

Abstract
Metastasis represents by far the most feared complication of prostate carcinoma and is the main cause of death for patients. The skeleton is frequently targeted by disseminated cancer cells and represents the sole site of spread in more than 80% of prostate cancer cases. Compatibility between select malignant phenotypes and the microenvironment of colonized tissues is broadly recognized as the culprit for the organ-tropism of cancer cells. Here, we review our recent studies showing that the expression of platelet-derived growth factor receptor alpha (PDGFRα ) supports the survival and growth of prostate cancer cells in the skeleton and that the soluble fraction of bone marrow activates PDGFRα in a ligand-independent fashion. Finally, we offer pre-clinical evidence that this receptor is a viable target for therapy.
Keywords: Platelet-derived growth factor receptor alpha, metastasis, prostate cancer, organ tropism
Eighty-five percent of patients are routinely diagnosed with prostate cancer in the absence of secondary tumors. However, depending on initial therapy, histologic grading and residual disease after surgery, many of these patients will eventually present cancer dissemination to bone. Skeletal metastases are responsible for a significant reduction in the quality of life and represent the main cause of death in patients with advanced prostate adenocarcinoma. Treatment for bone metastasis is mostly palliative and is unable to prevent skeletal dissemination or eradicate prostate cancer cells that colonize the bone microenvironment[1].
Metastasis is a process that requires the successful execution of several sequential steps by cancer cells[2],[3]. Many tumors show a propensity to colonize specific tissues in the body, a feature defined as organ-tropism[4]. It is widely recognized that the identification of factors responsible for promoting the adaptation of malignant prostate cells to the bone microenvironment will lead to more effective therapeutic strategies for advanced prostate cancer. However, the molecules and mechanisms determining the organ-tropism of cancer cells are vaguely defined[5]. Paget[6] assimilated the compatibility between migrating cancer cells and colonized organs to the required affinity between a seed and the specific soil. In support of this idea we have to date considerable evidence indicating that migration of cancer cells into a foreign tissue needs favorable conditions to survive and proliferate[5]. Cancer cells failing to receive appropriate support may remain dormant or undergo cell death[7], thereby exerting negligible clinical impact on the patient. This general paradigm has been proposed for skeletal metastasis and appropriate trophic factors in the bone appear to be crucial for initial cell survival, growth into small foci, and subsequent progression into macroscopic metastases[8]–[10]. Thus, disseminated cancer cells expressing the appropriate receptor arsenal for the trophic factors locally produced by the bone marrow stroma will have a major advantage in supporting their survival and growth into clinically evident tumors.
Expression of PDGFRα by Prostate Epithelial Cells
The presence of platelet-derived growth factor receptors (PDGFRs) and their ligands in the prostate were initially described by Fudge et al.[11]. More recently, Chott et al.[12] reported that primary prostate cancers and their skeletal metastases are positive for PDGFR expression, with PDGFRα being most represented. We confirmed this observation by showing that normal prostate expresses low levels of PDGFRα, which significantly increase upon malignant transformation. We detected a strong signal for PDGFRα in approximately 70% of tissue cores of human prostate and all the samples from skeletal metastases by immunohistochemistry[13]. Interestingly, a significant number of specimens showed dishomogeneous distribution of PDGFRα on the epithelial compartment, suggesting that cellular phenotypes with different expression patterns for PDGFRα co-exist in the same gland, a scenario replicated by the human prostate cancer cell lines commonly available. For instance, we found that PDGFRα is detectable only in cells derived from skeletal metastases, such as the widely used PC3 cell line. In contrast, cells obtained from lymph node metastases (LNCaP) or brain metastases (DU-145) in prostate cancer patients fail to express either the alpha or beta isoforms of PDGFR[14].
PDGFRα Expression and Bone-Metastasis Potential
A more direct correlation between PDGFRα expression levels and the propensity of prostate cancer cells to colonize the skeleton was derived from experiments that were conducted using two sub-lines originally obtained from the PC3 parental population. Employing an in vitro invasion assay, Wang et al.[15] obtained two sub-lines of invasive and non-invasive PC3 cells. These cells were subsequently evaluated for their metastatic potential in immunocompromised SCID mice through tail vein inoculation. The invasive cells demonstrated bone-metastatic abilities and are currently named PC3-ML, whereas the non-invasive cells also failed to produce macroscopic bone tumors and are currently named PC3-N. We decided to complement these initial experiments using an animal model of metastasis combining fluorescence stereomicroscopy, histologic analysis and digital imaging. We employed prostate cancer cells engineered to stably express enhanced green fluorescent protein (eGFP). The resulting emitted fluorescence facilitated their identification both at the single-cell stage and when growing as metastatic foci of progressively larger size in the skeleton of the inoculated mice[13]. When inoculated into mice via an intracardiac route, PC3-N and DU-145 cells were unable to produce macroscopic tumors in the skeleton or any other organ examined, whereas PC3-ML cells produced macroscopic tumors at 4 to 5 weeks post inoculation. However, when earlier time-points were investigated, we established that PC3-N and DU-145 cell lines were both capable of spreading to the skeleton through the blood circulation as effectively as the metastatic counterpart PC3-ML cells[13]. However, PC3-N could generally survive no longer than one week in the bone marrow and only a small number of mice showed small skeletal metastases at three weeks post inoculation. DU-145 cells could survive only for the first 72 h after arriving into the bone and were never detected at one week post inoculation (Figure 1). Thus, the disparity in metastatic potential of these three malignant prostate phenotypes is not related to the extent of their dissemination to bone. Instead, it appears to depend on their ability to survive in the bone marrow, in which they lodge after extravasating from the blood circulation. Interestingly, we have established that PC3-ML cells express significantly higher levels of PDGFRα than do their non-metastatic counterpart, PC3-N cells. If combined with the complete lack of PDGFRα expression observed in DU-145 cells, these results establish a positive correlation between the expression levels of PDGFRα and the progression of malignant prostate phenotypes in the bone marrow, and indicate that the survival of disseminated cells in a foreign microenvironment plays a key role in the overall metastatic potential of a specific prostate cell phenotype.
Figure 1. Survival and progression at the skeletal level of prostate cancer cell types expressing different levels of PDGFRα.

The PC3-ML sub-line expressed higher levels of the receptor and produced macroscopic skeletal metastases in mice inoculated with cancer cells in the hematogenous circulation via the left cardiac ventricle injection. PC3-N cells expressed lower levels of PDGFRα than did PC3-ML cells and could only survive two weeks in the bone after their dissemination. DU-145 cells were found negative to PDGFRα expression and disappeared from the skeleton between 72 h and one week post inoculation.
In light of this established correlation between PDGFRα expression and bone-metastatic potential, we tested whether the exogenous over-expression of receptor in PC3-N and DU-145 cells could sustain initial bone colonization and promote metastatic growth of these two malignant phenotypes. We found that the over-expression of PDGFRα in PC3-N cells conferred a bone-metastatic potential indistinguishable from PC3-ML cells in terms of number and size of skeletal tumors detected four weeks after inoculation. Interestingly, the ability of DU-145 cells to produce bone-metastatic tumors was unaffected by the expression of PDGFRα[13], suggesting that the pre-existing genetic background of a malignant phenotype may ultimately dictate the pro-metastatic role exerted by PDGFRα in prostate cancer cells.
Further studies revealed an unorthodox mechanism by which PDGFRα recruits downstream signaling pathways in prostate cancer cells. The experiments that fully epitomize this atypical signaling by PDGFRα were conducted by over-expressing a truncated form of the receptor, named αRΔX, in PC3-N cells. The receptor mutant, obtained from Dr. Kazlauskas and collaborators, lacks the extracellular ligand-binding domain and is therefore unable to bind or be activated by proper PDGF ligand(s) or other signaling molecules[16]. When PC3 (αRΔX) cells were tested for their metastatic potential in our animal model, they fully emulated the bone-metastatic behavior of both PC3-ML cells and PC3-N cells that over-express the full-length form of the receptor[17].
Evidence for Transactivation of PDGFRα by Human Bone Marrow
PDGFRs are tyrosine kinases and some of the best-studied growth factor receptors. Two structurally related forms of the receptor are PDGFRα and PDGFRβ. Their extracellular portion contains five immunoglobulin-like domains whereas the intracellular part of the molecule contains a kinase domain[18]. Five PDGF ligands, PDGF-AA, -BB, -AB, -CC and -DD, have been identified, which display different binding affinities for the different receptors[19]. Since PDGF ligands are dimeric molecules, they bind two receptors simultaneously. Upon binding, the two receptors dimerize, triggering reciprocal phosphorylation at tyrosine residues located at specific sites on the intracellular portion of each receptor[20]. This transphosphorylation of PDGFR upon ligand binding serves two important purposes. The phosphorylation of a tyrosine residue in the kinase domain increases its catalytic efficiency. In addition, the phosphorylation of tyrosine residues outside of the kinase domain creates docking sites for signaling molecules. Some of these molecules can function as enzymes, such as phosphatidylinositol 3′-kinase (PI3K), phospholipase C (PLC), the Src family of tyrosine kinases, the tyrosine phosphatase SHP-2, and a GTPase activating protein (GAP) for Ras. Other molecules lack enzymatic activity and function as adaptors, such as Grb2, Nck, Shc and others[18]. The biological functions of some of these signaling molecules have been characterized and are fundamental for cellular homeostasis. PI3K activates the downstream kinase Akt, which is richly implicated in promoting cell survival[21].
We initially reported that PDGFRα, in addition to its proper PDGF ligands, could activate downstream signaling pathways, such as PI3K/AW, when exposed to the soluble fraction of human bone marrow[22]. Interestingly, the phosphorylation of Akt in PC3-ML cells exposed to bone marrow could be reduced to less than 40% by AG1296, a putative specific inhibitor of PDGFRs[23]. More conclusive evidence for a direct activation of PDGFRα was obtained from the detection of its tyrosine-phosphorylation upon the bone marrow treatment of PC3-ML cells[22].
Considering these results, we decided to measure the concentration of PDGF in the bone marrow aspirates and to identify the isoform(s) of this growth factor responsible for Akt activation in PC3-ML cells. We found that bone marrow aspirates obtained from different donors contained both PDGF-AA and PDGF-BB in concentrations ranging from 400 pg/mL to 2 ng/mL. Our experiments were conducted employing bone marrow diluted twenty fold, thus containing PDGF ligands reaching a maximum concentration of 100 pg/mL. When PC3-ML cells were simultaneously exposed to 100 pg/mL (each) of PDGF-BB and PDGF-AA, the observed activation of Akt was minimal, representing less than 10% of the observed response when exposing these cells to bone marrow. In addition, similar concentrations of PDGF ligands were unable to reproduce tyrosine phosphorylation of the intracellular portion of PDGFRα generated by human bone marrow[17]. Notably, the phosphorylation of PDGFRα induced by bone marrow was of a lesser magnitude than that generated by the exposure of cells to PDGF-AA. However, the extent of Akt phosphorylation observed under these two conditions was remarkably similar, implying that the phosphorylation caused by bone marrow must predominantly or exclusively affect tyrosine residues on PDGFRα which are responsible for the recruitment and activation of PI3K.
Collectively, these observations strongly suggest that PDGFRα, in addition to being stimulated by PDGF ligands in a widely recognized fashion[24],[25], can also be recruited and activated via ligand-independent mechanisms in a phenomenon termed trans-activation that has been reported for several receptors, including PDGFR[26]–[28]. Accordingly, upon activation by the soluble fraction of bone marrow, the canonical dimerization of PDGFRα could not be observed[17]. This leads to hypothesize that the stimulation of other plasma membrane receptors could successively trigger the phosphorylation and signaling of monomeric forms of PDGFRα via intracellular mediators. As a role for SRC family kinases (SFK) by intracellular reactive oxygen species (ROS) observed in other systems[16] could be excluded, the implication of other receptor-pathway combinations is likely.
The conclusive evidence for PDGFRα transactivation was obtained using either DU-145 or PC3-N cells stably expressing αRΔ X. Both cell types responded to human bone marrow with strong Akt activation and tyrosine phosphorylation of αRΔ X [17]. These results complement the in vivo studies and show that PC3-N acquired a bone-metastatic potential comparable to that of PC3-ML cells when stably transfected with either the full-length or the truncated form of PDGFRα.
The possibility that the establishment and progression of prostate cancer in the bone could be independently supported by PDGFRα of direct ligand stimulation may have important translational implications. It can be inferred that anti-cancer therapeutics designed to block the ligand-binding domain of PDGFRα may not fully prevent downstream signaling in cells that have spread to the bone marrow. Alternatively, inducing the internalization of PDGFRα may provide a mean to prevent ligand-dependent and -independent activation and provide a better therapeutic option to counteract the growth of prostate cancer cells disseminated to the skeleton.
Targeting PDGFRα to Block Its Downstream Signaling
PDGFRα and PDGFRβ are involved in organism development, with PDGFRα playing a greater role during embryogenesis[29]. In the adult, both receptors cooperate in modulating cellular and physiological processes that largely overlap, including angiogenesis, wound healing and tissue homeostasis[19],[29]. PDGFRβ, however, plays a predominant role overall, as demonstrated in mice in which the cytoplasmic domains between PDGFRα and PDGFRβ were swapped. These experiments revealed that the PDGFRβ intracellular domain could fully substitute for the PDGFRα. In contrast, replacement of the PDGFRβ cytoplasmic domain with that of the α-receptor caused abnormalities in vascular smooth muscle cell development and function[30]. The use of the small-molecule inhibitor STI571 (imatinib mesylate or gleevec) has been reported to block PDGFRs and reduce the expansion of cancer cells within the bone[31],[32]. However, the inhibitory and pro-apoptotic effects of STI571 seem to be exerted prevalently on PDGFRβ expressed in endothelial cells of the tumor vasculature rather than directly affecting prostate cancer cells. Alternatively, the toxicity reported in phases I and II clinical trials, which in most cases had to be interrupted[33],[34], may explain the ability of STI571 to comparably block PDGFRα and PDGFRβ. In addition, pre-clinical animal studies investigating the survival role of PDGFRs for cancer cells and the effects exerted by STI571 were almost exclusively conducted using animal models in which bone tumors were produced by directly implanting cancer via an intra-osseous route. While this approach significantly shortens the duration of each experiment, it also bypasses the initial stages of lodging and colonization of the bone marrow. Therefore, the peculiar histopathologic features produced by this intra-osseous approach, as compared to naturally established and progressing skeletal metastases, might also explain the disappointing effects of STI571 in clinical trials.
It seems plausible that the selective inactivation of PDGFRα, employing a monoclonal antibody rather than a broad-range inhibitor such as STI571, could limit the survival of malignant cells that depend on it while causing limited side effects, due to the largely duplicate role exerted by PDGFRβ[35]. However, in the event that PDGFRα in prostate cancer cells undergoes transactivation when in the bone marrow microenvironment, an antibody that would target the extracellular ligand-binding domain would fail to completely block signaling. Conversely, an antibody that could induce the internalization of PDGFRα would remove from the plasma membrane an important target for the transactivation of cancer cells exerted by the bone marrow. With this goal in mind, we decided to test IMC-3G3, a humanized monoclonal antibody against PDGFRα. This antibody has been extensively characterized both in vitro and in vivo and was shown to block both PDGF-AA and PDGF-BB from binding PDGFRα, with a Kd of 40 pmol/L. Also, the binding kinetic of IMC-3G3 to human PDGFRα was defined by BIAcore analysis as well as flow Cytometry employing human cells. A significant neutralizing activity of IMC-3G3 against PDGFRα was also observed in mitogenic and phosphorylation assays and this antibody inhibited subcutaneous xenografts in nude mice[36].
In experiments in which PC3-ML cells were exposed to bone marrow, IMC-3G3 was consistently able to reduce downstream Akt phosphorylation in a time-dependent manner. Interestingly, cell-surface biotinylation experiments showed that the inhibitory effect of IMC-3G3 on PDGFRα downstream signaling was tightly correlated to the internalization of this receptor. This event affected more than 80% of the initial levels of PDGFRα after two hours of IMC-3G3 incubation[22]. Furthermore, by using experimental conditions that halt receptor internalization while preserving IMC-3G3 neutralization of the ligand-binding domain of PDGFRα, we could block Akt phosphorylation by PDGF-AA but not by bone marrow[22].
Hence, evidence strongly suggests that IMC-3G3 could be effective in our pre-clinical model of bone metastases to counteract the survival and progression prostate cancer cells disseminated to the skeleton.
Targeting PDGFRα Effectively Counteracts Skeletal Metastases in Animal Models
We initially confirmed the species-specificity of IMC-3G3 in vitro, observing that the antibody blocked signaling by human PDGFRα while leaving the mouse form of the receptor unaffected[17]. Following, we used IMC-3G3 according to a prophylactic protocol, in which SCID mice were inoculated in the blood circulation with PC3-ML cells and received a first loading dose of IMC-3G3 followed by subsequent maintenance doses of the antibody bi-weekly, all administered by intraperitoneal injection. When animals were euthanized four weeks later, the number of bone-tumors per mice as well as the number of animals presenting with skeletal metastases in the IMC-3G3-treated groups were significantly lower than those in the saline-treated groups[13]. Similar results were obtained when animals were euthanized two weeks post inoculation, in which bone metastases were reduced by 70% as compared to control groups[13].
Successively, we employed a curative protocol in which mice were inoculated with PC3-ML cells and left untreated for either 7 or 14 days, thus providing a time interval for metastatic tumor growth. Following this first period, treatment with IMC-3G3 began as previously described and continued until the fourth week post inoculation. We found that the skeletal lesions in animals administered IMC-3G3 were significantly reduced in size than those in the control groups receiving either saline or human immunoglobulins of the IgG1 subclass as the IMC-3G3 antibody[37] (Figure 2).
Figure 2. The monoclonal antibody for human PDGFR inhibited skeletal tumor growth.

IMC-3G3 is effective in counteracting the progression of established skeletal metastases. After mice were inoculated with prostate cancer cells, treatment was withheld from mice for either one or two weeks, after which treatment of IMC-3G3 was maintained for the remainder of the experiment. When mice were euthanized at four weeks, their tibiae and femora showed a significant reduction in the average size of bone tumors as compared to controls[37]. This figure is reprinted with permision from Russell et al. [37], Clinical Cancer Research, 2010, 16 (2):5002–5010. Copyright© 2010 by American Association for Cancer Research.
Since it had been previously reported that stromal PDGFRα could support tumor growth and local angiogenesis when stimulated by locally produced PDGF ligands (i.e. PDGF-AA and PDGF-CC), we decided to investigate the contribution of PDGFRα expressed by stromal cells (osteoblasts and mesenchymal bone stromal cells) on the skeletal colonization and metastatic progression of PC3-ML cells. Thus, we used IMC-1E10, a humanized monoclonal antibody selected for binding to mouse PDGFRα and otherwise sharing an identical structure with IMC-3G31[38]. IMC-3G3, IMC-1E10, or a combination of the two antibodies was used to treat mice that had been inoculated with PC3-ML for 4 weeks. We found that the animals treated with IMC-1E10 showed no decrease in tumor size as compared to control, whereas mice treated with IMC-3G3 either alone or in combination with IMC-1E10 showed a significant reduction in the size of skeletal tumor foci[37].
The current standard of care for patients with advanced metastatic prostate cancer includes the administration of bisphosphonates[39]. These molecules are very effective inhibitors of bone-matrix degradation caused by osteoclasts located in skeletal metastatic lesions[40]. The resorption of mineralized bone and consequent mobilization of growth factors has been shown to support cancer cell growth and survival, while also causing significant pain to patients and increasing the risk for skeletal-related events (SREs) such as pathological fractures and spinal-cord compression[41]. Zoledronic acid (ZA) shows a potent analgesic effect that can significantly delay the time to SREs[42]. However, while bisphophonates are credited for a significant palliative role, a recent clinical trial in which ZA was compared to placebo in 422 advanced prostate cancer patients failed to show significant differences in disease progression, performance status and quality of life among the groups[43]. Similar results were provided by pre-clinical studies in which the progression of the bone metastatic disease from breast cancer cells was transiently delayed, and at later stages the total tumor burden per animal became equivalent to that in vehicle-treated animals[44],[45].
To understand whether the palliative effect exerted by bisphosphonates could be complemented by the anti-metastatic role of IMC-3G3, we investigated whether animals with metastatic bone lesions would respond to the combined administration of ZA and IMC-3G3 with an increase in overall survival. We found that the treatment with IMC-3G3 alone and more significantly in combination with ZA was able to extend survival[37] (Figure 3).
Figure 3. Survival curves for various groups of animals with prostate cancer.
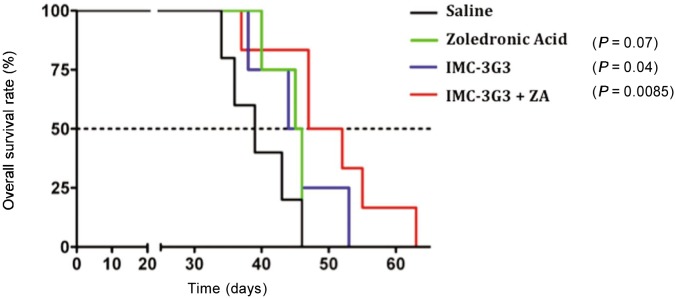
The Kaplan-Meier graphs show that targeting PDGFRα with IMC-3G3 induced a significant extension of overall survival in mice inoculated with prostate cancer cells, either alone or administered in combination with zoledronic acid (ZA). In contrast, ZA alone failed to prolong mean survival time[37]. This figure is reprinted with permision from Russell et al.[37], Clinical Cancer Research, 2010, 16(2):5002–5010. Copyright© 2010 by American Association for Cancer Research.
Conclusions
The series of studies presented here strongly support an important role of PDGFRα in facilitating the initial lodging and subsequent progression of prostate cancer cells in the bone microenvironment. In addition to an expected stimulation by PDGF ligands, the effect of PDGFRα is exerted through transactivation events initiated by activating signaling molecules present in the soluble fraction of human bone marrow. Importantly, the selective targeting of PDGFRα with monoclonal antibodies such as IMC-3G3, while still allowing PDGFRα to exercise its numerous physiological roles, can effectively counteract the growth of prostate cancer cells at the skeletal level. Finally, based on the positive results obtained with IMC-3G3 in combination with ZA in animal survival studies, a similar combination therapy approach could be envisioned in the clinic for prostate cancer patients.
Acknowledgments
We thank Drs. Olimpia Meucci and Mark E. Stearns (Drexel College of Medicine) and Dr. Andrius Kazlauskas (Harvard Medical School) for invaluable advices and discussion, and Drs. Nathan G. Dolloff, Michael R. Russell and Whitney L. Jamieson for their crucial contributions to our studies.
The work from our laboratory was supported by the W.W. Smith Charitable Trust and Department of Defense (CDMRP) grants W81XWH-09-1-0593 and W81XWH-09-1-0724.
References
- 1.Ross RW, Oh WK, Hurwitz M, et al. Neoplasm of the prostate [M] In: Kuff DW, Bast RC, Hait WN, et al., editors. Cancer Medicine. 7th ed. Hamilton, Ontario, Canada: BC Decker Inc; 2006. pp. 1431–1461. [Google Scholar]
- 2.Chambers AF, Groom AC, Macdonald IC. Metastasis: Dissemination and growth of cancer cells in metastatic sites [J] Nat Rev Cancer. 2002;2(8):563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 3.Mego M, Mani SA, Cristofanilli M. Molecular mechanisms of metastasis in breast cancer-clinical applications [J] Nat Rev Clin Oncol. 2010;7(12):693–701. doi: 10.1038/nrclinonc.2010.171. [DOI] [PubMed] [Google Scholar]
- 4.Fidler IJ. The pathogenesis of cancer metastasis: the “seed and soil” hypothesis revisited [J] Nat Rev Cancer. 2003;3(6):453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 5.Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective [J] Cancer Res. 2010;70(14):5649–5669. doi: 10.1158/0008-5472.CAN-10-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paget S. The distribution of secondary growths in cancer of the breast. 1889 [J] Cancer Metastasis Rev. 1989;8(2):98–101. [PubMed] [Google Scholar]
- 7.Luzzi KJ, MacDonald IC, Schmidt EE, et al. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases [J] Am J Pathol. 1998;153(3):865–873. doi: 10.1016/S0002-9440(10)65628-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chackal-Roy M, Niemeyer C, Moore M, et al. Stimulation of human prostatic carcinoma cell growth by factors present in human bone marrow [J] J Clin Invest. 1989;84(1):43–50. doi: 10.1172/JCI114167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keller ET, Zhang J, Cooper CR, et al. Prostate carcinoma skeletal metastases: cross-talk between tumor and bone [J] Cancer Metastasis Rev. 2001;20(3–4):333–349. doi: 10.1023/a:1015599831232. [DOI] [PubMed] [Google Scholar]
- 10.Mundy G. Metastasis to bone: causes, consequences and therapeutic opportunities [J] Nat Rev Cancer. 2002;2(8):584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 11.Fudge K, Bostwick DG, Stearns ME. Platelet-derived growth factor a and b chains and the and receptors in prostatic intraepithelial neoplasia [J] Prostate. 1996;29(5):282–286. doi: 10.1002/(SICI)1097-0045(199611)29:5<282::AID-PROS2>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 12.Chott A, Sun Z, Morganstem D, et al. Tyrosine kinases expressed in vivo by human prostate cancer bone marrow metastases and loss of the type 1 insulin-like growth factor receptor [J] Am J Pathol. 1999;155(4):1271–1279. doi: 10.1016/S0002-9440(10)65229-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Russell MR, Jamieson WL, Dolloff NG, et al. The alpha-receptor for platelet-derived growth factor as a target for antibody-mediated inhibition of skeletal metastases from prostate cancer cells [J] Oncogene. 2009;28(3):412–421. doi: 10.1038/onc.2008.390. [DOI] [PubMed] [Google Scholar]
- 14.Dolloff NG, Shulby SS, Nelson AV, et al. Bone-metastatic potential of human prostate cancer cells correlates with Akt/PKB activation by alpha platelet-derived growth factor receptor [J] Oncogene. 2005;24(45):6848–6854. doi: 10.1038/sj.onc.1208815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang M, Stearns ME. Isolation and characterization of PC-3 human prostatic tumor sublines which preferentially metastasize to select organs in S.C.I.D. mice [J] Differentiation. 1991;48(2):115–125. doi: 10.1111/j.1432-0436.1991.tb00250.x. [DOI] [PubMed] [Google Scholar]
- 16.Lei H, Velex G, Hovland P, et al. Growth factors outside the PDGF family drive experimental PVR [J] Invest Ophthalmol Vis Sci. 2009;50(7):3394–3403. doi: 10.1167/iovs.08-3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Russell MR, Liu Q, Lei H, et al. The alpha-receptor for platelet-derived growth factor confers bone-metastatic potential to prostate cancer cells by ligand- and dimerization-independent mechanisms [J] Cancer Res. 2010;70(10):4195–4203. doi: 10.1158/0008-5472.CAN-09-4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor [J] Physiol Rev. 1999;79(4):1283–1316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- 19.Hoch RV, Soriano P. Roles of PDGF in animal development [J] Development (Cambridge, England) 2003;130(20):4769–4784. doi: 10.1242/dev.00721. [DOI] [PubMed] [Google Scholar]
- 20.Bishayee S, Majundar S, Khire J, et al. Ligand-induced dimerization of the platelet-derived growth factor receptor. Monomer-dimer interconversion occurs independent of receptor phosphorylation [J] J Biol Chem. 1989;264(20):11699–11705. [PubMed] [Google Scholar]
- 21.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts [J] Genes Dev. 1999;13(22):2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 22.Dolloff N, Russell MR, Loizos N, et al. Human bone marrow activates the Akt pathway in metastatic prostate cells through transactivation of the alpha-platelet-derived growth factor receptor [J] Cancer Res. 2007;67(2):555–562. doi: 10.1158/0008-5472.CAN-06-2593. [DOI] [PubMed] [Google Scholar]
- 23.Rice AB, Moonaw CR, Morgan DL, et al. Specific inhibitors of platelet-derived growth factor or epidermal growth factor receptor tyrosine kinase reduce pulmonary fibrosis in rats [J] Am J Pathol. 2010;155(1):213–221. doi: 10.1016/S0002-9440(10)65115-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heldin CH. Structural and functional studies on platelet-derived growth factor [J] EMBO J. 1992;11(12):4251–4259. doi: 10.1002/j.1460-2075.1992.tb05523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heldin CH, Ostman A, R?nnstrand L. Signal transduction via platelet-derived growth factor receptors [J] Biochim Biophys Acta. 1998;1378(1):F79–F113. doi: 10.1016/s0304-419x(98)00015-8. [DOI] [PubMed] [Google Scholar]
- 26.Prenzel N, Zwick E, Daub H, et al. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF [J] Nature. 1999;402(6764):884–888. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- 27.Lei H, Kazlauskas A. Growth factors outside of the platelet-derived growth factor (PDGF) family employ reactive oxygen species/Src family kinases to activate PDGF receptor alpha and thereby promote proliferation and survival of cells [J] J Biol Chem. 2009;284(10):6329–6336. doi: 10.1074/jbc.M808426200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herrlich A, Daub H, Knebel A, et al. Ligand-independent activation of platelet-derived growth factor receptor is a necessary intermediate in lysophosphatidic, acid-stimulated mitogenic activity in L cells [J] Proc Natl Acad Sci USA. 1998;95(15):8985–8990. doi: 10.1073/pnas.95.15.8985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Betsholtz C. Insight into the physiological functions of PDGF through genetic studies in mice [J] Cytokine Growth Factor Rev. 2004;15(4):215–228. doi: 10.1016/j.cytogfr.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 30.Yarden Y, Escobedo JA, Kuang WJ, et al. Structure of the receptor for platelet-derived growth factor helps define a family of closely related growth factor receptors [J] Nature. 1986;323(6085):226–232. doi: 10.1038/323226a0. [DOI] [PubMed] [Google Scholar]
- 31.Uehara H, Kim SJ, karashima T, et al. Effects of blocking platelet-derived growth factor-receptor signaling in a mouse model of experimental prostate cancer bone metastases [J] J Natl Cancer Inst. 2003;95(6):458–470. doi: 10.1093/jnci/95.6.458. [DOI] [PubMed] [Google Scholar]
- 32.Kim SJ, Uehara H, Yazici S, et al. Targeting platelet-derived growth factor receptor on endothelial cells of multidrug-resistant prostate cancer [J] J Natl Cancer Inst. 2006;98(11):783–793. doi: 10.1093/jnci/djj211. [DOI] [PubMed] [Google Scholar]
- 33.Rao K, Goodin S, Levitt MJ, et al. A phase II trial of imatinib mesylate in patients with prostate specific antigen progression after local therapy for prostate cancer [J] Prostate. 2005;62(2):115–122. doi: 10.1002/pros.20130. [DOI] [PubMed] [Google Scholar]
- 34.Lin A, Rini BI, Weinberg V, et al. A phase II trial of imatinib mesylate in patients with biochemical relapse of prostate cancer after definitive local therapy [J] BJU Int. 2006;98(4):763–769. doi: 10.1111/j.1464-410X.2006.06396.x. [DOI] [PubMed] [Google Scholar]
- 35.Shah GD, Loizos N, Youssoufian H, et al. Rationale for the development of IMC-3G3, a fully human immunoglobulin G subclass 1 monoclonal antibody targeting the platelet-derived growth factor receptor α [J] Cancer. 2010;116(S4):1018–1026. doi: 10.1002/cncr.24788. [DOI] [PubMed] [Google Scholar]
- 36.Loizos N, Xu Y, Huber J, et al. Targeting the platelet-derived growth factor receptor alpha with a neutralizing human monoclonal antibody inhibits the growth of tumor xenografts: implications as a potential therapeutic target [J] Mol Cancer Ther. 2005;4(3):369–379. doi: 10.1158/1535-7163.MCT-04-0114. [DOI] [PubMed] [Google Scholar]
- 37.Russell MR, Liu Q, Fatatis A. Targeting the {alpha} receptor for platelet-derived growth factor as a primary or combination therapy in a preclinical model of prostate cancer skeletal metastasis [J] Clin Cancer Res. 2010;16(20):5002–5010. doi: 10.1158/1078-0432.CCR-10-1863. [DOI] [PubMed] [Google Scholar]
- 38.Shen J, Vil MD, Jimenez X, et al. Single variable domain-IgG fusion. A novel recombinant approach to Fc domain-containing bispecific antibodies [J] J Biol Chem. 2006;281(16):10706–10714. doi: 10.1074/jbc.M513415200. [DOI] [PubMed] [Google Scholar]
- 39.Saad F, Schulman CC. Role of bisphosphonates in prostate cancer [J] European Urology. 2004;45(1):26–34. doi: 10.1016/j.eururo.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 40.Bagi CM. Targeting of therapeutic agents to bone to treat metastatic cancer [J] Adv Drug Deliv Rev. 2005;57(7):995–1010. doi: 10.1016/j.addr.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 41.Coleman RE, Guise TA, Lipton A, et al. Advancing treatment for metastatic bone cancer: consensus recommendations from the Second Cambridge Conference [J] Clin Cancer Res. 2008;14(20):6387–6395. doi: 10.1158/1078-0432.CCR-08-1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Costa L, Major PP. Effect of bisphosphonates on pain and quality of life in patients with bone metastases [J] Nat Clin Pract Oncol. 2009;6(3):163–174. doi: 10.1038/ncponc1323. [DOI] [PubMed] [Google Scholar]
- 43.Saad F, Gleason DM, Murray R, et al. A randomized, placebo-controlled trial of zoledronic acid in patients with hormone-refractory metastatic prostate carcinoma [J] J Natl Cancer Inst. 2002;94(19):1458–1468. doi: 10.1093/jnci/94.19.1458. [DOI] [PubMed] [Google Scholar]
- 44.van der Pluijm G, Que I, Sijmons B, et al. Interference with the microenvironmental support impairs the de novo formation of bone metastases in vivo [J] Cancer Res. 2005;65(17):7682–7690. doi: 10.1158/0008-5472.CAN-04-4188. [DOI] [PubMed] [Google Scholar]
- 45.Thudi NK, Martin CK, Nadella MV, et al. Zoledronic acid decreased osteolysis but not bone metastasis in a nude mouse model of canine prostate cancer with mixed bone lesions [J] Prostate. 2008;68(10):1116–1125. doi: 10.1002/pros.20776. [DOI] [PMC free article] [PubMed] [Google Scholar]