

Abstract
Objective
We have previously found abrogated ischemia-induced coronary collateral growth in Zucker obese fatty rats (ZOF) compared to Zucker lean rats (ZLN). Because ZOF have structural abnormalities in their mitochondria suggesting dysfunction, and also show increased production of O2ׄ−, we hypothesized that mitochondrial dysfunction, caused by oxidative stress impairs coronary collateral growth in ZOF.
Methods and Results
Increased levels of ROS were observed in aortic endothelium and smooth muscle cells in ZOF compared to ZLN. ROS levels were decreased by the mitochondria-targeted antioxidants MitoQuinone (MQ) and MitoTempol (MT) as assessed by MitoSox Red and DHE staining. Lipid peroxides (a marker of oxidized lipids) were increased in ZOF by ∼47 % compared to ZLN. The elevation in oxidative stress was accompanied by increased antioxidant enzymes, except GPx-1, and by increased uncoupling protein-2 in ZOF vs ZLN. In addition, elevated respiration rates were also observed in the obese compared to leans. Administration of MQ significantly normalized the metabolic profiles and reduced lipid peroxides in ZOF to the same level observed in leans. The protective effect of MQ also suppressed the induction of UCP-2 in the obese rats. Resolution of mitochondrial oxidative stress by MQ or MT restored coronary collateral growth to the same magnitude observed in ZLN in response to repetitive ischemia.
Conclusions
We conclude that mitochondrial oxidative stress and dysfunction play a key role in disrupting coronary collateral growth in obesity and the metabolic syndrome, and elimination of the mitochondrial oxidative stress with MQ or MT rescues collateral growth.
Keywords: MitoQuinone, MitoTempol, metabolic syndrome, arteriogenesis, uncoupling protein-2, lipid peroxidation
Introduction
In prosperous societies, there is a rapid increase in the incidence of metabolic syndrome (MS), a condition characterized by abdominal obesity, hyperglycemia, insulin resistance and hyperinsulinemia. People with MS are particularly at increased risk for ischemic heart disease (IHD) and the deleterious consequences of myocardial infarction. Importantly, a well-developed coronary collateral circulation reduces infarct size and the incidence of sudden death in patients suffering from IHD.1
The myocardium, which has high energy requirements, relies on mitochondrial aerobic metabolism to maintain a high ATP/ADP ratio. A by-product of mitochondrial bioenergetic activity is the generation of reactive oxygen species (ROS), including the superoxide anion (O2ׄ-), the hydroxyl radical (HOׄ) and hydrogen peroxide (H2O2).2 Mitochondrial O2ׄ- is normally neutralized by superoxide dismutase 2 (SOD-2; also known as MnSOD or mitochondrial SOD), which is located within the mitochondrial matrix, and also by superoxide dismutase 1 (SOD-1; also known as Cu/ZnSOD or cytosolic SOD-1), which is found in the cytosol and also in the intermembrane space between the inner and outer mitochondrial membranes.3 Both SOD-1 and 2 dismutate O2ׄ- to H2O2 and O2. The H2O2 is then converted to O2 and H2O by the antioxidant enzymes glutathione peroxidase-1 (GPx-1), peroxiredoxins and catalase.4 However, if there is an imbalance between the mitochondrial pro-oxidant generation and anti-oxidant defenses, mitochondrial oxidative stress may ensue.
Due to the presence of large numbers of mitochondria and the relatively high oxygen tension, the myocardium is prone to oxidative damage from ROS produced by the mitochondrial electron transport chain (ETC). Chronic increases in mitochondrial ROS can lead to mitochondrial DNA fragmentation, functional decline, progression of myocardial remodeling and heart failure.5 Moreover, mice null for SOD-2 show damage to cardiac myocytes one week after birth and die of cardiomyopathy within three weeks of birth.6
In view of the above reports, and results showing that diabetes and metabolic syndrome are associated with mitochondrial abnormalities,7, 8 we hypothesized that mitochondrial oxidative stress leads to mitochondrial dysfunction, and compromises coronary collateral growth (CCG) in ZOF rats (a rat model of human metabolic syndrome). In this study, excessive lipid peroxidation was observed in ZOF rats as compared to their lean littermates, indicating oxidative stress. We further demonstrated induction of antioxidant enzymes (except GPx-1) and uncoupling proteins, as well as enhanced electron transfer activities in ZOF rats. Treatment of ZOF rats with MitoQuinone (MQ), a mitochondria–targeted antioxidant, normalized lipid peroxides, uncoupling protein-2 (UCP-2) and metabolic rate of the obese rats to levels comparable to the lean control animals. In addition, attenuation of mitochondrial and cytosolic ROS production was observed in aortic endothelium and smooth muscle cells in ZOF rats. The protective effect of MQ treatment partially restored CCG to levels comparable to control ZLN rats. Another mitochondria-targeted free radical scavenger, MitoTempol (MitoT), also restored collateral growth. Our data provide evidence linking mitochondrial oxidative stress and dysfunction to impaired coronary collateral growth in ZOF rats and indicate that mitochondria-targeted antioxidants may be a useful therapeutic strategy to decrease mitochondrial oxidative stress, improve bioenergetics, and restore collateral growth in the heart in this model of metabolic syndrome.
Materials and Methods
Animal model
Male Zucker lean (Fa/Fa or Fa/fa) (ZLN) and obese (fa/fa) (ZOF) rats, of 6 to 8-months-old, were obtained from the Charles River Laboratory Inc (Wilmington, MA) and housed under pathogen-free conditions. Rats that were homozygous for the fa gene (fa/fa) were obese and had metabolic syndrome, whereas those that were heterozygous (Fa/fa) or homozygous normal (Fa/Fa) were lean and metabolically normal. The lean rats provided a metabolically and pathophysiologically normal control for the obese fa/fa rats. The use of animals and experimental procedures were approved by the Institutional Animal Care and Use Committee at NEOMED (Rootstown, OH). The animal room was maintained on a reversed 12h: 12h light: dark cycle with temperature maintained at 25 °C.
MitoQuinone (MQ) and MitoTempol (MT) administration
ZOF rats were housed individually and were placed on normal chow diet. In brief, 50 μM MQ or 3 mM MT was given to the ZOF rats in drinking water for 10 to 14 days after the surgery. Water bottles, containing either MQ or MT, were covered with aluminium foil and all bottles were refilled every 3 to 4 days. Control ZOF and ZLN rats were supplied with water alone. All rats were monitored daily.9 Importantly the treatments did not affect weight gain in the obese rats (Supplement Figure 1).
Rat heart mitochondrial and cytosolic fractions isolation
For some molecular studies, crude mitochondria were prepared using mitochondrial extraction buffer (Mitosciences Inc., Eugene, OR). For bioenergetics studies, mitochondria were isolated according to the protocol by Miyamoto et al (2008) with slight modifications.10 Briefly, left ventricular tissues were minced, rinsed and homogenized in MSHE buffer containing 210 mM mannitol, 70 mM sucrose, 5 mM HEPES, 1 mM EGTA and 0.1 % fatty acid free BSA (pH7.2). The homogenate was centrifuged at 27000Xg for 10 min. The supernatant was collected as cytosolic fraction. The pellet was supplemented with nargarse protease (1 mg/g wet weight tissue) for 4 min on ice and rehomogenized. The homogenate was centrifuged at 400Xg for 10 min to remove nuclei and debris. The supernatant was poured through cheese cloth. The pellet obtained was subjected to the same homogenization and centrifugation processes, and the supernatant was poured through cheese cloth. The resulting supernatant was combined and centrifuged at 13,000Xg for 10 min. The mitochondria extract was finally resuspended in MSH buffer. Protein concentration was determined by BCA protein assay (Pierce, Rockford, IL).
Cytosolic lipid peroxidation assay
Thiobarbituric acid reactive substances (TBARS) assay was determined according to Buege and Aust (1978),11 Ohkawa et al (1979)12 and manufacturer's protocol (Zeptometrix Corp., Buffalo, NY) with modifications. In brief, 60 μl in volume of either MDA standards or 500 μg homogenate were added to 540 μl Thiobarbituric acid. The solution was heated at 95 °C for 1 hr, chilled on ice and centrifuged at 3000 rpm for 15 min. The supernatant was collected and absorbance was read at 532 nm in 200 μl duplicates.
Mitochondrial protein carbonyl groups detection
Excessive ROS production and/or defective anti-oxidant defense leads to oxidation of protein, lipids and DNA. Carbonyl (CO) groups (aldehydes and ketones) are produced on protein side chains, especially on Pro, Arg, Lys and Thr, when they are oxidized.13 To detect carbonylated proteins, carbonyl groups in protein side chains were derivatized with 2,4-dinitrophenylhydrazine (DNPH) to form the 2,4-dinitrophenylhydrazone (DNP) derivative (Oxyblot Protein Oxidation Detection Kit, Millipore, Billerica, MA). The lysates were then denatured with 12 % SDS, incubated with DNPH and mixed with neutralization solution and 5 % β-mercaptoethanol. The same protein samples also underwent the protein carbonyl detection procedure without the derivitization step as a negative control. The levels of carbonylated proteins were detected with anti-DNP antibody. Bands were visualized by Enhanced Chemiluminescence (ECL) (Pierce, Rockford, IL).
Immunoblot of mitochondrial and cytosolic proteins
Equal amounts of mitochondrial proteins from left ventricular tissue of ZLN and ZOF were estimated by the BCA protein assay (Pierce, Rockford, IL), electrophoresed on SDS-PAGE gel (Biorad, Hercules, CA) and transferred to a PVDF membrane (Millipore, Billerica, MS). Membranes were incubated with SOD-1, SOD-2 (Stressgen, Ann Arbor, MI), GPx-1, catalase, UCP-2, UCP-3, VEGF, Prd-3, and Prd-6 (Abcam, Cambridge, MA), porin (Mitosciences Inc., Eugene, OR) and GAPDH (Cell Signaling Technology, Danvers, MA) antibodies. Signals of the immunoreactive bands were visualized using ECL.
Mitochondrial function analysis
A XF24 extracellular flux analyzer (Seahorse Bioscience, North Billerica, MA) was used to measure intact isolated mitochondria function/bioenergetics according to protocol established by Rogers et al (2011)14 and Sauerbeck et al (2011)15 with modifications. First, a 24-well dual-analyte sensor cartridge was primed with calibration solution at 37 °C incubator without CO2 overnight. Prior to the start of experiment, the injection ports on the sensor cartridge were loaded with the appropriate mitochondrial substrates or inhibitors at 10X concentrations. The sensor cartridge was placed into the XF24 analyzer for automated calibration. During the calibration, isolated mitochondria were then seeded in XF24 V7 cell culture microplate (except for background correction well). Following centrifugation of the plate at 2000Xg for 20 min at 4 °C, MSHE with initial experiment conditions was gently added into the wells containing mitochondria and the plate was placed at 37 °C incubator without CO2 for 5 min. The plate was then transferred to the XF24 analyzer and the experiment initiated.
For electron flow assay, mitochondria (2.5 μg/well) were supplemented with electron flow initial media containing 10 mM pyruvate, 2 mM malate and 4 uM FCCP. Injections (10X concentration of either mitochondrial substrates or inhibitors) were made as follows: 20 μM rotenone, 100 mM succinate, 20 μM antimycin A, and 100 mM ascorbate + 1 mM TMPD, respectively. Typical mix, measure and mix cycles for the electron flow assay was 30 s, 3 min and 1 min, respectively.
All data were analyzed using the XF software and displayed as “point-to-point” oxygen consumption rates (pMole O2/min/well), or absolute oxygen tension (mmHg O2). Data were presented as the average of 3 to 5 wells/condition. For most measurements, either the highest or lowest point-to-point rates were taken. For oligomycin rate, an average of the last three point-to-point rates were taken since the effect of oligomycin requires at least 1 min to reach maximal effect and an average of the last 30 s of the measure provides the most consistent result15.
Rat model of coronary collateral growth
ZLN and ZOF rats, ∼ 6- to 8-months of age, were used for chronic (10 days) implantation of a pneumatic occluder over the left anterior descending coronary artery (LAD) as described by Toyota et al. (2005)16 to produce repetitive ischemia (RI). The RI protocol for rat consisted of eight 40-s occlusions, 1 every 20 min over 2 hr and 20 min followed by a period of ‘rest’ for 5 hr and 40 min. This 8-hr cycle was repeated three times per day over a period of 10 days.
MitoSox red and dihydroethidine (DHE) detection of mitochondrial and cytosolic ROS on vascular endothelial and smooth muscle cells ex-vivo
Vascular mitochondrial and cytosolic ROS productions were determined using MitoSox Red and DHE. MitoSox Red, a live cell permeant and derivative of hydroethidine, is rapidly and selectively targeted to mitochondria with positively charged triphenylphosphonium (TPP+). Once in the mitochondria, the reagent is intercalated into the mitochondrial DNA, oxidized by O2ׄ- and exhibited red fluorescence (Em∼580 nm). Freshly isolated aortic rings were incubated with MitoSox Red or DHE (Molecular Probes, Carlsbad, CA) in Krebs/HEPES buffer.17, 18 Fluorescent images were captured using an Olympus IX71 fluorescent microscope (Melville, NY) at 20× magnification and were further analyzed with NIH imaging software.
Collateral blood flow measurement
Flow to the collateral-dependent zone was measured by neutron-activated microspheres (2.5 × 106microspheres/ml) labeled with either Gold or Samarium (Biopal Inc., Worcester, MA).19 Microspheres were dispersed by agitation for 1 min and injected into the LV in 200 μl during LAD occlusion using a 25-G needle over 20 s. Gold microspheres were administered during surgery and Samarium microspheres were given at the time of harvesting. The normal (NZ) and collateral-dependent (CZ) zones were divided and total weight of each was measured. Radioactivity was measured following neutron bombardment and collateral blood flow was measured as a ratio of the cpm/g of the two regions, i.e., CZ/NZ flow ratio. Blood flow was also quantified by comparing the tissue counts per gram to the counts in an arterial blood sample obtained at 0.4 ml/min. In brief flow to the tissue is calculated as:
Statistical analysis
Data were normalized to the respective controls values and were expressed as mean ± SEM. Statistical analysis of data was performed by one-way ANOVA followed by post-hoc Bonferroni's test, as appropriate. p< 0.05 was considered statistically significant.
Results
Lipid peroxidation and protein carbonyls
Mitochondrial proteins were derivatized with DNPH and then monitored with DNP antibody for protein carbonyl content. As shown in Figure 1A (left and right panels), ZOF showed greater levels of mitochondrial protein carbonyl moieties (∼70%, p<0.05) compared to the lean controls, indicating that mitochondrial oxidative stress was higher in ZOF as compared to ZLN rats. Left ventricular oxidized lipid content is shown in Figure 1B. Basal TBARS level in ZOF rats was significantly elevated compared to ZLN (32.3 ± 1.2 vs 52.3 ± 6.0 nmoles/ml/mg protein). MQ treatment significantly reduced peroxidation of lipids in ZOF rats to the level similar to the lean (27.3 ± 0.9 nmoles/ml/mg protein; *p<0.05, n= 4 pairs).
Figure 1. Protein carbonyls and lipid peroxides contents.
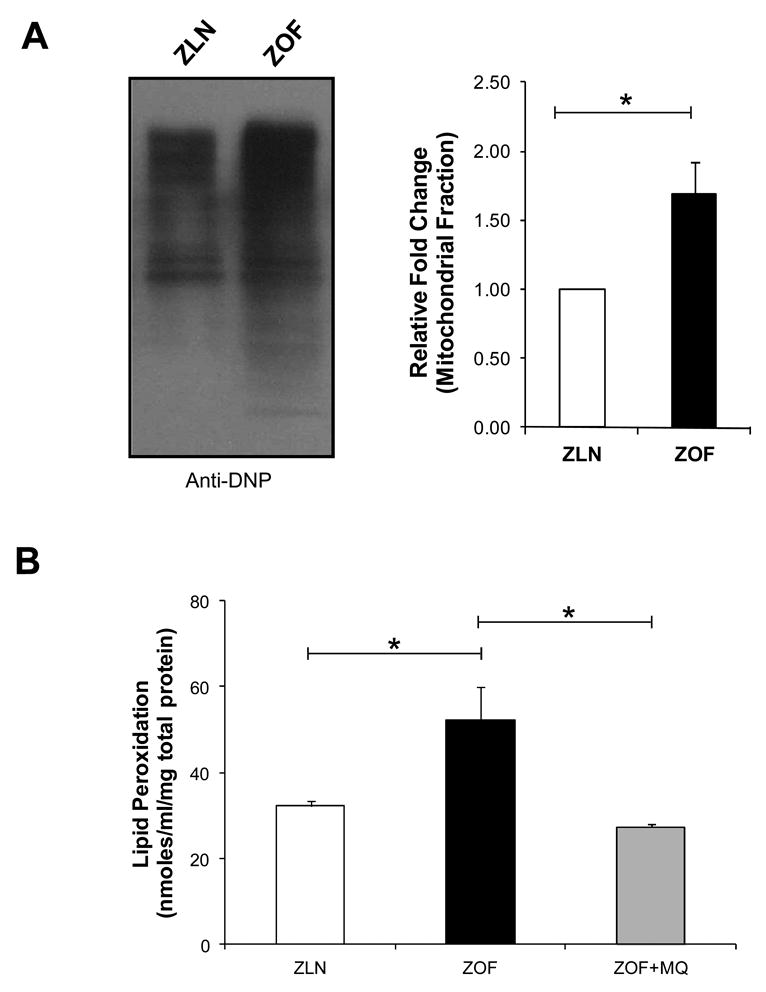
(A) Left--Immunoblot showing significantly higher total carbonyls content (total immunoreactive carbonyls) in ZOF compared to ZLN rats. Right—Carbonyl content was increase by ∼70 % in ZOF vs ZLN (*p<0.05). (B) Lipid peroxidation assay as an index of oxidative stress from cytosolic fraction from left ventricles. Higher lipid peroxidation content was observed in ZOF rats (∼47 vs ∼32 nmoles/ml/mg total protein of TBARS in ZOF vs ZLN; *p<0.05; n=4 pairs). MQ treatment significantly attenuated this effect in ZOF rats, indicating reduced oxidative stress in ZOF rats (*p<0.05; n=4 pairs).
MitoQuinone improved cytosolic and mitochondrial anti-oxidant defense in ZOF rats
Expression of cytosolic and mitochondrial anti-oxidant proteins from the left ventricles of Zucker rats were evaluated using immunoblots against SOD-1, SOD-2, GPx-1, catalase, Prd-3 and Prd-6. Superoxide dismutase-1, which is located in the cytosol and between the inner and outer-membranes of mitochondria, was significantly increased in ZOF (∼ 80 %; *p<0.05; n= 4 to 6 pairs) and ZOF+MQ (∼110 %; *p<0.05; n=4 to 6 pairs) as compared to the leans (Figure 2A). Expression level of SOD-2, which is located within the matrix, was also significantly higher in the ZOF (∼59 %; *p< 0.05; n=4 to 6 pairs) and ZOF+MQ (∼33 %; *p<0.05; n=4 to 6 pairs) as compared to the leans (Figure 2B). Interestingly, MQ did not improve GPx-1 expression, which was found to be low in the obese rats, suggesting a reduced catabolism of H2O2 by GPxs (∼50 % reduction in ZOF and ZOF+MQ vs ZLN; *p<0.05; n=4 to 6 pairs) (Figure 2C). In contrast, enhanced expression of catalase, Prd-3 and Prd-6, was found in ZOF (∼20 %, n=4 to 6 pairs) and ZOF+MQ (∼40 %, *p<0.05; n=4 to 6 pairs) compared to the leans (Figure 2D, E and F, respectively).
Figure 2. Expression of antioxidant proteins.
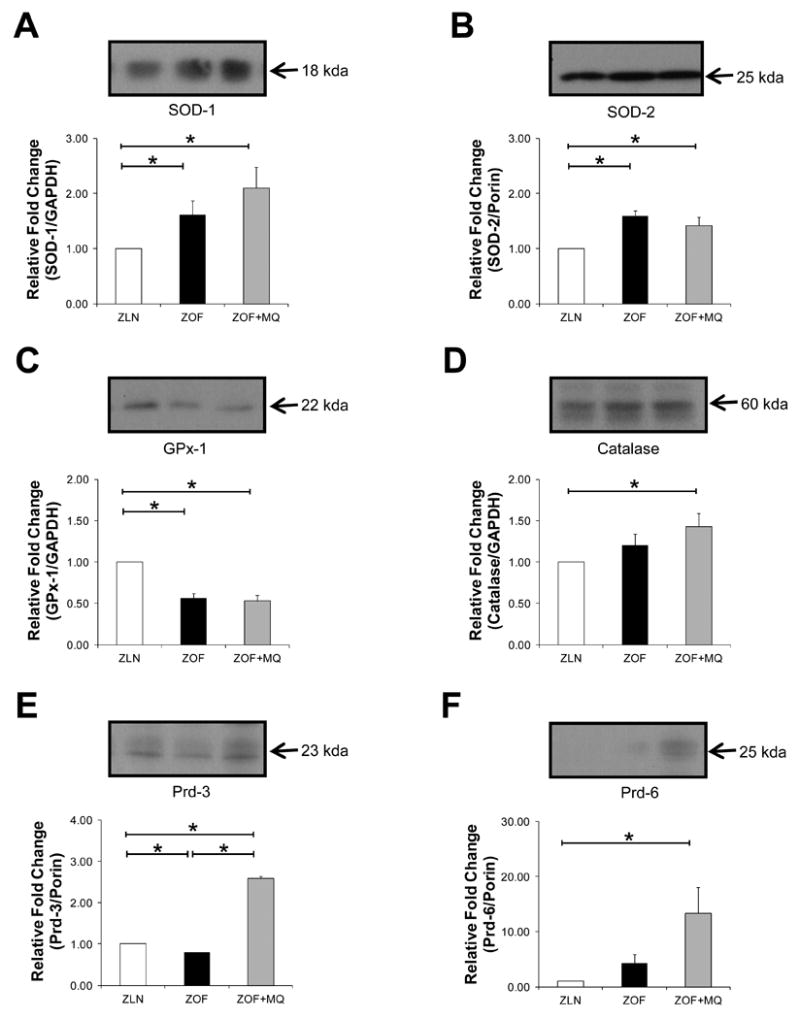
Immunoblots showing anti-oxidants profile from cytosolic and mitochondrial fractions from left ventricles. Cytosolic and mitochondrial anti-oxidant proteins were immunoblotted with SOD-1 (A), SOD-2 (B), GPx-1 (C), and catalase (D) antibodies with bar graph summarizing immunocaptured signals at the bottom panel, respectively. ZOF and ZOF+MQ rats showed significant higher SOD-1 (A) and SOD-2 (B) expression as compared to the lean (*p<0.05; n=4 to 6 pairs), indicating higher demands of SODs to scavenge excessive superoxide of the fat rats. MQ did not improve GPx-1 expression in the obese rats as compares to the lean (C) (*p<0.05; n=4 to 6 pairs). In contrast, catalase, Prd-3 and Prd-6 expressions appeared to be increased in the MQ-treated ZOF compared to the lean, suggesting a possible compensatory mechanism for catabolism of hydrogen peroxide due to lack of GPx-1 (D, E and F) (*p<0.05; n=4 to 6 pairs).
MitoQuinone improved mitochondrial function and bioenergetics profiles of ZOF rats
Figure 3 illustrates the respiration profiles of mitochondria isolated from Zucker rats. In the presence of FCCP as an uncoupler, mitochondria from the obese rats exhibited higher respiration rates with successive exposure to the following substrates and/or inhibitors: pyruvate+malate (∼ 39 %; complex I-dependent respiration; *p<0.05; n=6 pairs; 3 to 4 wells/condition) (A), rotenone+succinate (∼30 %, complex II-dependent respiration; n=6 pairs; 3 to 4 wells/condition) (B) and antimycin+TMPD+ascorbate (∼ 28 %; n=6 pairs; 3 to 4 wells/condition) (C) as compared to the lean. MitoQuinone treatment to the obese rats in vivo normalized the respiration rates of the subsequently isolated mitochondria to the level similar to those found in the lean.
Figure 3. Respiration profiles from isolated mitochondria.

In the presence of submaximal dose of FCCP, the obese rats exhibited higher respiration rates with successive exposure to the following substrates and/or inhibitors: pyruvate+malate (∼ 39 %; complex I-dependent respiration; *p<0.05; n=6 pairs; 3 to 4 wells/condition) (A), rotenone+succinate (∼30 %, complex II-dependent respiration; n=6 pairs; 3 to 4 wells/condition) (B) and antimycin+TMPD+ascorbate (∼ 28 %; n=6 pairs; 3 to 4 wells/condition) (C) as compared to the lean. MitoQuinone treatment to the obese normalized the respiration rates to the level similar to the lean.
MitoQuinone reduced the expression of uncoupling proteins in ZOF
To elucidate the mechanisms responsible for enhanced respiration rates in ZOF rat mitochondria and its reduction after MQ treatment, immunoblots on uncoupling proteins (UCPs) were performed (Figure 4). Uncoupling protein-2 expression was significantly higher in ZOF rats (∼37 %; ZOF vs ZLN; *p<0.05; n=4 pairs). UCP-2 and UCP-3 protein expressions in ZOF+MQ group were significantly lower as compared to the ZOF controls rats (∼ 31 % for UCP-2, *p<0.05; and 21 % for UCP-3, n=4 pairs).
Figure 4. Expression of uncoupling proteins.
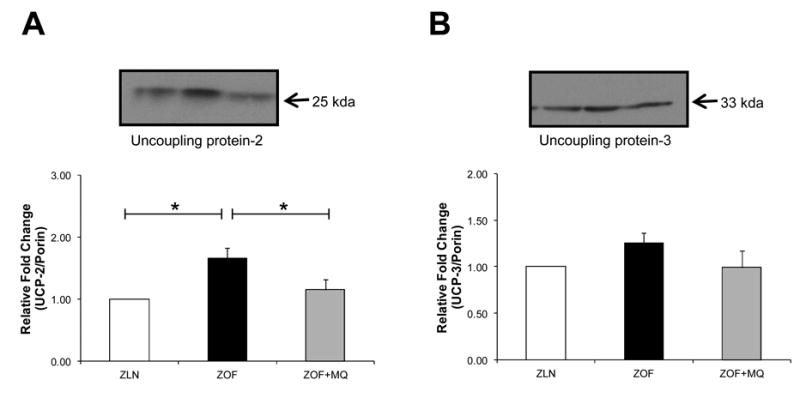
Mitochondrial uncoupling proteins were immunoblotted with UCP-2 (A), and UCP-3 (B) antibodies with bar graph summarizing immunocaptured signals at the bottom panel, respectively. Uncoupling protein-2 expression was significantly higher in ZOF rats (∼ 37 %; ZOF vs ZLN; *p<0.05; n=4 pairs) (A), and that MQ treatment to the obese rats lowered UCP-2 (∼31 % ZOF vs ZOF+MQ, *p<0.05; n=4 pairs) (A) and UCP-3 expressions (∼ 21 % ZOF vs ZOF+MQ; n=4 pairs) (B). Reduction in uncoupling proteins after MQ treatment appears to be in line with the respiration profiles, indicating that improvement in bioenergetics profile after MQ treatment appear to be regulated through reduction of oxidative stress and uncoupling proteins.
MitoQuinone and MitoTempol decreased mitochondrial and cytosolic ROS production in the aorta
Steady state mitochondrial and cytosolic O2ׄ- production in the aortic wall was significantly higher (>2-fold) in ZOF than in ZLN rats (Figure 5A and B for Mitosox Red ZLN vs ZOF, *p<0.05, n=3 pairs; Figure 5E and F for DHE ZLN vs ZOF, *p<0.05, n=3 pairs). Conversely, treatment with MQ and MT (50 μM and 3 mM, respectively) supplemented in their drinking water for ∼10 to 14 days significantly reduced mitochondrial and cytosolic ROS throughout the wall in ZOF to the similar level observed in the lean rats (Figure 5C and D compared to 5A for Mitosox red; Figure 5G and H compared to 5E for DHE). These findings suggest that mitochondria are one of the major sources of ROS in the obese rats.
Figure 5. MitoSox Red and DHE stainings for mitochondrial and cytosolic ROS from freshly isolated aorta.
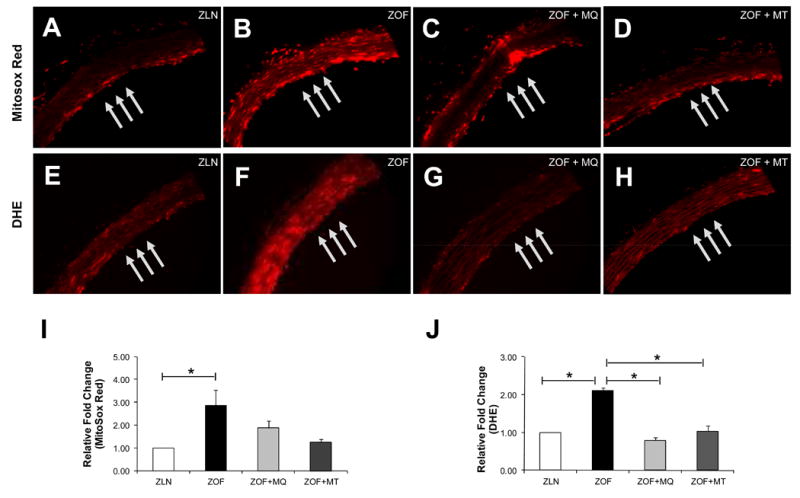
Representative fluorescent images showing mitochondrial and cytosolic ROS productions in aortic endothelium and smooth muscle cells from ZLN (A/E), ZOF (B/F), ZOF+MQ (C/G) and ZOF+MT (D/H). There were ∼ 2-fold increases in intensity for both the MitoSox Red (*p< 0.05) and DHE (*p<0.05) stainings for ZOF as compared to ZLN, indicating oxidative stress in the obese vessels. MQ and MT treatment (∼10 to 14 days) ameriolated mitochondrial and cytosolic ROS productions in ZOF to the level similar as observed in ZLN rats, suggesting that the mitochondria are one of the major sources of ROS and may be the major source, in the obese vessels. Bar graph summarizing MitoSox Red and DHE relative fluorescent intensities are shown in (I) and (J), respectively.
MitoQuinone and MitoTempol treatment induced coronary collateral growth in ZOF
Coronary collateral growth expressed in empirical units of flow (ml/min/g tissue) or as the ratio of perfusion between the collateral-dependent zone (CZ) and the normal zone (NZ) was shown in Figure 6. In ZLN, collateral flows increased from 0.23±0.02 to 0.84±0.02 ml/min/g (before and after RI), but in sham operated ZLN collateral flows did not change (0.23±0.03 to 0.24±0.02 ml/min/g) from the beginning to the end of the protocol. In ZOF rats, either sham (0.09±0.01 to 0.12±0.01 ml/min/g) or control ZOF exposed to RI (0.08±0.01 to 0.15±0.05 ml/min/g, collateral flows did not change. In ZOF rats treated with MQ or with MT, collateral flow increased from 0.08±0.03 to 0.72±0.01 ml/min/g and from 0.08±0.01 to 0.64±0.03 ml/min/g, respectively (Figure 6A). The CZ/NZ ratios also mimicked these changes with significant increases in the ratio occurring in ZLN exposed to RI (∼ 0.81), and in ZOF rats subjected to RI with MQ (∼0.72) or MitoT (∼0.64). In the sham groups or in ZOF rats subjected to RI, the CZ/NZ ratio did not significantly change (Figure 6B). Surprisingly, VEGF expression was significantly lower in the obese as compared to the lean, and that MQ treatment did not salvage this deficiency (Figure 6C, *p<0.05, n=4 pairs).
Figure 6.
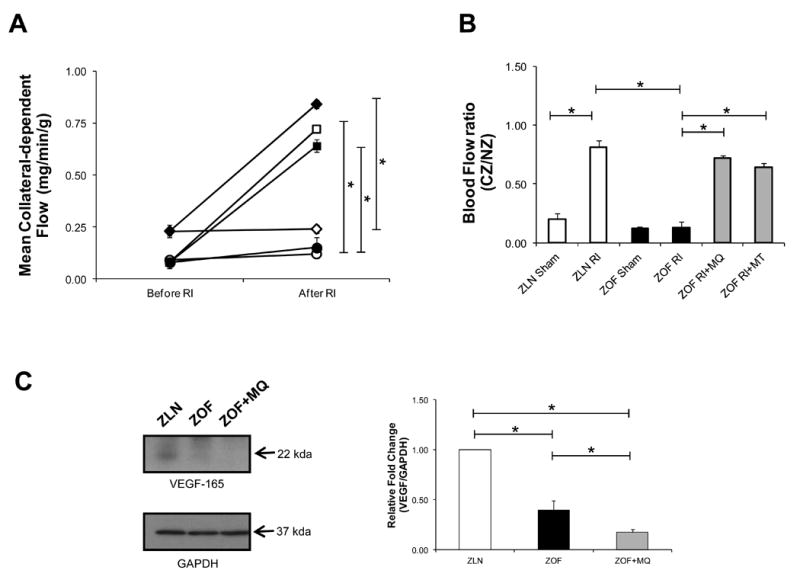
Collateral blood flow (A) and the ratio of flow between the collateral and normal zones (CZ/NZ, B) in the groups of ZLN and ZOF rats. In ZLN, collateral flows increased from 0.23±0.02 to 0.84±0.02 ml/min/g (before and after RI, closed diamond), but in sham ZLN collateral flows did not change (0.23±0.03 to 0.24±0.02 ml/min/g, opened diamond) from the beginning to the end of the protocol. In ZOF rats, either sham (0.09±0.01 to 0.12±0.01 ml/min/g, opened circle) or control ZOF exposed to RI (0.08±0.01 to 0.15±0.05 ml/min/g, closed circle) collateral flows did not change. In ZOF rats treated with MQ or with MT, collateral flow increased from 0.08±0.03 to 0.72±0.01 ml/min/g (opened square) and from 0.08±0.01 to 0.64±0.03 ml/min/g (closed square), respectively. The CZ/NZ ratios also mimicked these changes with a large increase in the ratio occurring in ZLN exposed to RI, and in the ZOF rats subjected to RI with MT or MQ. In the sham groups or in ZOF rats subjected to RI, the CZ/NZ ratio did not significantly change.
Discussion
Over the years, NADPH oxidase, xanthine oxidase and eNOS uncoupling are the three major sources of cytosolic ROS that have been studied extensively in cardiovascular research.4, 5,20 In line with the findings from other research groups,21, 22 our laboratory had previously demonstrated blunted endothelial function and cytosolic oxidative stress in ZOF rats,23,24 a rat model which presents many characteristics of metabolic syndrome, including obesity, insulin resistance, hyperlipidemia, hyperinsulinemia, hypertriglyceridemia and hyperphagia.25 We further reported compromised CCG of ZOF rats in response to episodic ischemia.26 When VEGF was transfected into smooth muscle cells and introduced into coronary circulation, we did not observe significant improvement in CCG in ZOF rats. However, correction of oxidative stress with ecSOD coupled with the same VEGF smooth muscle-based gene delivery system restored coronary collateral development partially in the obese rats. These results indicated that amelioration of oxidative stress may help restore redox-dependent signaling in CCG of ZOF rats26. To further decipher the role of redox-dependent signaling in mediating coronary collateral development, we hypothesized a role for mitochondrial dysfunction and oxidative stress in our rat model of MS. This hypothesis was in part based on literature showing mitochondrial dysfunction in the hearts of OVE26 mouse model of type I diabetes.27 Our hypothesis was also supported by unpublished observation from our own laboratory and others that ZOF rats were known to develop structural abnormalities of the mitochondria,28, 29 suggesting a possible role for oxidative stress. Thus we focused our studies on the mitochondria.
Our major observations from this study were that in ZOF rats compared to lean controls: (1) lipid peroxides were higher in the myocardium, (2) antioxidant enzymes and uncoupling proteins (UCP-2 in particular) were induced in the myocardium, (3) maximal respiration rates were elevated in the myocardium, and (4) both mitochondrial and cytosolic ROS were increased throughout the vascular wall suggesting an increase in ROS in both the endothelium and in smooth muscle. Administration of the mitochondrial-targeted antioxidant, MQ, ameliorated lipid peroxidation in the myocardium and ROS generation in the vascular cells. MitoQuinone also normalized the metabolic rates and UCP-2 protein to levels comparable to the lean animals. The improvement in bioenergetic profile of the ZOF rats by MQ restored of CCG in response to repetitive ischemia. Another mitochondrial directed antioxidant, MT, also reduced ROS levels throughout the vascular wall and restored CCG. Collectively, our data indicate that mitochondrial dysfunction and oxidative stress play key roles in the maladaptive response of the myocardium to ischemia, as shown by impaired coronary collateral growth.
In this study, we also characterized mitochondrial bioenergetics in lean and obese Zucker rats and evaluated the metabolic profile after treatment with MQ. During uncoupling with FCCP, mitochondria from obese rats had a significantly higher increase in oxygen consumption rate compared to those from leans. Chronic treatment with MQ prevented this increase. Moreover, MQ induced a protective effect in mitochondria from ZOF rats by normalizing the maximal respiration rates to the levels observed in the leans.
Metabolic syndrome and the pre-diabetic myocardium
Metabolic syndrome is a constellation of metabolic abnormalities that markedly increases the risk of developing ischemic heart disease (IHD), atherosclerosis, cardiomyopathy and type II diabetes.30 Development of MS is tightly associated with impaired mitochondrial function and increased mitochondrial oxidative stress.31-34 Mitochondrial O2ׄ- production occurs primarily at discrete points in the ETC at CI and CIII,35 and in components of the tricarboxylic acid (TCA) cycle, including α–ketoglutarate dehydrogenase.36 Mitochondrial-derived H2O2 diffuses to the cytoplasm and ultimately leads to oxidation of cytoplasmic proteins, and vice versa. Chronic exposures of ROS result in oxidative damage to DNA, proteins, and lipids, and inactivation of TCA cycle aconitase and the ETC at CI, CII and CIII.37, 38
In this study, we observed enhanced lipid peroxidation in ZOF rats as compared to the lean. One possible explanation could be due to excessive leak of electrons, which are passing down the respiratory chain prior to the reduction of oxygen to water at cytochrome c oxidase. These released electrons form superoxide with molecular oxygen. Superoxide and lipid peroxidation products, such as hydroxynonenal, activate UCPs. Activation of UCPs, UCP-2 in particular, have been proposed to act as a negative feedback loop to induce mild uncoupling that subsequently lowers ROS production.39 This suggestion is further supported by the observation of decreased lipid peroxidation, as well as attenuated levels of UCP-2 in the obese on MQ treatment as compared with ZOF rats. However, whether mitochondrial electron or proton leak is the cause or effect of impaired mitochondrial function in metabolic syndrome or vice versa, remain elusive.
MitoQuinone as a potential therapy for diseases related to mitochondrial oxidative stress and dysfunction
MQ and MT are promising anti-oxidants that have been successfully targeted to mitochondria.40, 41 Of these two compounds, MQ has been used more widely to treat a variety of conditions associated with oxidative stress40. It contains a covalently attached lipophilic triphenylphosphonium cation (TPP+) that, due to the large negative potential (∼150 to 180 mV) across the inner mitochondrial membrane, leads to MQ being concentrated several hundred-fold within mitochondria. Once adsorbed to the matrix face of the mitochondrial inner membrane, MQ accepts electrons from CII, to form the quinol reduction product, which is an effective antioxidant. It should be noted that the ubiquinone/ubiquinol pool exists mainly in the reduced ubiquinol form in vivo, and ubiquinol has been reported to function as an anti-oxidant by donating a hydrogen atom to a lipid peroxyl radical, thereby decreasing lipid peroxidation within the mitochondrial inner membrane.9, 42, 43 The ubiquinol radical formed in this process rapidly disproprotionates and ETC subsequently recycles ubiquinone back to ubiquinol, restoring the anti-oxidant function.
The effects of long-term ad libitum oral administration of 500 μM MQ on the behavior, metabolism, gene expression and accumulation of oxidative damage markers of young C57BL/6 mice has been investigated and shown to have no effect on redox cycling or cause uncoupling in vivo9. In addition, MQ has been used in a wide range of animal studies where it has shown beneficial effects. These include cardiac ischemia/reperfusion injury,44, 45 damage to endothelial cells in vivo associated with chronic exposures to nitroglycerin,46 increase in blood pressure in a spontaneously hypertensive rat model in which the increase in blood pressure is thought to arise from elevated mitochondrial oxidative damage in endothelial cells,47 sepsis,48, 49 and against heart damage associated with the anti-cancer compound Adriamycin.50
As mentioned previously, our former work using smooth muscle cells to deliver ecSOD and VEGF had modest effects on increasing coronary collateral flows in ZOF rats.26 However, a striking result of the present study was the nearly complete restoration of coronary collateral flow in the ZOF rats by mitochondrial directed antioxidants- MQ and MT. Thus, it seems that mitochondrial oxidative stress plays a key role in abrogating CCG in response to ischemia. Our connection between mitochondrial oxidative stress and the abrogation of collateral growth is somewhat tenuous, but we believe that altered mitochondrial bioenergetics and decreases in high energy phosphates play a critical role. Therefore, it may be possible that mitochondria-targeted antioxidants may be worth exploring as therapies in patients with this disorder. In this regard, it is useful to note that MQ has undergone two human clinical trials, one in Parkinson's disease and another in protecting against liver inflammation.51, 52 These trials showed that MQ could be given safely to patients for up to a year and that it was effective in decreasing liver damage.
Conclusion
There is increasing evidence to support the concept that mitochondria play a critical role in IHD and various cardiovascular diseases. However, the question of whether abnormalities leading to mitochondrial oxidative stress and dysfunction are the cause or response to various cardiovascular diseases is far from resolved. A better understanding of the redox-dependent mitochondrial signal transduction pathways, availability of pharmacological agents that can alter the production and scavenging mitochondrial ROS and various animal models with impaired mitochondrial function would aid in resolving the question. In the context of our study, we conclude that mitochondrial dysfunction, induced by oxidative stress due to accelerated electron or proton leak, abrogates the process of coronary collateral growth in a rat model of obesity and insulin resistance, and that resolution of the oxidative burden rescues this impaired phenotype as illustrated in Figure 8.
Supplementary Material
Figure 7. Proposed mechanism for increased coronary collateral development in the obese and the pre-diabetic myocardium.

In the insulin resistant animals augmented fatty acid oxidation would increase the delivery of reducing equivalents to the electron transport chain, leading to excessive superoxide production. The enhanced production of superoxide and lipid peroxides induce expression of uncoupling protein-2. Increased levels of uncoupling protein-2 lead to enhanced oxygen consumption, without an increment or even a reduction in the ATP synthesis. Reduced ATP synthesis and levels potentially contribute to compromised arteriogenesis/angiogenesis in response to repeated ischemic insults insofar as reduced energy levels would prevent a cell from undergoing a phenotypic switch which requires energy. Scavenging of mitochondrial-derived free radicals (MQ and MT treatment) would normalize the electron transfer activities in the obese rats to the level in leans, leading to reduced lipid peroxides. Reduction in lipid peroxides, which is the known agonist for uncoupling proteins, appears to improve bioenergetics profile in the obese myocardium. Improvement in bioenergetics is essential in mediating the phenotypic switch of vascular cells during arteriogenesis/angiogenesis.
Acknowledgments
This work was supported by NIH grants HL32788, HL83366 and RC1HL100828 (to WMC); and by an AHA post-doctoral fellowship 09POST2290021 (to YFP).
Abbreviations
- ANT
adenine nucleotide translocase
- CCG
coronary collateral growth
- CZ
collateral-dependent zone
- DNP
2,4-dinitrophenylhydrazone
- DNPH
2,4-dinitrophenylhydrazine
- ecSOD
extracellular superoxide dismutase
- ETC
electron transport chain
- GPx-1
glutathione peroxidase 1
- H2O2
hydrogen peroxide
- IHD
ischemic heart disease
- LAD
left anterior descending artery
- MQ
MitoQuinone
- MT
MitoTempol
- MS
metabolic syndrome
- NZ
normal zone
- O2ׄ-
superoxide
- Prd-3
peroxiredoxin-3
- Prd-6
peroxiredoxin-6
- RI
repetitive ischemia
- ROS
reactive oxygen species
- SOD-1
superoxide dismutase-1 or CuZnSOD
- SOD-2
superoxide dismutase-2 or MnSOD
- TBARS
Thiobarbituric acid reactive substances
- TCA
tricarboxylic acid cycle
- TPP+
triphenylphosphonium
- UCPs
uncoupling proteins
- VDAC
voltage-dependent anion channel
- VEGF
vascular endothelial growth factor
- ZLN
Zucker lean rat
- ZOF
Zucker obese fatty rat
References
- 1.Nathoe HM, Koerselman J, Buskens E, van Dijk D, Stella PR, Plokker TH, Doevendans PA, Grobbee DE, de Jaegere PP. Determinants and prognostic significance of collaterals in patients undergoing coronary revascularization. Am J Cardiol. 2006;98:31–35. doi: 10.1016/j.amjcard.2006.01.050. [DOI] [PubMed] [Google Scholar]
- 2.Raha S, Robinson BH. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem Sci. 2000;25:502–508. doi: 10.1016/s0968-0004(00)01674-1. [DOI] [PubMed] [Google Scholar]
- 3.Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (sod) in rat liver: Cu,zn-sod in mitochondria. J Biol Chem. 2001;276 doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- 4.Pung YF, Chilian WM. Corruption of coronary collateral growth in metabolic syndrome: Role of oxidative stress. World J Cardiol. 2010;2:421–427. doi: 10.4330/wjc.v2.i12.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsutsui H. Oxidative stress in heart failure: The role of mitochondria. Intern Med. 2001;40:1177–1182. doi: 10.2169/internalmedicine.40.1177. [DOI] [PubMed] [Google Scholar]
- 6.Lebovitz RM, Zhang H, Vogel H, Cartwright JJ, Dionne L, Lu N, Huang S, Matzuk MM. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci U S A. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duncan JG, Fong JL, Medeiros DM, Finck BN, Kelly DP. Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-alpha/pgc-1alpha gene regulatory pathway. Circulation. 2007;115:909–917. doi: 10.1161/CIRCULATIONAHA.106.662296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han X, Yang J, Cheng H, Yang K, Abendschein DR, Gross RW. Shotgun lipidomics identifies cardiolipin depletion in diabetic myocardium linking altered substrate utilization with mitochondrial dysfunction. Biochemistry. 2005;44:16684–16694. doi: 10.1021/bi051908a. [DOI] [PubMed] [Google Scholar]
- 9.Rodriguez-Cuenca S, Cocheme HM, Logan A, Abakumova I, Prime TA, Rose C, Vidal-Puig A, Smith AC, Rubinsztein DC, Fearnley IM, Jones BA, Pope S, Heales SJ, Lam BY, Neogi SG, McFarlane I, James AM, Smith RA, Murphy MP. Consequences of long-term oral administration of the mitochondria-targeted antioxidant mitoq to wild-type mice. Free Radic Biol Med. 2010;48:161–172. doi: 10.1016/j.freeradbiomed.2009.10.039. [DOI] [PubMed] [Google Scholar]
- 10.Miyamoto S, Murphy AN, Brown JH. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-ii. Cell Death Differ. 2008;15:521–529. doi: 10.1038/sj.cdd.4402285. [DOI] [PubMed] [Google Scholar]
- 11.Buege JA, Aust SD. Microsomal lipid peroxidation. Methods Enzymol. 1978;52:302–310. doi: 10.1016/s0076-6879(78)52032-6. [DOI] [PubMed] [Google Scholar]
- 12.Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- 13.Madian AG, Regnier FE. Proteomic identification of carbonylated proteins and their oxidation sites. J Proteome Res. 2010;9:3766–3780. doi: 10.1021/pr1002609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rogers GW, Brand MD, Petrosyan S, Ashok D, Elorza AA, Ferrick DA, Murphy AN. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One. 2011;6:e21746. doi: 10.1371/journal.pone.0021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sauerbeck A, Pandya J, Singh I, Bittman K, Readnower R, Bing G, Sullivan P. Analysis of regional brain mitochondrial bioenergetics and susceptibility to mitochondrial inhibition utilizing a microplate based system. J Neurosci Methods. 2011;198:36–43. doi: 10.1016/j.jneumeth.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toyota E, Warltier DC, Brock T, Ritman E, Kolz C, O'Malley P, Rocic P, Focardi M, Chilian WM. Vascular endothelial growth factor is required for coronary collateral growth in the rat. Circulation. 2005;112:2108–2113. doi: 10.1161/CIRCULATIONAHA.104.526954. [DOI] [PubMed] [Google Scholar]
- 17.Csiszar A, Labinskyy N, Zhao X, Hu F, Serpillon S, Huang Z, Ballabh P, Levy RJ, Hintze TH, Wolin MS, Austad SN, Podlutsky A, Ungvari Z. Vascular superoxide and hydrogen peroxide production and oxidative stress resistance in two closely related rodent species with disparate longevity. Aging Cell. 2007;6:783–797. doi: 10.1111/j.1474-9726.2007.00339.x. [DOI] [PubMed] [Google Scholar]
- 18.Ungvari Z, Labinskyy N, Mukhopadhyay P, Pinto JT, Bagi Z, Ballabh P, Zhang C, Pacher P, Csiszar A. Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am J Physiol Heart Circ Physiol. 2009;297:H1876–1881. doi: 10.1152/ajpheart.00375.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carrão AC, Chilian WM, Yun J, Kolz C, Rocic P, Lehmann K, van den Wijngaard JP, van Horssen P, Spaan JA, Ohanyan V, Pung YF, Buschmann I. Stimulation of coronary collateral growth by granulocyte stimulating factor: Role of reactive oxygen species. Arterioscler Thromb Vasc Biol. 2009;29:1817–1822. doi: 10.1161/ATVBAHA.109.186445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 21.Zhou X, Ma L, Habibi J, Whaley-Connell A, Hayden MR, Tilmon RD, Brown AN, Kim JA, Demarco VG, Sowers JR. Nebivolol improves diastolic dysfunction and myocardial remodeling through reductions in oxidative stress in the zucker obese rat. Hypertension. 2010 doi: 10.1161/HYPERTENSIONAHA.109.145136. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Serpillon S, Floyd BC, Gupte RS, George S, Kozicky M, Neito V, Recchia F, Stanley W, Wolin MS, Gupte SA. Superoxide production by nad(p)h oxidase and mitochondria is increased in genetically obese and hyperglycemic rat heart and aorta before the development of cardiac dysfunction. The role of glucose-6-phosphate dehydrogenase-derived nadph. Am J Physiol Heart Circ Physiol. 2009;297:H153–162. doi: 10.1152/ajpheart.01142.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Picchi A, Gao X, Belmadani S, Potter BJ, Focardi M, Chilian WM, Zhang C. Tumor necrosis factor-alpha induces endothelial dysfunction in the prediabetic metabolic syndrome. Circ Res. 2006;99:69–77. doi: 10.1161/01.RES.0000229685.37402.80. [DOI] [PubMed] [Google Scholar]
- 24.Gao X, Xu X, Belmadani S, Park Y, Tang Z, Feldman AM, Chilian WM, Zhang C. Tnf-alpha contributes to endothelial dysfunction by upregulating arginase in ischemia/reperfusion injury. Arterioscler Thromb Vasc Biol. 2007;27:1269–1275. doi: 10.1161/ATVBAHA.107.142521. [DOI] [PubMed] [Google Scholar]
- 25.Bray GA. The zucker-fatty rat: A review. Fed Proc. 1977;36:148–153. [PubMed] [Google Scholar]
- 26.Hattan N, Chilian WM, Park F, Rocic P. Restoration of coronary collateral growth in the zucker obese rat: Impact of vegf and ecsod. Basic Res Cardiol. 2007;102:217–223. doi: 10.1007/s00395-007-0646-3. [DOI] [PubMed] [Google Scholar]
- 27.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED. Mitochondrial energetics in the heart in obesity-related diabetes: Direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–2466. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- 28.Holloway GP, Snook LA, Harris RJ, Glatz JF, Luiken JJ, Bonen A. In obese zucker rats, lipids accumulate in the heart despite normal mitochondrial content, morphology and long-chain fatty acid oxidation. J Physiol. 2011;589:169–180. doi: 10.1113/jphysiol.2010.198663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holloway GP, Gurd BJ, Snook LA, Lally J, Bonen A. Compensatory increases in nuclear pgc1alpha protein are primarily associated with subsarcolemmal mitochondrial adaptations in zdf rats. Diabetes. 2010;59:819–828. doi: 10.2337/db09-1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bugger H, Abel ED. Molecular mechanisms for myocardial mitochondrial dysfunction in the metabolic syndrome. Clin Sci (Lond) 2008;114:195–210. doi: 10.1042/CS20070166. [DOI] [PubMed] [Google Scholar]
- 31.Hu F, Liu F. Mitochondrial stress: A bridge between mitochondrial dysfunction and metabolic diseases? Cell Signal. 2011;23:1528–1533. doi: 10.1016/j.cellsig.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Finocchietto PV, Holod S, Barreyro F, Peralta JG, Alippe Y, Giovambattista A, Carreras MC, Poderoso JJ. Defective leptin-amp-dependent kinase pathway induces nitric oxide release and contributes to mitochondrial dysfunction and obesity in ob/ob mice. Antioxid Redox Signal. 2011 doi: 10.1089/ars.2010.3857. [DOI] [PubMed] [Google Scholar]
- 33.Ren J, Pulakat L, Whaley-Connell A, Sowers JR. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J Mol Med (Berl) 2010;88:993–1001. doi: 10.1007/s00109-010-0663-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mercer JR, Cheng KK, Figg N, Gorenne I, Mahmoudi M, Griffin J, Vidal-Puig A, Logan A, Murphy MP, Bennett M. DNA damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome. Circ Res. 2010;107:1021–1031. doi: 10.1161/CIRCRESAHA.110.218966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muller FL, Liu Y, Van Remmen H. Complex iii releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem. 2004;279:49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 36.Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reddy PH, Beal MF. Are mitochondria critical in the pathogenesis of alzheimer's disease? Brain Res Brain Res Rev. 2005;49:618–632. doi: 10.1016/j.brainresrev.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 38.Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 39.Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD. Mitochondrial proton and electron leaks. Essays Biochem. 2010;47:53–67. doi: 10.1042/bse0470053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 41.Kelso GF, Porteous CM, Coulter CV, Hughes G, Porteous WK, Ledgerwood EC, Smith RA, Murphy MP. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J Biol Chem. 2001;276:4588–4596. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- 42.Ernster L, Forsmark P, Nordenbrand K. The mode of action of lipid-soluble antioxidants in biological membranes: Relationship between the effects of ubiquinol and vitamin e as inhibitors of lipid peroxidation in submitochondrial particles. Biofactors. 1992;3:241–248. [PubMed] [Google Scholar]
- 43.Maroz A, Anderson RF, Smith RA, Murphy MP. Reactivity of ubiquinone and ubiquinol with superoxide and the hydroperoxyl radical: Implications for in vivo antioxidant activity. Free Radic Biol Med. 2009;46:105–109. doi: 10.1016/j.freeradbiomed.2008.09.033. [DOI] [PubMed] [Google Scholar]
- 44.Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. Faseb J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 45.Neuzil J, Widen C, Gellert N, Swettenham E, Zobalova R, Dong LF, Wang XF, Lidebjer C, Dalen H, Headrick JP, Witting PK. Mitochondria transmit apoptosis signalling in cardiomyocyte-like cells and isolated hearts exposed to experimental ischemia-reperfusion injury. Redox Rep. 2007;12:148–162. doi: 10.1179/135100007X200227. [DOI] [PubMed] [Google Scholar]
- 46.Esplugues JV, Rocha M, Nunez C, Bosca I, Ibiza S, Herance JR, Ortega A, Serrador JM, D'Ocon P, Victor VM. Complex i dysfunction and tolerance to nitroglycerin: An approach based on mitochondrial-targeted antioxidants. Circ Res. 2006;99:1067–1075. doi: 10.1161/01.RES.0000250430.62775.99. [DOI] [PubMed] [Google Scholar]
- 47.Graham D, Huynh NN, Hamilton CA, Beattie E, Smith RA, Cocheme HM, Murphy MP, Dominiczak AF. Mitochondria-targeted antioxidant mitoq10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension. 2009;54:322–328. doi: 10.1161/HYPERTENSIONAHA.109.130351. [DOI] [PubMed] [Google Scholar]
- 48.Supinski GS, Murphy MP, Callahan LA. Mitoq administration prevents endotoxin-induced cardiac dysfunction. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1095–1102. doi: 10.1152/ajpregu.90902.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lowes DA, Thottakam BM, Webster NR, Murphy MP, Galley HF. The mitochondria-targeted antioxidant mitoq protects against organ damage in a lipopolysaccharide-peptidoglycan model of sepsis. Free Radic Biol Med. 2008;45:1559–1565. doi: 10.1016/j.freeradbiomed.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 50.Chandran K, Aggarwal D, Migrino RQ, Joseph J, McAllister D, Konorev EA, Antholine WE, Zielonka J, Srinivasan S, Avadhani NG, Kalyanaraman B. Doxorubicin inactivates myocardial cytochrome c oxidase in rats: Cardioprotection by mito-q. Biophys J. 2009;96:1388–1398. doi: 10.1016/j.bpj.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O'Sullivan JD, Fung V, Smith RA, Murphy MP, Taylor KM. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant mitoq as a disease-modifying therapy in parkinson's disease. Mov Disord. 2010;25:1670–1674. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- 52.Gane EJ, Weilert F, Orr DW, Keogh GF, Gibson M, Lockhart MM, Frampton CM, Taylor KM, Smith RA, Murphy MP. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase ii study of hepatitis c patients. Liver Int. 2010;30:1019–1026. doi: 10.1111/j.1478-3231.2010.02250.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.