on the sides of the gels. Asterisks in lanes 2 and 3 mark positions of radiolabeled bands excised for phosphopeptide mapping. B and C, two-dimensional thin layer electrophoresis (1st), thin layer chromatography (2nd; two-dimensional TLE/TLC) phosphopeptide mapping. Open arrowheads denote origins. Proteins in gels similar to A were electrophoretically transferred to nitrocellulose. Bands corresponding to full-length 12xHIS-Spc110–201p(756Δ) and 12xHIS-Spc110p-(1–225) (asterisks in panel A) were excised from the nitrocellulose support after comparison to an autoradiogram of the same membrane. Phosphopeptides were then subjected to two-dimensional TLE/TLC after digestion of the proteins off of the nitrocellulose membrane using the protease trypsin as described in (43). Shown are the tryptic phosphopeptide maps of 12xHIS-Spc110–201p(756Δ) (B) and 12xHIS-Spc110-(1–225) (C) after phosphorylation by GST-Mps1p. D, graphical representation of the recombinant proteins with respect to full-length Spc110p (containing 944 residues). The shaded area represents the central coiled-coil domain (residues 155–798 as predicted by PAIRCOIL). Fusions containing the C-terminal domain were insoluble and could not be tested. However, serine and threonine residues within the C-terminal 116 amino acids have been shown previously not to be involved in the mitosis-specific phosphorylation of Spc110p (26), leaving eight serines and/or threonines between residues 756 and 828 that have not been tested.
Trypsin digestion followed by two-dimensional TLE/TLC phosphopeptide mapping was used to determine the complexity of Spc110p phosphorylation by GST-Mps1p. The two-dimensional phosphopeptide maps of 12xHIS-Spc110–201p(756Δ) and 12xHIS-Spc110p-(1–225) were nearly identical, exhibiting three major 32P-labeled phosphopeptides and varying only in weak background phosphorylation (Fig. 4, B and C). Thus, the major phosphorylation of Spc110p by GST-Mps1p resides in the N-terminal globular domain of Spc110p. This domain of Spc110p resides at the inner plaque of the SPB and associates with the microtubule-organizing Tub4p complex containing Spc97p, Spc98p, and Tub4p (23-25).
Ser60, Thr64, and Thr68 in Spc110p Are Phosphorylated by GST-Mps1p in Vitro
We used an E. coli-expressed GST-Spc110p -(1–183) fusion protein containing the first 183 residues of Spc110p as a substrate for GST-Mps1p to determine the in vitro sites of phosphorylation by GST-Mps1p within the N-terminal globular domain of Spc110p. Stoichiometry of phosphorylation in three experiments ranged from 0.9 to 1.7 mol of phosphate per mol of GST-Spc110p-(1–183), and this phosphorylation produced a mobility shift of GST-Spc110p-(1–183) on SDS-PAGE (Fig. 5A).
Fig. 5. Mps1p phosphorylates Spc110p at residues Ser60, Thr64, and Thr68 in vitro.
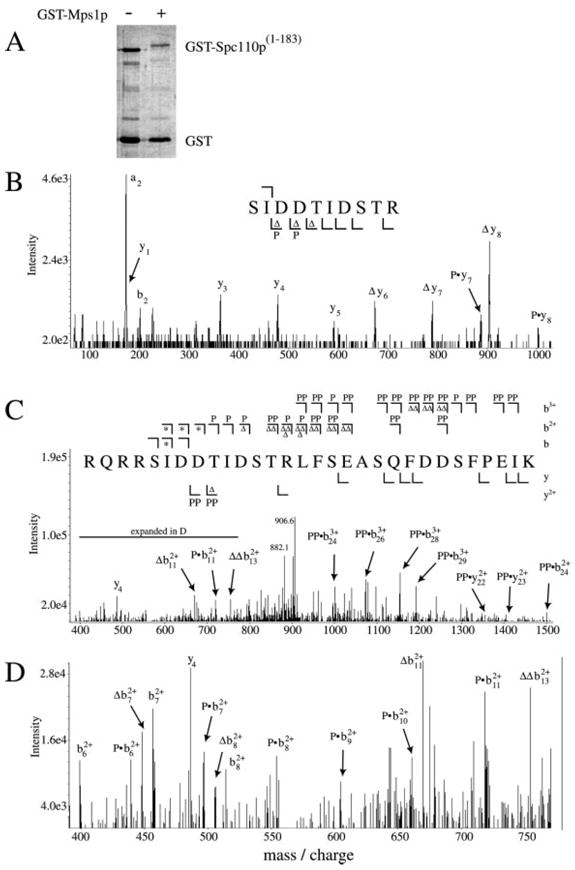
A, GST-Mps1p in vitro kinase assay. GST-Spc110p-(1–183) was purified from E. coli and subjected to phosphorylation by GST-Mps1p in vitro as described under “Experimental Procedures.” Lane 1, GST-Spc110p-(1–183) (0.2 μg) with no GST-Mps1p kinase added to the reaction. Lane 2, same as lane 1 with the addition of GST-Mps1p as described under “Experimental Procedures.” The GST-Spc110p-(1–183) mobility shift is accompanied by 1.4 mol/mol of phosphate incorporation. Tryptic or endoLys-C peptides were obtained from phosphorylated GST-Spc110p-(1–183) after incubation with protease as described under “Experimental Procedures,” and each peptide mixture was analyzed by ESI-LC/MS in positive ion mode. Scans across the entire LC range were surveyed for each peptide mass including mass-shifts of 80 Da to account for phosphopeptides. B and C, MS/MS peptide sequencing of two phosphopeptides. Peptide bond cleavages generating b ions (containing the N terminus) and y ions (containing the C terminus) are illustrated. A summary of the observed fragmentation ions is shown for the two spectra, including m/z shifts consistent with phosphorylation (P and PP) and neutral loss of H3PO4 (Δ and ΔΔ). Singly, doubly, or triply charged b ions (b, b2+, and b3+) are displayed above the sequence (⌉), and singly or doubly charged y ions (y and y2+) are displayed below (⌊). B, MS/MS spectrum of tryptic fragment 60SIDDTIDSTR69 + 80 Da (one phosphate; (M+2H)2+ = 602.0 Da/e). This spectrum represents the summation of nine scans that were acquired as the phosphopeptide eluted from the HPLC column. Singly charged y1, y3, y4, and y5 ions were found at their expected masses based on the amino acid sequence (175.4, 362.8, 478.6, and 590.2 Da/e, respectively). y6, y7, and y8 ions were also found, but only with m/z shifts consistent with phosphorylation (Py7 = 888.2 Da/e; Py8 = 1002.8 Da/e) or neutral loss of H3PO4 (Δy6 = 675.4 Da/e; Δy7 = 788.8 Da/e; Δy8 = 904.6 Da/e). Ions consistent with phosphorylation appear in the y-ion series beginning with ion y6, indicating Thr64 as the phosphorylated residue in this peptide. C, MS/MS spectrum of endoLys-C peptide 56RQRRSIDDTIDSTRLFSEASQFDDSFPEIK85 + 160 Da (two phosphates; (M+4H)4+ = 931.0 Da/e). The m/z of the two H3PO4 neutral loss products of the parent ion are labeled (906.6 and 882.1 Da/e). For doubly charged fragment ions, symbols indicate ions shifted in m/z from that predicted by the amino acid sequence by +40, +80, −9, or −18 Da, representing the addition of one or two phosphates (P or PP) or the neutral loss of one or two H3PO4 groups (Δ or ΔΔ). The same changes in the triply charged fragment ions create m/z shifts of +26.7, +53.3, or −12 Da, representing the addition of one or two phosphates (P or PP) or the neutral loss of two H3PO4 groups (ΔΔ). Doubly and triply charged b ion cleavages past Thr68 are found in either the mono- or di-phosphorylated forms only. Asterisks indicate both phosphorylated and unmodified ions were observed, as detailed in D. Ion signals stronger than 2.5× background noise are included in the summary above the spectrum. Weaker fragment ions indicating phosphorylation at Thr68 and not Ser67 were found but not labeled. D, expansion of the spectrum shown in C from 400 to 800 Da/e detailing ions consistent with a mixture of phosphorylated and unphosphorylated Ser60. Symbols indicate doubly charged ions shifted in m/z from that predicted by the amino acid sequence by +40, −9, or −18 Da, representing the addition of phosphate (P) or the neutral loss of one (Δ) or two (ΔΔ) H3PO4 groups. The expected mass of the unmodified doubly charged ion b72+ was found (456.5 Da/e) along with ions consistent with phosphorylation (Pb72+, 496.9 Da/e) and neutral loss of H3PO4 (Δb72+, 448.0 Da/e). The same pattern was found for b82+ ions (505.3, 514.2, and 554.2 Da/e for ions Δb82+, b82+, and Pb82+, respectively). Also shown are b62+ (398.9 Da/e) and Pb62+ (439.2 Da/e). Cleavages creating the b92+, b102+, and b112+ ions include both Ser60 and Thr64, and only the mono-phosphorylated forms are found for these ions (Pb92+ = 604.6 Da/e, Pb102+ = 661.3 Da/e, Pb112+ = 718.8 Da/e, Δb112+ = 670.1 Da/e). Shown is the double-neutral loss ion ΔΔb132+ at 755.0 Da/e. Nearly every peak is accounted for despite ion signals being close to 2.5× background noise in this region.
The phosphorylated GST-Spc110p-(1–183) was digested with trypsin or endoLys-C, and the peptides were analyzed by MALDI-TOF and ESI-LC/MS mass spectrometry. This analysis accounted for every serine, threonine, and tyrosine residue within the first 183 residues of Spc110p, and identified four tryptic peptides and two endoLys-C peptides that appeared to be phosphopeptides based on mass increases of 80 Da (or multiples thereof) over the expected masses for these peptides. All candidate phosphopeptides encompassed the residues 60SIDD-TIDSTR69 within the N-terminal globular domain of Spc110p. Two representative fragmentation spectra of these phosphopeptides are described below.
The tryptic fragment 60SIDDTIDSTR69 (Tp60–69) was found to contain a single phosphate. Sequencing this peptide by collision-induced dissociation during ESI-LC/MS/MS confirmed this assignment and mapped the site of phosphorylation to residue Thr64 (Fig. 5B). This peptide, containing phosphorylation at Thr64, was also identified from the lowest of the three spots in the two-dimensional phosphopeptide maps shown in Fig. 3 (data not shown). In the endoLys-C digest, mono- and di-phosphorylated forms of the peptide 56RQRRSIDDTIDSTRLFSEASQFDDSFPEIK85 (K56–85) were observed. The fragmentation spectrum of the di-phosphorylated form of K56–85 by ESI-LC/MS/MS showed heterogeneity in phosphorylation at any two of the three residues Ser60, Thr64, and Thr68 (Fig. 5, C and D). Several fragment ions indicated both phosphorylated (addition of HPO4 or neutral loss of H3PO4) as well as unmodified Ser60 (denoted by the asterisk in Fig. 5C and detailed in Fig. 5D). Fragment ions containing all three residues Ser60, Thr64, and Thr68 were found in the mono- or di-phosphorylated forms, but never at their expected, unmodified masses. Thus, a mixture of di-phosphorylated forms of this peptide (phosphorylated at any two of these three sites) co-eluted from the reverse phase resin and were fragmented simultaneously during the LC/MS/MS experiment. A tri-phosphorylated form of K56–85 was not found.
Sites of in vitro Spc110p phosphorylation by Mps1p were confirmed by repeating the kinase assays using GST-Spc110p-(1–183) fusions containing combinations of alanine substitutions at residues Ser60, Thr64, and Thr68 (Fig. 6). The level of phosphate incorporation for each single mutant was decreased relative to the wild-type level (80%, 40%, and 50% that of the wild-type level for the single mutants S60A (not shown in Fig. 6), T64A, and T68A, respectively). The level of phosphate incorporation into the T64A,T68A double mutant was 20% that of the wild-type level, and the level for the triple mutant S60A,T64A,T68A was reduced to near background levels (Fig. 6).
Fig. 6. Mutation of Spc110p phosphorylation sites abolishes phosphorylation by Mps1p in vitro.
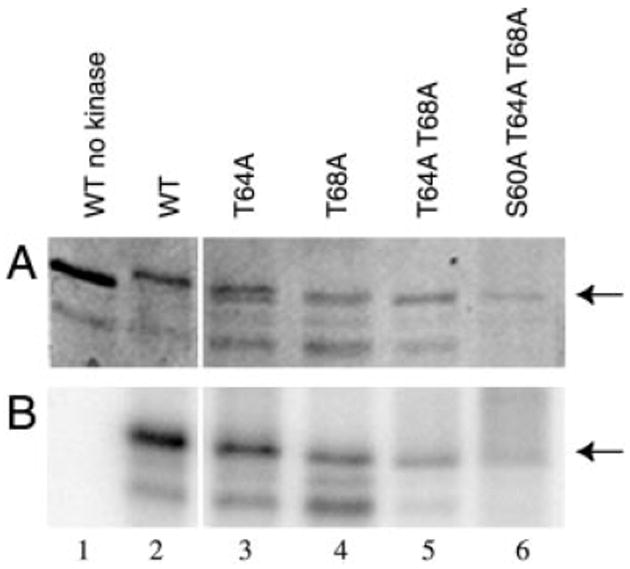
GST-Spc110p-(1–183) containing alanine substitutions at the indicated residues were constructed and purified as described under “Experimental Procedures.” Approximately 0.2 μg were used as substrates for in vitro GST-Mps1p kinase assays as described under “Experimental Procedures,” after which reactions were boiled for 4 min in SDS-PAGE buffer prior to separation through a 10% SDS-polyacrylamide gel (42). The gel was stained with Coomassie Brilliant Blue (A), dried, and exposed to a PhosphorImager screen (B). Arrows indicate the mobility of full-length GST-Spc110p-(1–183). Lane 1, wild-type GST-Spc110p-(1–183) (pDV29) without GST-Mps1p. Lanes 2–6: GST-Mps1p kinase assay using as substrate wild-type GST-Spc110p-(1–183) (pDV29, lane 2), GST-Spc110p-(1–183) containing the T64A mutation (pJK4, lane 3), the T68A mutation (pJK2, lane 4), the T64A,T68A double mutation (pJK7, lane 5), or the triple S60A,T64A,T68A mutation (pJK20, lane 6).
The kinase assays using mutant GST-Spc110p-(1–183) fusions as substrates confirmed phosphorylation at Thr68. The MS/MS spectrum for K56–85 does not clearly distinguish between phosphorylation at Ser67 and Thr68 (Fig. 5C and data not shown). However, phosphorylation of GST-Spc110p-(1–183) containing the T68A mutation was reduced relative to the wild-type protein as stated above, whereas the same fusion protein containing the S67A mutation was indistinguishable from the wild-type protein in this assay (data not shown). In addition, the mobility shift of GST-Spc110p-(1–183) observed during the in vitro kinase assay was dependent upon the presence of Thr68 (Fig. 6).
S60A, T64A, or T68A Mutations, in Conjunction with an S36A Mutation, Are Synthetically Lethal with Mutations in SPC97
The spc110–221 allele, which contains mutations in the N-terminal globular domain of Spc110p (19), is synthetically lethal with alleles of SPC97 and SPC98 (25). We find that full-length Spc110p containing any of the S60A, T64A, or T68A mutations, in conjunction with an alanine substitution at Ser36, fails to complement the synthetic lethality between spc110-221 and spc97-62 or spc97-114 (Table III, lines 5–9). We have also found that Ser36 is phosphorylated in Spc110p when expressed in insect cells.2 The S36A substitution alone was still able to complement spc110-221 spc97 synthetic lethality, as was the triple Mps1p site substitution (Table III, lines 2–4). Full-length Spc110p containing aspartate substitution at Ser36, Ser60, Thr64, and Thr68 was also able to complement the synthetic lethality (Table III, lines 10–11), suggesting that the effects due to the alanine substitutions were not due to an overall perturbation of protein structure. In contrast, the synthetic lethality between spc110-221 and an allele of Spc98p (spc98-63) was still suppressed by the quadruple alanine substitution Spc110p (Table III, line 9).
Table III.
Complementation of synthetic lethal interactions
| Spc110p | Plasmid | TNY155–26A, pHS26 (spc110–221, spc97–62), 37 °C | TNY137, pHS26 spc110–221, spc97–114, 30 °C | TNY76–1C, pHS26 spc110–221, spc98–63, 37 °C | |
|---|---|---|---|---|---|
| 1 | WT | pHS31 | Functional | Functional | Functional |
| 2 | S36A | pJK8 | Functional | Functional | |
| 3 | T64A/T68A | pJK12 | Functional | Functional | |
| 4 | S60A/T64A/T68A | pJK22 | Functional | Functional | |
| 5 | S36A/S60A | pJK31 | Not functional | Not functional | |
| 6 | S36A/T64A | pJK33 | Not functional | Not functional | |
| 7 | S36A/T68A | pJK34 | Not functional | Not functional | |
| 8 | S36A/T64A/T68A | pJK11 | Not functional | Not functional | |
| 9 | S36A/S60A/T64A/T68A | pJK21 | Not functional | Not functional | Functional |
| 10 | S36D/T64D/T68D | pJK16 | Functional | Functional | |
| 11 | S36D/S60D/T64D/T68D | pJK29 | Functional | Functional |
Plasmids containing mutations in the phosphorylated residues of Spc110p were transformed into the indicated strains and plated on SD-uracil low adenine medium at 30 °C and 37 °C. The requirement for uracil maintains selection on the plasmids carrying alleles of SPC110. If the mutant alleles are not functional, then these cells require the presence of plasmid pHS26 (expressing wild-type SPC110) and form solid red colonies. However, if these mutant alleles are functional, then the cells form red colonies with white sectors.
These results were confirmed by integrating the spc110-4A allele containing the S36A, S60A, T64A, and T68A mutations and testing directly for synthetic lethal interactions with spc97-114 (“Experimental Procedures”). The double mutant (spc110-4A and spc97-114) was not viable at 32 °C, whereas neither allele alone (spc110-4A or spc97-114) conferred a temperature-sensitive phenotype.
These mutant proteins were assayed by Western blot analysis when expressed in strain HSY2-12C to assess their effect on the Spc110p mitosis-specific SDS-PAGE mobility shift. An asynchronous wild-type culture exhibits both Spc110p isoforms (Fig. 2, lanes 1 and 6; Fig. 3, lane 1; Fig. 7 lane 1), whereas the mitosis-specific mobility shift of Spc110p containing alanine substitutions at the three Mps1p phosphorylation sites (S60A,T64A,T68A) was abolished (Fig. 7, lane 4). The complete loss of the slower-migrating mitosis-specific isoform was dependent upon simultaneous mutation of all three Mps1p phosphorylation sites (Fig. 7, lanes 4 and 5 and data not shown) and was independent of alanine substitution at Ser36. The mobility shift of the quadruple alanine substitution mutant S36A,S60A,T64A,T68A Spc110p was similarly abolished (Fig. 7, lane 3, and Fig. 3, lane 2), and alanine substitution at only Ser36 did not markedly affect the slower-migrating mitosis-specific isoform (Fig. 7, lane 6). Simultaneous aspartate substitution at these same four residues resulted in the complete shift of Spc110p into a slower-migrating form (Fig. 7, lane 2), but otherwise did not affect Spc110p function.
Fig. 7. Mutation of phosphorylation sites in full-length Spc110p abolishes the mitosis-specific mobility shift in vivo.

Western blot analysis using affinity-purified anti-Spc110p antibodies and preparation of total cell protein extracts was performed as in Fig. 1. Plasmids expressing full-length spc110 alleles encoding the indicated mutations of serine and threonine residues to alanine or aspartate were transformed into yeast strain HSY2-12C using a red/white plasmid shuffle scheme (17). Thus plasmid-encoded Spc110p is the only source of Spc110p in HSY2-12C cells. Lane 1, plasmid pHS31 (WT). Lane 2, plasmid pJK29 (S36D,S60D,T64D,T68D). Lane 3, plasmid pJK21 (S36A,S60A,T64A,T68A). Lane 4, plasmid pJK22 (S60A,T64A,T68A). Lane 5, plasmid pJK12 (T64A,T68A). Lane 6, plasmid pJK8 (S36A). 120- (p120) and 112-kDa (p112) Spc110p isoforms are indicated.
DISCUSSION
We have shown previously that the mitosis-specific serine/threonine phosphorylation of Spc110p occurs after SPB duplication is completed as the mitotic spindle first forms, and persists up to the metaphase/anaphase transition (26). We show here that the dual-specificity kinase Mps1p phosphorylates Spc110p and that this phosphorylation is necessary for the production of the mitosis-specific Spc110p isoform in vivo. Mitosis-specific Spc110p phosphorylation can occur in the absence of proper SPB duplication but cannot occur in the absence of Mps1p activity, and Mps1p production during G1 arrest can drive formation of the mitosis-specific Spc110p isoform. Spc110p is phosphorylated at residues Ser60, Thr64, and Thr68 by GST-Mps1p in vitro, and alanine substitution at these sites abolishes the mitosis-specific phosphorylation in vivo. These sites of phosphorylation are in the N-terminal globular domain of Spc110p, which resides at the inner plaque of the SPB and interacts with members of the microtubule-nucleating Tub4p complex (19, 23-25).
In vivo, alanine substitution at the Mps1p phosphorylation sites in Spc110p is synthetically lethal with alleles of SPC97, which encodes a component of the Tub4p complex. This synthetic lethality also requires an additional alanine substitution at residue Ser36. Ser36 falls within an (S/T)PX(R/K) consensus sequence for phosphorylation by cdc2p/Cdc28p cyclin-dependent kinase (32, 33), and Ser36 is phosphorylated when Spc110p is purified from insect cells.2 We have shown previously that Ser36 does not contribute to the mitosis-specific Spc110p isoform (26), and those findings are reiterated in this study (Fig. 7, lane 6). However, the S36A mutation is required for synthetic lethality with alleles of SPC97, and, in the presence of the S36A mutation, alanine substitution at any one of the three Mps1p sites is all that is required to produce the synthetic lethal phenotype.
Simultaneous aspartate substitution at Ser36 and the Mps1p sites does not interfere with Spc110p function, including the ability to complement synthetic lethality between alleles of spc110 and spc97. Whereas alanine substitution at serine and threonine residues is thought to prohibit side-chain phosphorylation without perturbing overall protein structure, aspartate substitutions at these residues are thought to mimic phosphorylation owing to the bulky, negatively charged aspartate residue side chain. Thus the phenotype associated with the alanine substitution Spc110p is not due simply to an overall perturbation of protein structure, but more likely to a specific defect due to the loss of phosphorylation at these residues.
The mitosis-specific phosphorylation of Spc110p occurs at a time when spindles first form (26), and alleles containing alanine substitutions at Ser36 and the Mps1p sites perturb the interaction between Spc110p and Spc97p. It is tantalizing to suggest that Spc110p phosphorylation is important for proper interaction with the Tub4p complex. However, it is important to note that the spc97-114 allele exhibits a number of genetic interactions both with mutations in SPC110 and with genes encoding components of the Tub4p complex (25). Thus the synthetic lethality between spc110-4A and spc97-114 reflects a defect in Spc110p function but does not specify which function of Spc110p is compromised. In contrast, the spc110-4A allele was still able to complement synthetic lethality between spc110-221 and spc98-63 (Table III, line 9). The fact that the spc98-63 allele tested here was unperturbed by the spc110-4A allele is not surprising, because spc98-63 has defects that are specific to the spc110-221 and spc110–222 alleles and exhibits no genetic interactions with other mutations in SPC110 or with mutations in the Tub4p complex components (25).3
Mps1p has a demonstrated role in both SPB duplication and the mitotic spindle checkpoint (6, 28, 29). A role for Mps1p during mitotic spindle formation has been suggested by genetic interactions with the CIN8-encoded kinesin-like protein (40), and the spindle component encoded by DAM1 (41). Our results here confirm a third role for Mps1p activity during spindle formation and suggest that it is important for Spc110p function. Consistent with this third Mps1p role demonstrated herein, Mps1p kinase isolated from synchronized cycling cells exhibits a peak of activity at the same time Spc110p phosphorylation occurs as the mitotic spindle forms.4
Mps1p phosphorylation of the Spc110p N-terminal globular domain may modulate the interaction between the microtubule-nucleating Tub4p complex and the SPB. The regions encompassing sites of Spc110p phosphorylation may form a docking site for the Tub4p complex or provide access to the docking site when phosphorylated. Another possible role for this phosphorylation is modulation of microtubule dynamics during mitotic spindle formation, which may also occur through additional phosphorylation of Tub4p complex components by Mps1p. Indeed, the Tub4p complex component Spc98p is also a phosphoprotein that exhibits mitosis-specific phosphorylation similar to that seen for Spc110p, and GST-Mps1p can phosphorylate Spc98p in vitro (30). Phosphorylation of Spc98p may complement the phosphorylation of Spc110p in the binding of the Tub4p complex such that mutations in the phosphorylation sites of both of these binding partners would have catastrophic consequences. Overall, Mps1p phosphorylation of proteins involved in microtubule nucleation during mitosis may be a mechanism by which Mps1p contributes to the assembly and stability of the SPB and the mitotic spindle that it forms.
Acknowledgments
We thank Ken Winter, Nancy Cyrus, Julian Watts, and Reudi Aebersold for technical advice and assistance.
Footnotes
This work was supported in part by National Institutes of Health (NIH) Grant GM40506 (to T. N. D.), NIH Grant GM-51312 (to M. W.), and NIH Grant AR39730 (to K. A. R.); by the Howard Hughes Medical Institute (to N. G. A.); and by NCRR, NIH Grant P41RR11823 (to J. Y.). Mass spectra presented were generated at the University of Colorado, Boulder.
The abbreviations used are: SPB, spindle pole body; GST, glutathione S-transferase; PAGE, polyacrylamide gel electrophoresis; TLE/T-LC, thin layer electrophoresis, thin layer chromatography; DTT, dithiothreitol; ESI-LC/MS, electrospray ionization, liquid chromatography mass spectrometry; ESI-LC/MS/MS, ESI liquid chromatography tandem mass spectrometry; HPLC, high performance liquid chromatography; MALDI-TOF, matrix-assisted laser desorption ionization, time-of-flight; endoLys-C, endoproteinase Lys-C.
D. B. N. Vinh, D. B. Friedman, and T. N. Davis, unpublished observations.
T. Nguyen and T. N. Davis, unpublished results.
E. Steiner and M. Winey, unpublished observations.
References
- 1.Vandr’e DD, Borisy GG. J Cell Sci. 1989;94:245–258. doi: 10.1242/jcs.94.2.245. [DOI] [PubMed] [Google Scholar]
- 2.Masuda H, Sevik M, Cande WZ. J Cell Biol. 1992;117:1055–1066. doi: 10.1083/jcb.117.5.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Engle DB, Doonan JH, Morris NR. Cell Motil Cytoskelet. 1988;10:434–437. doi: 10.1002/cm.970100310. [DOI] [PubMed] [Google Scholar]
- 4.Centonze VE, Borisy GG. J Cell Sci. 1990;95:405–411. doi: 10.1242/jcs.95.3.405. [DOI] [PubMed] [Google Scholar]
- 5.Winey M, Goetsch L, Baum P, Byers B. J Cell Biol. 1991;114:745–754. doi: 10.1083/jcb.114.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lauz’e E, Stoelcker B, Luca FC, Weiss E, Schutz AR, Winey M. EMBO J. 1995;14:1655–1663. doi: 10.1002/j.1460-2075.1995.tb07154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glover DM, Leibowitz MH, McLean DA, Parry H. Cell. 1995;81:95–105. doi: 10.1016/0092-8674(95)90374-7. [DOI] [PubMed] [Google Scholar]
- 8.Lacey KR, Jackson PK, Stearns T. Proc Natl Acad Sci U S A. 1999;96:2817–2822. doi: 10.1073/pnas.96.6.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hinchcliffe EH, Li C, Thompson EA, Maller JL, Sluder G. Science. 1999;283:851–854. doi: 10.1126/science.283.5403.851. [DOI] [PubMed] [Google Scholar]
- 10.Matsumoto Y, Hayashi K, Nishida E. Curr Biol. 1999;9:429–432. doi: 10.1016/s0960-9822(99)80191-2. [DOI] [PubMed] [Google Scholar]
- 11.Fry AM, Mayor T, Meraldi P, Stierhof YD, Tanaka K, Nigg EA. J Cell Biol. 1998;141:1563–1574. doi: 10.1083/jcb.141.7.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hisamoto N, Sugimoto K, Matsumoto K. Mol Cell Biol. 1994;14:3158–3165. doi: 10.1128/mcb.14.5.3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Black S, Andrews PD, Sneddon AA, Stark MJ. Yeast. 1995;11:747–759. doi: 10.1002/yea.320110806. [DOI] [PubMed] [Google Scholar]
- 14.MacKelvie SH, Andrews PD, Stark MJ. Mol Cell Biol. 1995;15:3777–3785. doi: 10.1128/mcb.15.7.3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vandr’e DD, Wills VL. J Cell Sci. 1992;101:79–91. doi: 10.1242/jcs.101.1.79. [DOI] [PubMed] [Google Scholar]
- 16.Kilmartin JV, Dyos SL, Kershaw D, Finch JT. J Cell Biol. 1993;123:1175–1184. doi: 10.1083/jcb.123.5.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sundberg HA, Goetsch L, Byers B, Davis TN. J Cell Biol. 1996;133:111–124. doi: 10.1083/jcb.133.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spang A, Grein K, Schiebel E. J Cell Sci. 1996;109:2229–2237. doi: 10.1242/jcs.109.9.2229. [DOI] [PubMed] [Google Scholar]
- 19.Sundberg HA, Davis TN. Mol Biol Cell. 1997;8:2575–2590. doi: 10.1091/mbc.8.12.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kilmartin JV, Goh PY. EMBO J. 1996;15:4592–4602. [PMC free article] [PubMed] [Google Scholar]
- 21.Elliott S, Knop M, Schlenstedt G, Schiebel E. Proc Natl Acad Sci U S A. 1999;96:6205–6210. doi: 10.1073/pnas.96.11.6205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adams IR, Kilmartin JV. J Cell Biol. 1999;145:809–823. doi: 10.1083/jcb.145.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knop M, Schiebel E. EMBO J. 1997;16:6985–6995. doi: 10.1093/emboj/16.23.6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knop M, Schiebel E. EMBO J. 1998;17:3952–3967. doi: 10.1093/emboj/17.14.3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen T, Vinh DBN, Crawford DK, Davis TN. Mol Biol Cell. 1998;9:2201–2216. doi: 10.1091/mbc.9.8.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friedman DB, Sundberg HA, Huang EY, Davis TN. J Cell Biol. 1996;132:903–914. doi: 10.1083/jcb.132.5.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stirling DA, Stark MJ. Biochem Biophys Res Commun. 1996;222:236–242. doi: 10.1006/bbrc.1996.0728. [DOI] [PubMed] [Google Scholar]
- 28.Weiss E, Winey M. J Cell Biol. 1996;132:111–123. doi: 10.1083/jcb.132.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hardwick KG, Weiss E, Luca FC, Winey M, Murray AW. Science. 1996;273:953–956. doi: 10.1126/science.273.5277.953. [DOI] [PubMed] [Google Scholar]
- 30.Pereira G, Knop M, Schiebel E. Mol Biol Cell. 1998;9:775–793. doi: 10.1091/mbc.9.4.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wigge PA, Jensen ON, Holmes S, Soues S, Mann M, Kilmartin JV. J Cell Biol. 1998;141:967–977. doi: 10.1083/jcb.141.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langan TA, Gautier J, Lohka M, Hollingsworth R, Moreno S, Nurse P, Maller J, Sclafani RA. Mol Cell Biol. 1989;9:3860–3868. doi: 10.1128/mcb.9.9.3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shenoy S, Choi JK, Bagrodia S, Copeland TD, Maller JL, Shalloway D. Cell. 1989;57:763–774. doi: 10.1016/0092-8674(89)90791-5. [DOI] [PubMed] [Google Scholar]
- 34.Davis TN. J Cell Biol. 1992;118:607–617. doi: 10.1083/jcb.118.3.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geiser JR, van Tuinen D, Brockerhoff SE, Neff MM, Davis TN. Cell. 1991;65:949–959. doi: 10.1016/0092-8674(91)90547-c. [DOI] [PubMed] [Google Scholar]
- 36.Ito H, Fukuda Y, Murata K, Kimura A. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boeke JD, Trueheart J, Natsoulis G, Fink GR. Methods Enzymol. 1987;154:164–175. doi: 10.1016/0076-6879(87)54076-9. [DOI] [PubMed] [Google Scholar]
- 38.Schutz AR, Winey M. Mol Biol Cell. 1998;9:759–774. doi: 10.1091/mbc.9.4.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baum P, Furlong C, Byers B. Proc Natl Acad Sci U S A. 1986;83:5512–5516. doi: 10.1073/pnas.83.15.5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geiser JR, Schott EJ, Kingsbury TJ, Cole NB, Totis LJ, Bhattacharyya G, He L, Hoyt MA. Mol Biol Cell. 1997;8:1035–1050. doi: 10.1091/mbc.8.6.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones MH, Bachant JB, Castillo AR, Giddings TH, Jr, Winey M. Mol Biol Cell. 1999;10:2377–2391. doi: 10.1091/mbc.10.7.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 43.Watts JD, Affolter M, Krebs DL, Wange RL, Samelson LE, Aebersold R. J Biol Chem. 1994;269:29520–29529. [PubMed] [Google Scholar]
- 44.Geiser JR, Sundberg HA, Chang BH, Muller EG, Davis TN. Mol Cell Biol. 1993;13:7913–7924. doi: 10.1128/mcb.13.12.7913. [DOI] [PMC free article] [PubMed] [Google Scholar]