

Abstract
Tumor-induced osteomalacia (TIO), or oncogenic osteomalacia (OOM), is a rare acquired Paraneoplastic disease characterized by renal phosphate wasting and hypophosphatemia. Recent evidence shows that tumor-overexpressed fibroblast growth factor 23 (FGF23) is responsible for the hypophosphatemia and osteomalacia. The tumors associated with TIO are usually phosphaturic mesenchymal tumor mixed connective tissue variants (PMTMCT). Surgical removal of the responsible tumors is clinically essential for the treatment of TIO. However, identifying the responsible tumors is often difficult. Here, we report a case of a TIO patient with elevated serum FGF23 levels suffering from bone pain and hypophosphatemia for more than three years. A tumor was finally located in first metacarpal bone by octreotide scintigraphy and she was cured by surgery. After complete excision of the tumor, serum FGF23 levels rapidly decreased, dropping to 54.7% of the preoperative level one hour after surgery and eventually to a little below normal. The patient's serum phosphate level rapidly improved and returned to normal level in four days. Accordingly, her clinical symptoms were greatly improved within one month after surgery. There was no sign of tumor recurrence during an 18-month period of follow-up. According to pathology, the tumor was originally diagnosed as “glomangioma” based upon a biopsy sample, “proliferative giant cell tumor of tendon sheath” based upon sections of tumor, and finally diagnosed as PMTMCT by consultation one year after surgery. In conclusion, although an extremely rare disease, clinicians and pathologists should be aware of the existence of TIO and PMTMCT, respectively.
Keywords: Tumor-induced osteomalacia (TIO), fibroblast growth factor 23 (FGF23), phosphaturic mesenchymal tumors mixed connective tissue variants (PMTMCT), hypophosphatemia
Tumor-induced osteomalacia (TIO), also known as oncogenic osteomalacia (OOM), is a rare acquired Paraneoplastic disease. TIO was first described in 1947, and nearly 300 cases have been reported in the literature[1]. TIO is characterized by renal phosphate wasting and hypophosphatemia[2],[3]. Muscle weakness and bone pain are usually the first clinical manifestations [4]. Surgical removal of the responsible tumors is clinically essential for the treatment of TIO[3]; however, identifying the responsible tumors is often difficult because they predominantly consist of small, slow-growing benign neoplasms[5]. Recent evidence has shown that tumors associated with TIO overexpress fibroblast growth factor 23 (FGF23)[3], which was identified as the last member of the FGF family. Excessive FGF23 action is known to cause several hypophosphatemic diseases, whereas deficient FGF23 activity has been found to result in hyperphosphatemic tumoral calcinosis[3].
To locate tumors responsible for TIO, octreotide scintigraphy has been helpful in most patients[5]. TIO-associated tumors are usually reported to be phosphaturic mesenchymal tumor mixed connective tissue variants (PMTMCT)[1]. However, most clinicians and pathologists are not aware of the existence of this type of tumor, and it is often misdiagnosed as another type of mesenchymal tumor[4]. Here, we describe a TIO patient suffering from bone pain and hypophosphatemia for more than three years and having an elevated serum FGF23 level. A tumor was found in the first metacarpal bone by octreotide scintigraphy and cured by surgery. Upon pathologic assessment, the tumor was originally misdiagnosed as “glomangioma” based on biopsy, “proliferative giant cell tumor of tendon sheath” based on tumor sections, and finally diagnosed as PMTMCT by consultation one year after surgery.
Case Report
Case history
The 45-year-old female patient first noted pain in lower sternocostal part of the left side in July 2006. It was a dull pain, with no pressing pain and no radiation. She was diagnosed with “costal chondritis” at the local hospital and treated with corticosteroid (local blockade) and vitamin B12 (subcutaneous injection), but the pain was gradually aggravating; she experienced sternocostal pain of both sides and vertebral pain. Over time, she lost her muscle strength and had difficulty walking. In March 2007, with serious hip pain and loss of mobility, she was diagnosed with “bilateral femoral neck fractures with bilateral aseptic necrosis of femoral heads. ” Despite being treated in several large hospitals in Beijing, China and lying in bed for two years, the patient's pain became progressively worse and she developed severe osteoporosis. In October 2009, owing to “polyuria and thirsty with no weight losing” as well as with the fasting serum glucose of 16 mmol/L, she was diagnosed with type 2 diabetes. On November 26th, 2009, she was admitted into the Department of Endocrinology of our hospital for “metabolic bone disease, diabetes, bilateral aseptic necrosis of femoral heads and osteoporosis. ”
On admission to the Department of Endocrinology, the clinical findings were bone pain of whole body (especially the back and hip), loss of mobility (the patient even could not turn over in bed by herself), atrophy of lower extremity muscles, and a swollen left basal thumb. Abnormal laboratory findings are presented in Table 1. Other laboratory tests, including urine phosphate, urine calcium, rheumatoid factor (RF), parathyroid hormone (PTH), tumor markers, immunoglobulins, antinuclear antibody, and adrenocorticotropic hormone, were normal. The patient also received various radiological examinations. Chest X-ray indicated osteoporosis and wedge-like variation of the vertebral bodies; pelvic X-ray and computed tomography (CT) revealed osteoporosis and fuzzy sacroiliac joint space; ankle X-ray showed osteoporosis; hip X-ray (November 27th, 2009, Figure 1A) revealed bilateral aseptic necrosis of femoral heads, bilateral obsolete femoral neck fractures, and severe osteoporosis; and wrist X-ray (November 30th, 2009, Figure 1B) indicated expansive destruction of the first metacarpal bone, as well as thin cortical bone with even density soft tissues inside and swelling soft tissues around. 99mTc methylene diphosphonate (99mTc-MDP) labeled emission CT scanning for bone revealed slight radioactive accumulation in the spine, ribs, sacroiliac joint, and femoral heads, as well as reduced radioactivity on long bones of the whole body.
Table 1. Abnormal laboratory findings for a patient with tumor-induced osteomalacia (TIO) on admission.
| Abnormal laboratory finding | Result | Reference range |
| Hypophosphatemia (mmol/L) | 0.35 ± 0.05 | 0.89–1.60 |
| High serum alkaline phosphatase (U/L) | 177.5 ± 15.1 | 0–130 |
| Hypocalcemia (mmol/L) | 2.14 ± 0.08 | 2.25–2.75 |
| Hypopotassemia (mmol/L) | 3.01 ± 0.10 | 3.5–5.5 |
| High urine protein quantity (g/24 h) | 1.102 | <0.15 |
| Urine phosphate test | (+) | (–) |
| High serum erythrocyte sedimentation rate (mm/h) | 49 | 0–20 |
| High serum osteocalcin (µg/L) | 7.56 | 1.97–6.45 |
Figure 1. Radiology results for a female patient with tumor-induced osteomalacia.

A, pelvic X-ray image (November 27th, 2009) shows bilateral aseptic necrosis of femoral heads, bilateral obsolete femoral neck fractures, severe osteoporosis, and fuzzy sacroiliac joint space. B, wrist X-ray image (November 30th, 2009) shows expansive destruction of the first metacarpal bone, thin cortical bone with even density soft tissues inside and swelling soft tissues around. C, 99mTc-0CT-guided octreotide scintigraphy (December 12th, 2009) shows high expression of somatostatin receptor on the first metacarpal bone of left hand (posterior and anterior projection).
In the Department of Endocrinology, the patient was treated with symptomatic therapies, including caltrate D, calcitriol, and salmon calcitonin for osteoporosis, neutral phosphate supplement for hypophosphatemia, and oral hypoglycemic agents for diabetes, as well as hepatoprotective treatments and analgesics. However, the patient's symptoms continued to worsen. A provisional diagnosis of TIO was made on the basis of clinical, laboratory, and radiological results, and attempts were made to locate the associated tumor even though wrist X-ray showed the destruction of first metacarpal bone. Octreotide scanning showed high expression of somatostatin receptor, growth hormone release inhibiting hormone receptor (GIHR), on the first metacarpal bone (December 12th, 2009, Figure 1C). Eventually, the patient was confirmed to have TIO, and the tumor was located on the first metacarpal bone of left hand. She was transferred to the Department of Orthopedics for surgery.
In order to identify the tumor (benign or malignant), ultrasonic guided biopsy was taken and the result showed glomangioma. Although a benign pathology, the wrist X-ray showed an invasive lesion. Considering that the muscles of the thenar eminence were infiltrated by the tumor and that it was difficult to preserve the thumb, instead of a reconstruction operation, an intact resection was performed by disarticulation of the thumb by first carpometacarpal joint on December 22th, 2009. The patient recovered well, and notably, the bone pain reduced the second day after surgery. The patient was discharged from hospital on the 40th day after hospitalization. One month after surgery, clinical symptoms, such as bone pain, greatly improved. Three months after surgery, the patient could walk by herself and the pain completely disappeared. During an 18-month period of follow-up, no sign of tumor recurrence occurred.
Serum FGF23 measurement
The patient's blood samples were taken before and after surgery then centrifugated, and the serum was preserved below –80°C. Serum C-terminal FGF23 was measured in duplicate using the Human FGF23 (C-term) enzyme-linked immunosorbent assay (ELISA) kits (Immutopics Inc, USA). The level of serum FGF23 was higher before surgery [(417.2 ± 52.7) RU/mL] than the reference level measured with five normal controls [(54.0 ± 26.5) RU/mL]. The serum FGF23 level dropped dramatically after removal of the tumor, measuring (239.3 ± 64.4) RU/mL (54.7% of the preoperative level) 1 h after surgery, (120.0 ± 21.9) RU/mL 12 h after surgery, and (47.9 ± 3.7) RU/mL 24 h after surgery, which is a little lower than normal level according to the literature[6]. Hypophosphatemia was rapidly improved and serum phosphate level returned to normal within four days (reference range, 0.89–1.60 mmol/L), whereas serum alkaline phosphatase (ALP) level was evaluated, and the mild hypocalcemia was not improved (Table 2 and Figure 2).
Table 2. Changes of the serum phosphate, serum FGF23, serum alkaline phosphatase (ALP), serum phosphate (P), and serum calcium before and after surgery.
| Time | FGF23 (RU/mL) [reference, 54.9 ± 26.5] | ALP (U/L) [reference, 0–130] | P (mmol/L) [reference, 0.89–1.60] | Ca (mmol/L) [reference, 2.25–2.75] | |
| Before surgery | 417.2 ± 52.7 | 177.5 ± 15.1 | 0.35 ± 0.05 | 2.14 ± 0.08 | |
| After surgery | 1 hour | 239.3 ± 64.4 | NA | NA | NA |
| 0.5 day | 120.9 ± 21.9 | NA | NA | NA | |
| 1 day | 47.9 ± 3.7 | NA | 0.39 | 2.05 | |
| 1.5 days | 39.0 ± 3.1 | NA | NA | NA | |
| 2 days | 29.4 ± 7.9 | NA | NA | NA | |
| 3 days | NA | NA | 0.76 | 2.17 | |
| 4 days | NA | NA | 0.94 | 2.19 | |
| 6 days | 31.9 ± 11.2 | 221.6 | 1.04 | 2.13 | |
| 9 days | NA | NA | 1.20 | 2.15 | |
| 14 days | NA | 225.4 | 1.34 | 2.13 |
NA, not available.
Figure 2. Changes of the serum phosphate, serum FGF23, alkaline phosphatase (ALP), and serum calcium before and after surgery.

Serum FGF23 levels dropped dramatically after removal of the tumor, falling to 54.7% of the preoperative level 1 h after surgery and to slightly lower than normal level over time. Hypophosphatemia was rapidly improved, and phosphate levels returned to normal within 4 days. However, serum ALP level was evaluated, and the mild hypocalcemia was not improved in the short term.
Histopathology
By histological assessment, the tumor was originally diagnosed as “glomangioma” according to biopsy, “proliferative giant cell tumor of tendon sheath” according to tumor sections, and finally “PMTMCT” by consultation of the same HE staining section one year after surgery. The tumor measured 3 cm x 3 cm x 3 cm, appeared gray, and exhibited moderate hardness. Hematoxylin and eosin staining results showed vascular proliferation and uniformly oval- to spindle-shaped macrophage-like mononuclear cells without atypia embedded within a distinctive collagenous matrix. Osteoclast-like giant cells were scattered in the tumor tissue. Mitotic figures were rarely observed (Figure 3). Immunohistochemical staining was also performed, and the results are as follows: CD31 (–), CD34 (–), CD68 (positive cells were scattered), Desmin (–), Ki-67 (positive rate <25%), S-100 (–), vimentin (+), F8 (–), and actin (pan) (–) (Figure 4).
Figure 3. Hematoxylin and eosin staining of the tumor-induced osteomalacia.

This tumor was originally diagnosed as “proliferative giant cell tumor of tendon sheath” and finally diagnosed as PMTMCT one year after surgery. Highly vascular proliferation and oval- to spindle-shaped macrophage-like mononuclear cells without atypia formed most of the background cells and were embedded in a distinctive smudgy matrix. The osteoclast-like giant cells were scattered in the tumor. A, black arrowheads indicate neovessels, whereas the white arrowhead indicates uniform spindle tumor cells. B, black arrowheads indicate osteoclast-like giant cells, whereas white arrowheads indicate scattered hemorrhage. C, the black arrowhead indicates uniform spindle tumor cells, whereas the white arrowhead indicates cartilage formation. D, the black arrowhead indicates osteoid-like matrix, whereas the white arrowhead indicates uniform spindle tumor cells.
Figure 4. Immunohistochemisty for CD34, CD68, and vimentin of the tumor.
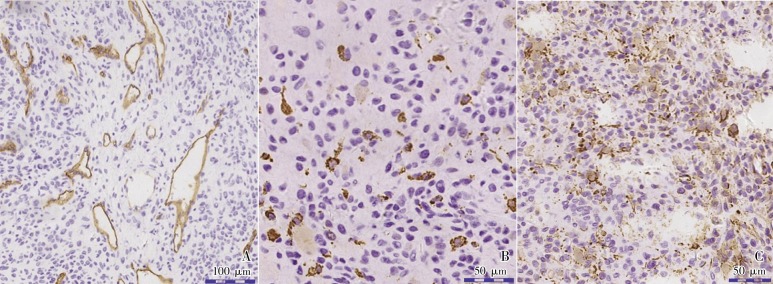
A, the tumor contains a large number of small capillaries that mimic those observed in hemangiopericytoma, most of which are positive for CD34 staining (brown) whereas tumor cells are negative. B, CD68 (+) indicates the scattered distribution, morphology and density of monocytes/macrophages (brown). C, as a mesenchymal cell marker, vimentin is positive in most of the tumor cells, indicating the tumor originated from mesenchymal tissues (brown).
Amplification of FGF23 from genomic DNA and detection of mutations
The tumor sample was collected during the surgery and preserved below –80°C. Genomic DNA was extracted from the resected tumor tissue using the Genomic DNA Purification Kit (Promega Corporation). Reported primers that spanned the intron/exon boundaries were synthesized by Invitrogen Corporation and used to amplify the three exons of FGF23[7]. The polymerase chain reaction products were sequenced (by Biosune Biotechnology), and no mutations in FGF23 were detected (compared to GenBank accession NM_0220638). Notably, mutations previously described in FGF23 involving nucleotides encoding arginines at codons 176 and 179[8] were absent.
Discussion
TIO patients are characterized by renal phosphate wasting and hypophosphatemia[2],[3], and the associated clinical and laboratory findings are bone pain, atrophy of proximal muscles, gait disturbance and hypophosphatemia. About 46% of reported cases of TIO have occurred in females and 54% in males, with a mean age of 45.3 years when definitive diagnosis was made. Of all cases, 42.9% could be originated from soft tissues and 57.1% from the bone. Most tumors were reported to occur in the thigh and femur (22.7%), facies crania (20.7%), ankle and foot (8.8%), pelvis (8.2%), tibia and fibula (6.5%), and arm (6.5%). The less common locations were the vertebra, knee, hand, chest, abdomen, groin, perineum, and gluteal region[1]. Some tumors were even located in organs such as the liver[9], tongue[10], thyroid[1] and lung[1]. Thus, these tumors have occurred almost anywhere in the body[1].
TIO located in the hand
As presented in Table 3, there were only six other cases of TIO located in the hand reported by literature. Including our case, all reported TIO cases have involved four females and three males, mean age 47.4, with three cases in the metacarpal bone, two in the finger, one in soft tissue of the palm, and one of unclear origin. While present in a superficial location, most tumors can be palpated earlier than the clinical manifestations of osteomalacia. However, none of the patients could be diagnosed and treated at an early stage. The seven patients all suffered muscle weakness and bone pain, with a mean osteomalacia duration of 3.8 years. All the patients had low serum phosphate levels, high or normal phosphate serum alkaline phosphatase, and normal urinary phosphate levels. They were treated by surgery and no signs of recurrence occurred. The case described here is the first report of a tumor in the first metacarpal bone and is the largest reported tumor in the hand. With more serious clinical manifestations and lower serum phosphate level, our patient developed into severe osteoporosis, unilateral aseptic necrosis of femoral heads, and bilateral femoral neck fractures.
Table 3. Reported cases of TIO located in the hand.
| Authors and year | Age/gender | Tumor location | Tumor size (cm3) | Mass found before surgery (years) | Duration of osteo-malacia | Clinical manifestations | Serum phosphate (mmol/L, reference 0.89–1.6) | Serum alkaline phosphatase U/L) [reference 0–130] | Urinary phosphate (mmol/24 h) [reference 23–48] | Recovered from osteomalacia after surgery (months) | Follow-up result | Original pathologic diagnosis | Revised pathologic diagnosis |
| Yoshikawa et al., 1977[36] | 18/M | Fourth metacarpal | NA | 5 | 5 | Difficulty in walking, pain in the lumbar region and knees | 0.45 | 63 | 17.1 | 3 | No recurrence | Osteoblastoma | Osteoblastoma and osteosarcoma |
| Weidner et al., 1987[18] | 34/M | Mid palm | 9 | 5 | 1 | Back stiffness, lower extremity weakness, waddling gait | 0.51 | Elevated | 24.32 | 3 | No recurrence in 2 years | Giant cell variant of soft part chondroma | PMTMCT |
| Folpe et al., 2004[1] | 61/F | Hand | 12 | NA | 6 | NA | NA | NA | NA | NA | No recurrence in 4 years | Giant cell tumor of tendon sheath | PMTMCT |
| 54/F | 4th finger | Small | NA | 5–6 | NA | NA | NA | NA | NA | No recurrence in 3 years | Atypical enchondroma | Enchondroma with cytologic atypia | |
| 61/F | Metacarpal | NA | NA | NA | NA | NA | NA | NA | NA | NA | Atypical giant cell tumor | PMTMCT | |
| Jacob et al., 2007[37] | 59/M | Index finger | 2 | 2 | 2 | Pain in the lumbar spine, sacroiliac joints, and hips; difficulty in rising and climbing stairs | 0.58 | 436 | 25.6 | 6 | No recurrence | PMTMCT | PMTMCT |
| Current case | 45/F | First metacarpal | 14 | 4 | 3.5 | Serious bone pain of whole body, immobility (could nottumoverinbed), and pathological fractures of the hip | 0.35 | 178 | 28.62 | 1 | No recurrence in 1.5 years | Giant cell tumor of tendon sheath | PMTMCT |
NA, not available. PMTMCT, phosphaturic mesenchymal tumor mixed connective tissue variants.
The role of FGF23 and mutation detection
Recently, FGF23 was shown to be responsible for the pathogenesis of TIO, especially the hypophosphatemia[3],[11]. As a circulating FGF produced by osteocytes and osteoblasts, FGF23 has been identified as the key regulator of phosphate metabolism. Excessive FGF23 action causes several hypophosphatemic diseases[3],[11], including TIO, X-linked hypophosphatemic rickets (XLHR), autosomal dominant and recessive hypophosphatemic rickets (ADHR and ARHR), whereas deficient FGF23 activity results in hyperphosphatemic tumoral calcinosis[3],[8]. Under physiological conditions, FGF23 is secreted predominantly by bone and undergoes degradation by the phosphate regulating endopeptidase homolog, X-linked (PHEX) and other proteolytic enzymes. Increased amounts of FGF23 may result from decreased degradation or increased production. Decreased degradation of FGF23 occurs in XLHR and ADHR due to mutations of the PHEX and FGF23 genes, respectively, that render FGF23 resistant to cleavage and thereby reduce its degradation[12]. Although ADHR, ARHR, and XLHR are biochemically indistinguishable from TIO, symptoms of ADHR, ARHR, and XLHR typically present in childhood, whereas symptoms of TIO present later in life. ADHR, ARHR, and XLHR are associated with deformities in lower extremities and short stature, and a definitive diagnosis can be made by genetic testing of the FGF23, dentin matrix protein 1 (DMP1) and PHEX genes, which are defective in ADHR, ARHR and XLHR, respectively[8]. In the present study, the three exons of FGF23 were amplified from genomic DNA, but no mutation was found compared to the wild type, nor was there detection of previously described mutations of FGF23 in TIO involving the nucleotides that encode arginines at codons 176 and 179[8].
Increased production of FGF23 occurs in TIO wherein PMTMCT secrete FGF23 at levels that are several hundred-fold greater than normal, thereby potentially overwhelming the FGF23 degradation pathway[12]. Therefore, TIO is purportedly caused by overexpression of FGF23 in the responsible tumor, which results in hypophosphatemia and renal phosphate wasting, reduced 1,25-dihydroxyvitamin D3 [1,25 (OH)2D3] synthesis, and osteomalacia[11]. Circulatory FGF23 levels are elevated in virtually all patients with TIO and rapidly decreases below normal after removal of the responsible tumor[6]. However, how the tumor cells overexpress FGF23 is still unknown.
Bone-kidney-parathyroid endocrine axes
A bone-kidney-parathyroid endocrine signaling axis controls the biological effects of FGF23 on renal phosphate and vitamin D metabolism, as shown in Figure 5. However, the mechanisms of FGF23's action in the kidney are unclear. Liu et al.[13] and Gattineni et al.[14] recently showed that FGFR1, but not FGFR3 or FGFR4, was the predominant receptor for the hypophosphatemic action of FGF23 in vivo and that the distal tubule may be an effector site of FGF23. Although the FGF23 co-receptor Klotho (KL) is expressed as both secreted and membrane-bound isoforms, only the membrane-bound isoform that localizes at the distal convoluted tubule (DCT) is capable of mediating FGF23 bioactivity[15]. Nevertheless, how KL can facilitate the FGF23-mediated phosphate transport process as sodium-dependent phosphate transporter 2a (NaPi-2a) and sodium-dependent phosphate transporter 2c (NaPi-2c) and localize at the proximal tubules (PT) is still unknown.
Figure 5. The bone-kidney-parathyroid endocrine axes mediated by FGF23 and 1,25(OH)2D3.

FGF23 is primarily produced in the bones (osteocyte). Through circulation, FGF23 is transferred to the kidney, where it interacts with the FGFRs in the presence of co-receptor Klotho[26]–[28], decreasing the expression of an electrogenic phosphate transporter (sodium-dependent phosphate transporter 2a, NaPi-2a)[29],[30] and an electroneutral phosphate transporter (NaPi-2c) on the apical surface of the proximal tubule and leading to inhibition of renal phosphate reabsorption and 1,25(OH)2D3 synthesis[15],[27]. This results in phosphaturia and reduced intestinal absorption of calcium and phosphate, eventually decreasing the plasma Pi (inorganic phosphate) and resulting in hypophosphatemia[31]. On the other hand, serum parathyroid hormone (PTH) stimulates while FGF23 inhibits 1,25(OH)2D3 production in the kidney[11],[32]. In turn, 1,25(OH)2D3 inhibits PTH production and secretion from the parathyroid glands and stimulates FGF23 production from the bone as a negative feedback mechanism[33],[34]. FGF23 can also decrease PTH secretion, and PTH can decrease the expression of NaPi-2a and NaPi-2c[35]. The red arrows indicate positive regulation, and the gray arrows indicate negative regulation.
It is not clear whether FGF23 is the only (or even the major) m ediator of reduced renal phosphate transport in oncogenic osteomalacia. Overexpression of other genes, such as matrix extracellular phosphoglycoprotein (MEPE), secreted frizzled related protein 4 (SFRP4), and FGF7, has been reported in oncogenic osteomalacia tumors[16]. It is possible that the phenotype of oncogenic osteomalacia may be due to multiple factors that work alone or together to inhibit phosphate reabsorption and/or 1,25(OH)2D3 production[17].
PMTMCT
The tumors associated with TIO are usually small and mesenchymal in origin, but the diversity of their location often complicates the detection of origin. Tumor associated with TIO are usually PMTMCT, which was first described in 1987 by Weidner et al.[18], who determined that most cases of TIO, unlike any other mesenchymal tumor, were histologically distinct and coined the name “phosphaturic mesenchymal tumor mixed connective tissue variants” (PMTMCT). However, most clinicians and pathologists are not aware of the existence of this type of tumor. Thus, as PMTMCT typically has a variegated appearance, it is often misdiagnosed as another type of mesenchymal tumor, such as hemangiopericytoma, osteosarcoma, giant cell tumor, or others[4]. As presented in Table 3, the TIO tumors in the hand were often misdiagnosed as giant cell tumors of tendon sheath, giant cell tumor chondroma, and so on.
More recently, a large study and detailed literature review by Folpe et al.[1] further established that more than 90% of TIO-associated mesenchymal tumors were, in fact, PMTMCT. Histologically, PMTMCT are composed of highly vascular spindled to stellate cells with low nuclear grade and low mitotic activity embedded in a distinctive myxoid to myxochondroid matrix and exhibiting “grungy” or flocculent calcification[7]. Other features that may be present include microcystic change, mature fat, chondroid or osteoid-like matrix, and woven bone[7]. The cellular constituents comprise of four types of cells: macrophage-like mononuclear cells, epithelioid histiocyte-like cells, osteoclast-like giant cells, and xanthomatous cells. At the molecular level, PMTMCT induces overexpression of FGF23 in patients with TIO; however, the underlying reason and mechanism for this is unknown. Clinicians and pathologists should be aware of the existence of PMTMCT, especially the nonphosphaturic or asymptomatic variants of PMTMCT, and a patient with a tumor that is identical to PMTMCT should be carefully monitored by both imaging and laboratory examination.
Tumor localization and FGF23 measurements
The tumors associated with TIO are usually difficult to locate as they are often typically small slow-growing benign tumors and can occur anywhere in the body. As a result, the time from osteomalacia to identifying the associated tumor averages a period of five years[19]. In the present case, the tumor occurred in the first metacarpal bone of the left hand, where it could be easily located compared to other parts of the body. Even though the patient suffered from systemic bone pain for more than three years and had a swollen thumb for almost five years, no clinicians could associate the little mass of the thumb with the serious systemic metabolic disease when she visited several hospitals of Beijing.
Although noninvasive imaging modalities, such as CT, magnetic resonance imaging (MRI), and positron emission tomography, may be able to detect the tumor except for the small TIO tumors, they all lack specificity[20]. In vitro studies have revealed that many TIO tumors express the somatostatin receptor subtype 2 (SST2)[21], and octreotide scintigraphy has been used successfully to detect TIO tumors[20],[21]. As shown in Figure 1C, octreotide scintigraphy revealed high expression of somatostatin receptor on the first metacarpal bone of the left hand. This observation combined with the high serum FGF23 level facilitated the diagnosis of TIO for the patient, after which the tumor was located definitely.
Serum FGF23 levels have been measured by three commercially available ELISA kits. One kit, Human C-term FGF23 (Immutopics Inc, USA), measures total FGF23, whereas the other two kits, Immutopics Intact (Immutopics Inc, USA) and FGF23 ELISA (Kainos, Japan), measure only the biologically active intact molecule. Measurements of FGF23 levels using both immutopics assays were altered in the presence of low circulating concentrations of serum ferritin, whereas this effect was not demonstrated with the Kainos intact assay [22]. Interestingly, Takeuchi et al.[6] located a TIO before surgery by testing venous blood sampling for FGF23, indicating that the serum FGF23 level was higher around the tumor than in other parts of the body. This method, combined with other modalities like MRI, was also reported by other authors as an approach to locate the small tumor responsible for TIO[5],[23].
Treatments
Most of the patients with TIO were reported to be resistant to treatment with high-dose vitamin D[24]. Oral phosphate therapy or parenteral phosphate may be insufficient, and the medical therapy may itself be associated with long-term complications[25]. Because humoral phosphaturic factors such as FGF23 produced by the tumor cause hypophosphatemia and osteomalacia, surgical removal of the associated tumor is essential for definitive treatment of TIO. Although a benign lesion in 90% of the cases, integrated resection of tumor is necessary due to the recurrence and metastases in over 10% of PMTMCT[12].
In the present case, serum FGF23 levels rapidly decreased after complete resection of the tumor, dropping to 54.7% of the preoperative level one hour after surgery, which was in accordance with the results in the literature[6]. The patient's hypophosphatemia rapidly improved and serum phosphate level returned to normal within four days. Accordingly, her clinical symptoms greatly improved within one month. She could walk by herself, and her pain completely disappeared after three months. There was no sign of tumor recurrence during an 18-month period of follow-up.
We believed that the tumor in the present case produced FGF23, resulting in hypophosphatemia and osteomalacia. The tumor was located by octreotide scintigraphy, and the patient was cured of this disease by surgery. Although an extremely rare disease, clinicians and pathologists should be aware of the existence of TIO and PMTMCT, respectively.
References
- 1.Folpe AL, Fanburg-Smith JC, Billings SD, et al. Most osteomalaciaassociated mesenchymal tumors are a single histopathologic entity: an analysis of 32 cases and a comprehensive review of the literature [J] Am J Surg Pathol. 2004;28(1):1–30. doi: 10.1097/00000478-200401000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Econs MJ, Drezner MK. Tumor-induced osteomalacia— unveiling a new hormone [J] N Engl J Med. 1994;330(23):1679–1681. doi: 10.1056/NEJM199406093302310. [DOI] [PubMed] [Google Scholar]
- 3.Ramon I, Kleynen P, Body JJ, et al. Fibroblast growth factor 23 and its role in phosphate homeostasis [J] Eur J Endocrinol. 2009;162(1):1–10. doi: 10.1530/EJE-09-0597. [DOI] [PubMed] [Google Scholar]
- 4.Yoshioka K, Nagata R, Ueda M, et al. Phosphaturic mesenchymal tumor with symptoms related to osteomalacia that appeared one year after tumorectomy [J] Intern Med. 2006;45(20):1157–1160. doi: 10.2169/internalmedicine.45.1797. [DOI] [PubMed] [Google Scholar]
- 5.Nasu T, Kurisu S, Matsuno S, et al. Tumor-induced hypophosphatemic osteomalacia diagnosed by the combinatory procedures of magnetic resonance imaging and venous sampling for FGF23 [J] Intern Med. 2008;47(10):957–961. doi: 10.2169/internalmedicine.47.0745. [DOI] [PubMed] [Google Scholar]
- 6.Takeuchi Y, Suzuki H, Ogura S, et al. Venous sampling for fibroblast growth factor-23 confirms preoperative diagnosis of tumor-induced osteomalacia [J] J Clin Endocrinol Metab. 2004;89(8):3979–3982. doi: 10.1210/jc.2004-0406. [DOI] [PubMed] [Google Scholar]
- 7.Bahrami A, Weiss SW, Montgomery E, et al. RT-PCR analysis for FGF23 using paraffin sections in the diagnosis of phosphaturic mesenchymal tumors with and without known tumor induced osteomalacia [J] Am J Surg Pathol. 2009;33(9):1348–1354. doi: 10.1097/PAS.0b013e3181aa2311. [DOI] [PubMed] [Google Scholar]
- 8.Liu S, Quarles LD. How fibroblast growth factor 23 works [J] J Am Soc Nephrol. 2007;18(6):1637–1647. doi: 10.1681/ASN.2007010068. [DOI] [PubMed] [Google Scholar]
- 9.Seijas R, Ares O, Sierra J, et al. Oncogenic osteomalacia: two case reports with surprisingly different outcomes [J] Arch Orthop Trauma Surg. 2009;129(4):533–539. doi: 10.1007/s00402-008-0808-2. [DOI] [PubMed] [Google Scholar]
- 10.Uramoto N, Furukawa M, Yoshizaki T. Malignant phosphaturic mesenchymal tumor, mixed connective tissue variant of the tongue [J] Auris Nasus Larynx. 2009;36(1):104–105. doi: 10.1016/j.anl.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Shimada T, Mizutani S, Muto T, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia [J] Proc Natl Acad Sci USA. 2001;98(11):6500–6505. doi: 10.1073/pnas.101545198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hannan FM, Athanasou NA, Teh J, et al. Oncogenic hypophosphataemic osteomalacia: biomarker roles of fibroblast growth factor 23, 1,25-dihydroxyvitamin D3 and lymphatic vessel endothelial hyaluronan receptor 1 [J] Eur J Endocrinol. 2008;158(2):265–271. doi: 10.1530/EJE-07-0485. [DOI] [PubMed] [Google Scholar]
- 13.Liu S, Vierthaler L, Tang W, et al. FGFR3 and FGFR4 do not mediate renal effects of FGF23 [J] J Am Soc Nephrol. 2008;19(12):2342–2350. doi: 10.1681/ASN.2007121301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gattineni J, Bates C, Twombley K, et al. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1 [J] Am J Physiol Renal Physiol. 2009;297(2):F282–F291. doi: 10.1152/ajprenal.90742.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farrow EG, Davis SI, Summers LJ, et al. Initial FGF23- mediated signaling occurs in the distal convoluted tubule [J] J Am Soc Nephrol. 2009;20(5):955–960. doi: 10.1681/ASN.2008070783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Beur SM, Finnegan RB, Vassiliadis J, et al. Tumors associated with oncogenic osteomalacia express genes important in bone and mineral metabolism [J] J Bone Miner Res. 2002;17(6):1102–1110. doi: 10.1359/jbmr.2002.17.6.1102. [DOI] [PubMed] [Google Scholar]
- 17.Imel EA, Econs MJ. Fibroblast growth factor 23: Roles in health and disease [J] J Am Soc Nephrol. 2005;16(9):2565–2575. doi: 10.1681/ASN.2005050573. [DOI] [PubMed] [Google Scholar]
- 18.Weidner N, Santa Cruz D. Phosphaturic mesenchymal tumors. A polymorphous group causing osteomalacia or rickets [J] Cancer. 1987;59(8):1442–1454. doi: 10.1002/1097-0142(19870415)59:8<1442::aid-cncr2820590810>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 19.Seufert J, Ebert K, Muller J, et al. Octreotide therapy for tumor-induced osteomalacia [J] N Engl J Med. 2001;345(26):1883–1888. doi: 10.1056/NEJMoa010839. [DOI] [PubMed] [Google Scholar]
- 20.Hesse E, Moessinger E, Rosenthal H, et al. Oncogenic osteomalacia: exact tumor localization by co-registration of positron emission and computed tomography [J] J Bone Miner Res. 2007;22(1):158–162. doi: 10.1359/jbmr.060909. [DOI] [PubMed] [Google Scholar]
- 21.Reubi JC, Waser B, Laissue JA, et al. Somatostatin and vasoactive intestinal peptide receptors in human mesenchymal tumors: in vitro identification [J] Cancer Res. 1996;56(8):1922–1931. [PubMed] [Google Scholar]
- 22.Durham BH, Joseph F, Bailey LM, et al. The association of circulating ferritin with serum concentrations of fibroblast growth factor-23 measured by three commercial assays [J] Ann Clin Biochem. 2007;44(Pt 5):463–466. doi: 10.1258/000456307781646102. [DOI] [PubMed] [Google Scholar]
- 23.Ogura E, Kageyama K, Fukumoto S, et al. Development of tumor-induced osteomalacia in a subcutaneous tumor, defined by venous blood sampling of fibroblast growth factor-23 [J] Intern Med. 2008;47(7):637–641. doi: 10.2169/internalmedicine.47.0761. [DOI] [PubMed] [Google Scholar]
- 24.Schapira D, Ben Izhak O, Nachtigal A, et al. Tumor-induced osteomalacia [J] Semin Arthritis Rheum. 1995;25(1):35–46. doi: 10.1016/s0049-0172(95)80016-6. [DOI] [PubMed] [Google Scholar]
- 25.Shields AT, Chesnut CH., 3rd Diagnosis of postmenopausal osteoporosis: reviews in endocrine and metabolic disorders [J] Rev Endocr Metab Disord. 2001;2(1):23–33. doi: 10.1023/a:1010050823176. [DOI] [PubMed] [Google Scholar]
- 26.Nakatani T, Sarraj B, Ohnishi M, et al. In vivo genetic evidence for klotho–dependent, fibroblast growth factor 23 (FGF23) –mediated regulation of systemic phosphate homeostasis [J] FASEB J. 2009;23(2):433–441. doi: 10.1096/fj.08-114397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuro-o M. Klotho as a regulator of fibroblast growth factor signaling and phosphate/calcium metabolism [J] Curr Opin Nephrol Hypertens. 2006;15(4):437–441. doi: 10.1097/01.mnh.0000232885.81142.83. [DOI] [PubMed] [Google Scholar]
- 28.Kuro-o M. Klotho in chronic kidney disease—what's new? [J] Nephrol Dial Transplant. 2009;24(6):1705–1708. doi: 10.1093/ndt/gfp069. [DOI] [PubMed] [Google Scholar]
- 29.Larsson T, Marsell R, Schipani E, et al. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1 (I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis [J] Endocrinology. 2004;145(7):3087–3094. doi: 10.1210/en.2003-1768. [DOI] [PubMed] [Google Scholar]
- 30.Shimada T, Hasegawa H, Yamazaki Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis [J] J Bone Miner Res. 2004;19(3):429–435. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- 31.Razzaque MS. FGF23-mediated regulation of systemic phosphate homeostasis: is klotho an essential player? [J] Am J Physiol Renal Physiol. 2009;296(3):F470–F476. doi: 10.1152/ajprenal.90538.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perwad F, Zhang MY, Tenenhouse HS, et al. Fibroblast growth factor 23 impairs phosphorus and vitamin d metabolism in vivo and suppresses 25-hydroxyvitamin D-1alpha-hydroxylase expression in vitro [J] Am J Physiol Renal Physiol. 2007;293(5):F1577–1583. doi: 10.1152/ajprenal.00463.2006. [DOI] [PubMed] [Google Scholar]
- 33.Liu S, Tang W, Zhou J, et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D [J] J Am Soc Nephrol. 2006;17(5):1305–1315. doi: 10.1681/ASN.2005111185. [DOI] [PubMed] [Google Scholar]
- 34.Saito H, Maeda A, Ohtomo S, et al. Circulating FGF-23 is regulated by 1alpha,25-dihydroxyvitamin D3 and phosphorus in vivo [J] J Biol Chem. 2005;280(4):2543–2549. doi: 10.1074/jbc.M408903200. [DOI] [PubMed] [Google Scholar]
- 35.Bikle D. Nonclassic actions of vitamin D [J] J Clin Endocrinol Metab. 2009;94(1):26–34. doi: 10.1210/jc.2008-1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoshikawa S, Nakamura T, Takagi M, et al. Benign osteoblastoma as a cause of osteomalacia. A report of two cases [J] J Bone Joint Surg Br. 1977;59(3):279–286. doi: 10.1302/0301-620X.59B3.893505. [DOI] [PubMed] [Google Scholar]
- 37.Jacob JJ, Finny P, Thomas M, et al. Oncogenic osteomalacia [J] J Assoc Physicians India. 2007;55:231–233. [PubMed] [Google Scholar]