

Abstract
Partitioning of specific proteins between soluble and insoluble forms because of aggregation, membrane attachment, and (or) association with senile plaques and neurofibrillary tangles is a major feature of several neurodegenerative disorders, including Alzheimer’s disease (AD). Ubiquitin carboxyl-terminal hydrolase L1 (UCH-L1) is an example of a neuron-specific protein which displays two different dimerization-dependent catalytic activities and can be farnesylated for membrane attachment, oxidized, and truncated. Decreased levels of soluble UCH-L1 are inversely proportional to the number of neurofibrillary tangles. Further assessment of a link between UCH-L1 function and the pathogenesis of AD requires an analytical method to separately quantify different UCH-L1 forms. In the present study, we have developed a multiple reaction monitoring (MRM) assay to measure UCH-L1 in the high-speed supernatant and pellet of frontal cortex homogenate. The well-characterized 15N-labeled quantification concatamer (QconCAT) carrying prototypic tryptic peptides of UCH-L1 was used as an internal standard. The composed protocol of frontal cortex processing includes solubilization and reduction/alkylation of proteins in the presence of 1% sodium dodecyl sulfate (SDS) and following with desalting/delipidation of the sample by chloroform/methanol precipitation with extra water washing of the protein pellet. The measurements were performed for frontal cortex samples from control and severe AD donors. The proposed workflow can be recommended for quantification of partitioning of other proteins of interest.
Ubiquitin carboxyl-terminal hydrolase L1 (UCH-L1) is an abundant neuronal protein in the brain.1 In vitro, UCH-L1 possesses hydrolase and dimerization-dependent ligase catalytic activities. Hydrolase activity of UCH-L1 causes hydrolysis of C-terminal ubiquityl esters and amides1 and presumably facilitates ubiquitin recycling. Ligase activity results in ubiquitin–ubiquitin ligation.2 Thus, UCH-L1 may play a role in recycling and substrate conjugation of free ubiquitin. It was also reported that UCH-L1 has a monoubiquitin stabilizing effect in vivo, which is independent from enzymatic activity.3 In addition to be important in ubiquitin–proteasome system, recent in vivo studies on UCH-L1-deficient mice indicated that UCH-L1 may be involved in other cellular processes.4 Overall, various roles and regulations of UCH-L1 indicates that UCH-L1 is essential for neurons to survive and to maintain their proper function.5 There are many different forms of UCH-L1 in neurodegenerative diseases. UCH-L1 was reported to undergo oxidation6,7 and N-terminal truncation.7 UCH-L1 can be associated with neurofibrillary tangles7 and farnesylated for association with membrane.8 It appears that multiple UCH-L1 forms with potentially different functions can exist simultaneously, and the relationship between these forms and neurodegenerative disease needs to be clarified. We thought that development of a workflow to separately quantify soluble and nonsoluble particulate forms of UCH-L1 will help further elucidate the molecular details that may link UCH-L1 to neurodegenerative diseases.
Multiple reaction monitoring (MRM) mass spectrometry in combination with isotope-labeled quantification concatamer (QconCAT) internal standard have been proven to be an effective method for quantification of specific proteins in biological samples.9–11 In the present study, we have expressed, purified, and characterized a QconCAT containing unique tryptic peptides of UCH-L1. We further optimized a sample processing protocol to quantify UCH-L1 in the supernatant and pellet fractions generated by high-speed centrifugation from frontal cortex homogenate. Measurements were performed for control and severe Alzheimer’s disease (AD) donors.
EXPERIMENTAL SECTION
Materials
The DC protein assay kit was from Bio-Rad Laboratories (Hercules, CA). Ammonium chloride (15N, 99%) was purchased from Cambridge Isotope Laboratories (Andover, MA). Nickel–nitrilotriacetic acid resin was from Qiagen (Valencia, CA). SpinTrap G-25 spin columns were from GE Healthcare (Waukesha, WI). Sequencing grade modified trypsin was obtained from Promega Corp. (Madison, WI). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Expression, Purification, and Characterization of 15N-Labeled QconCAT
A synthetic gene encoding 412 amino acids composed of the sequence MEVWTQRLHGGSAPLPQDRGFLVNQIKVDLVDENFTELRGEIAGPPDTPYEGGRYQLEIKIPETYPFNPPKVRFIGFDRNAVIVALSSKSWDVKRFKSHTDQLVLIFAGKILKDNQDRALSNLESIPGGYNALRRMYTYYLKHQFYPTVVYLTKSSPSVMGKVFFGQLRAAEMFSPRFVALFTLLLFLKCFHWFMERSPNISWLFHCRIVSLEELRALEGHERQHLEVLSRLGVAGQWRFVDVEELKGQEVSPKVYFMNQDKLGFEDGSVLKQFLSETEKMSPEGGARLGAGGGSPEKSPSAQELKEQGNAITRNPLVAVYYTNRALCYDCRRALELDGQSVKAHFFKEQRLNFGDDIPSALRIAKKECQRNHEGDEDDSHVRAQQAYDRKDIEEHLQRVGHFDPVTRSPLT was synthesized by Integrated DNA Technologies (Coralville, IA). Four prototypic tryptic peptides of UCH-L1 (Q-peptides) are underlined. The design of this QconCAT also includes prototypic Q-peptides from other ubiquitin-related proteins, such as ubiquitin-conjugating enzyme E2 K, ubiquilin-1, E3 ubiquitin-protein ligase synoviolin, and E3 ubiquitin-protein ligase CHIP. In addition, 4-amino acid long extensions from their natural sequences on both sides of the Q-peptides were included. The synthetic gene was cloned into the NdeI/HindIII restriction sites of pET21a expression vector in-frame to the C-terminal His6-tag. For expression, the plasmid was transformed into One Shot BL21(DE3) and cells were cultivated at 37 °C in M9 minimum medium containing 1 g/L 15NH4Cl as the sole nitrogen source. Initial inoculation started with 5 mL of LB media, and the cells were grown for 6–8 h at 37 °C. Cells were collected by centrifugation at 20 000g for 20 min and washed once by 10 mL of 15NH4Cl-containing M9 medium. Cells were then transferred to 50 mL of 15NH4Cl-containing M9 medium and grown for 12–14 h at 37 °C. Cells were collected by centrifugation at 20 000g for 20 min and washed twice by 100 mL of 15NH4Cl-containing M9 medium. Cells were then transferred to 500 mL of 15NH4Cl-containing M9 medium. The expression was induced with 1 mmol/L isopropyl β-D-1-thiogalactopyranoside at OD600 of 0.6–0.8 and incubation for an additional 3 h at 37 °C. Cells were divided into 10 portions and harvested by centrifugation at 20 000g for 30 min. One portion of cells was resuspended in 20 mL of lysis buffer (50 mmol/L Tris·HCl, pH 7.5). Cells were disrupted by sonication and centrifuged at 20 000g for 30 min. The supernatant was discarded. The pellet was resuspended in 3 mL of urea buffer (7 M urea/0.1 M NaH2PO4/0.01 M Tris·HCl, pH 8.0), and 15N-labeled QconCAT was purified on nickel–nitrilotriacetic acid resin in batch mode. Finally, the purified 15N-labeled QconCAT was loaded onto a SpinTrap G-25 spin column to exchange buffer into 25 mmol/L NH4HCO3 with 1% sodium dodecyl sulfate (SDS). The protein concentration was measured in the presence of 1% SDS using the DC protein assay kit and bovine serum albumin as a standard. The final purified 15N-QconCAT was aliquoted and kept frozen at −80 °C.
To determine the molecular mass of QconCAT, liquid chromatography–mass spectrometry (LC–MS) analyses were performed in positive ion mode with a Thermo LTQ Orbitrap Discovery (San Jose, CA) coupled with an Agilent 1200 HPLC (Palo Alto, CA). The protein was trapped on a C8 guard column (2.1 mm diameter × 1.25 cm length, Agilent) and eluted with a 9.5 min gradient operated at 200 μL/min flow rate. The gradient settings were 5–35% solvent B in 3 min, 35–70% solvent B in 5 min, 70–100% solvent B in 0.5 min, and isocratic flow at 100% solvent B for 0.5 min, then returned to 5% solvent B in 0.5 min. Solvent A was water containing 0.1% formic acid, and solvent B was 80% acetonitrile, 20% water containing 0.1% formic acid.
Human Tissues
Frozen samples of frontal cortex were received from the Washington University School of Medicine Alzheimer’s Disease Research Center. All brain specimens were collected from deidentified donors following informed consent of the respective families. Demographic information on the donors is summarized in the Supporting Information (Table S1). The Clinical Dementia Rating (CDR) listed in Supporting Information Table S1 is a numeric scale used to quantify the severity of symptoms of dementia. The composite score of CDR ranging from 0 through 3 with CDR equals 0 for no symptoms and CDR equals 3 for severe symptoms (http://www.biostat.wustl.edu/~adrc/cdrpgm/index.html).
Processing of Samples
The minced brain tissue was placed in 25 mmol/L NH4HCO3/1% SDS and homogenized by sonication at 30 W using five 10 s continuous cycles (Sonicator 3000, Misonix Inc., Farmingdale, NY). The homogenate was centrifuged at 2000g for 5 min to remove tissue debris. The supernatant (whole tissue homogenate) was used to measure total protein concentration in the presence of 1% SDS using the DC protein assay kit and bovine serum albumin as a standard. The whole tissue homogenate was then aliquoted to 0.2 mg of total tissue protein per tube. One set of tubes was frozen at −80 °C, whereas another set was subjected to centrifugation at 106 000g for 60 min. The resulting high-speed supernatants and pellets were individually frozen at −80 °C. Three types of fractions were obtained for subsequent processing steps: the whole tissue homogenate and the high-speed supernatant and pellet. During the following processing, each fraction was placed in 25 mmol/L NH4HCO3/1% SDS/20 mmol/L dithiothreitol and supplemented with various amounts of QconCAT, ranging from 1 to 25 pmol per sample. The mixture was incubated at room temperature for 60 min to allow reduction of cysteines and was then treated with 50 mmol/L iodoacetamide for another 60 min to alkylate the reduced cysteines. Alkylated samples were precipitated with chloroform/methanol.12 This step depleted salts, SDS, and biological lipids from the samples. To remove possible trace contaminates, the protein pellets were sonicated in 1 mL of water and precipitated again by centrifugation at 20 000g for 10 min. After washing with water, protein pellets were sonicated in 100 μL of 25 mmol/L NH4HCO3/0.1% RapiGest and treated with trypsin for 15 h at 37 °C. The substrate/trypsin ratio was 50:1 (m/m). After trypsin digestion, the peptide samples were treated with 0.5% trifluoroacetic acid for 30 min at 37 °C and centrifuged at 106 000g for 30 min to remove RapiGest and other byproducts not soluble at low pH. After centrifugation, the supernatants were dried using a vacuum centrifuge (Vacufuge, Eppendorf AG, Hamburg, Germany).
LC–MS/MS Analysis
Dried peptides were reconstituted in 10 μL of 3% acetonitrile/97% water (v/v) containing 0.1% formic acid, and 2 μL was used for a single LC–MS/MS run. Instrumental analyses were performed on a hybrid triple-quadrupole/linear ion trap mass spectrometer (4000 QTRAP, ABI/MDS-Sciex) coupled to an Eksigent nanoLC-2D system (Dublin, CA). Separation of peptides was performed with an Eksigent cHiPLC-nanoflex system equipped with a nano cHiPLC column, 15 cm × 75 μm, packed with ReproSil-Pur C18-AQ, 3 μm (Dr. Maisch, Germany). Peptides were eluted over a 29 min gradient from 15% to 35% acetonitrile, containing 0.1% formic acid at a flow rate of 300 nL/min. The column effluent was continuously directed into the nanospray source of the mass spectrometer. All acquisition methods used the following parameters: an ion spray voltage of 2200 V, curtain gas of 105 kPa (15 psi), source gas of 140 kPa (20 psi), interface heating temperature of 170 °C, declustering potential of 76 V for +2 precursor ions and 65 V for +3 precursor ions, collision energy of 30 V for +2 precursor ions and 22 V for +3 precursor ions, and collision cell exit potential of 16 V for +2 precursor ions and 13 V for +3 precursor ions. The dwell time for all transitions was 40 ms.
Quantitative Analysis and Validation
The initial list of MRM transitions was selected as previously described13 and was experimentally screened for the three most intensive transitions per peptide. These transitions were further used for quantification (summarized in the Supporting Information, Table S2). The identities of the measured peptides were confirmed based on the equivalence of retention time of the three MRM peaks from a given peptide and the ratio among the three MRM peaks. The mean and standard deviation (SD) of the protein concentration were calculated by treating the three transitions for each of the different target peptides and the three experimental replicates all as independent measurements.
RESULTS AND DISCUSSION
Validation of UCH-L1 Quantification
The scope of our study was to measure UCH-L1 partitioning between soluble and particulate fractions of brain homogenate using MRM assay. MRM assay relies on isotopically labeled internal standard that must be spiked into the biological sample prior to further sample processing. In addition, sample processing must include only those steps that do not change analyte/standard ratio. Therefore, we focused first on internal standard characterization, then followed by sample processing.
The artificial protein composed of concatenated tryptic peptides (QconCAT) that collectively act as internal standards for quantification of a panel of ubiquitin-related proteins was designed, expressed as 15N-labeled in E. coli with His6-tag, and purified on nickel–nitrilotriacetic acid resin. To ensure that efficiency of tryptic digestion will not vary between QconCAT and endogenous proteins, each prototypic peptide (Q-peptide) in the QconCAT was flanked by 4-amino acid long extensions from natural sequences of endogenous proteins. Figure 1A shows that QconCAT was highly purified. Thus, total protein concentration determined for the final preparation of QconCAT was used as “mg/mL concentration” of internal standard. To convert this concentration into the M concentration used for spiking brain samples, the full-length expression of QconCAT was first verified. Electrospray ionization (ESI) mass spectrometry (Figure 1B) was used to determine the average molecular mass of QconCAT. The measured average molecular mass (49 289.0 Da) was nearly close to the theoretical average molecular mass (49 299.0 Da) and confirms full-length expression of QconCAT. Accordingly, the theoretical average molecular mass was used to convert mg/mL concentration of QconCAT into the M concentration. Figure 1C shows extracted ion chromatograms of transitions monitored for a representative Q-peptide from UCH-L1 after trypsin digestion of QconCAT. No signals were observed from unlabeled peptide. This indicates a nearly complete labeling, and thus no correction for labeling efficiency was applied during data analysis.
Figure 1.

Characterization of 15N-labeled QconCAT. (A) A 10% SDS–PAGE of purified QconCAT. The molecular mass standards are shown on the left. (B) ESI mass spectrum of purified QconCAT. The inset shows deconvoluted spectrum with experimental average molecular mass of purified QconCAT. (C) Extracted ion chromatograms of transitions monitored for LGVAGQWR peptide from UCH-L1 after trypsin digestion of purified QconCAT: (green) 443.7/546.3 and 450.2/555.3; (red) 443.7/617.3 and 450.2/627.3; (blue) 443.7/716.4 and 450.2/727.4; (orange) 443.7/773.4 and 450.2/785.4.
After characterization of QconCAT, peptide performance in MRM assay was evaluated before quantification of UCH-L1 was carried out. QconCAT has four Q-peptides from UCH-L1, which were named by Q1 (LGVAGQWR), Q2 (GQEVSPK), Q3 (LGFEDGSVLK), and Q4 (QFLSETEK). Figure 2 shows that quantification based on different peptides can yield different outcomes ranging from reliable to no quantification at all. Reliable quantification was observed for Q1 and Q3. Both standard and analyte generate confidently integrated peaks at the same retention time and with nearly identical fragmention relative intensities of the light and heavy isotope-labeled versions of the peptide (Figure 2, parts A and C). Quantification based on Q4 experienced random interference from the biological sample, which changed the relative intensities of transitions. Quantification based on Q2 is impossible because no signals were observed from both standard and analyte. Therefore, all subsequent measurements were performed based on Q1 and Q3 only.
Figure 2.

Extracted ion chromatograms of transitions monitored for Q1, Q2, Q3, and Q4 from UCH-L1 in the whole frontal cortex homogenate. Light and heavy versions of each peptide are color-coordinated. Transitions are summarized in the Supporting Information, Table S2.
To determine the linearity range of the optimum transitions, we supplemented seven frontal cortex samples with varying amounts of QconCAT. Figure 3 shows response curves for Q1 and Q3. The peak area ratios were determined from the extracted ion chromatograms of the transitions. The results demonstrated that the concentration of UCH-L1 can be measured accurately over at least a 25-fold range of internal standard and indicated remarkably similar behavior of selected transitions.
Figure 3.
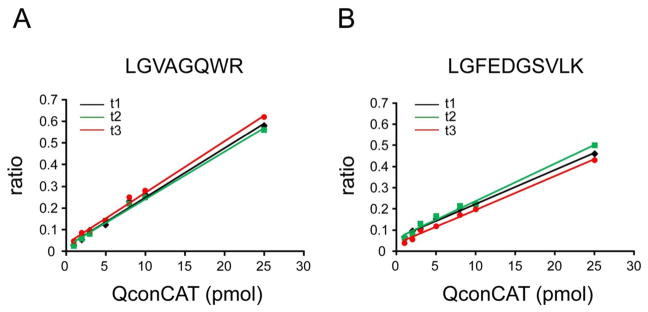
Response curves for quantification of UCH-L1 in the whole frontal cortex homogenate. The area ratio of a corresponding heavy peptide to light peptide was plotted vs amount of QconCAT added. (A) Three individual transitions (t1–t3) for Q1: (t1) 443.7/546.3 and 450.2/555.3; (t2) 443.7/617.3 and 450.2/627.3; (t3) 443.7/773.4 and 450.2/785.4. (B) Three individual transitions (t1–t3) for Q3: (t1) 532.8/618.3 and 538.3/625.3; (t2) 532.8/747.4 and 538.3/755.4; (t3) 532.8/951.5 and 538.3/961.4.
Partitioning of UCH-L1
High-speed centrifugation (106 000g, 60 min) is a gold standard for separation of the whole tissue homogenate into the supernatant with soluble proteins and pellet with particulate proteins, which include various forms of membrane-bound, aggregated, and low-solubility proteins. Partitioning of UCH-L1 between high-speed supernatant and pellet has potential relation to AD. Therefore, simultaneous quantification of UCH-L1 in both fractions may have a diagnostic or prognostic value to AD clinical research. Methodologically, it requires a universal sample processing protocol with complete solubilization of particulate forms of protein. SDS is the anionic surfactant of choice for complete solubilization, but depletion of SDS after solubilization is a prerequisite for the following trypsinolysis and mass spectrometric analysis. However, because of critical micelle concentration around 0.2% and aggregation number about 62,14 SDS solutions generate larger micelles, and complete removal of SDS in solution was thought to be impossible. We have recently described that SDS can be fully depleted from protein sample by chloroform/methanol precipitation.15 Using this observation as a starting point, here we developed a protocol for quantitative assessment of UCH-L1 partitioning between soluble and particulate tissue fractions. The key feature of the protocol (Experimental Section) is the combination of strong surfactant for complete solubilization and reduction/alkylation of proteins with following desalting and delipidation by chloroform/methanol precipitation. In addition, water washing of precipitated proteins before trypsinolysis was incorporated in the protocol to obtain better purity of peptides for mass spectrometry analysis.
Table 1 summarizes UCH-L1 quantification in the high-speed supernatant and pellet for control (n = 8) and AD (n = 10) groups. Analytical variation for individual donors (median CVs are about 10%) had less impact on measurements than donor-to-donor biological variation for consensus values (median CVs are about 26%), an expected source of error with human tissue samples. Unexpected source of error came from lower values based on Q1 in comparison to Q3. Both peptides passed preliminary selection by demonstrating perfect chromatographic and fragmentation properties, such as retention time, fragment ion relative intensities, and slope/r2 of signal responses (Figures 2 and 3). Nevertheless, quantification results based on Q1 and Q3 were not identical. One possible explanation for the lower values based on Q1 is that Q1 undergoes post-translational modification in the native UCH-L1. For example, oxidation of Trp in Q1 may reduce the abundance of nonoxidized Q1, which is selected for MRM analysis. However, this is an assumption and we do not have experimental data validating post-translational modification in Q1. Another reasonable explanation is that digestion efficiency of internal standard and analyte with trypsin is not fully identical for individual sequences and this adds variability to quantification. Our sample processing protocol includes SDS and chloroform/methanol treatments and has supposedly resulted in completely unfolded proteins. Nevertheless, it seems that measurements on complex biological samples, such as brain tissue, can have additional features that affect digestion efficiency of specific sequences with trypsin. In this content, it is important to emphasize that the goal of most MRM applications is comparison of protein levels, rather than assessment of absolute amounts.16 While the accuracy of MRM analysis may be uncertain, the measurements are reasonable since deviations are evenly distributed across the measurements in all sample groups. Figure 4 shows replotted consensus data from Table 1. Despite the different values based on Q1 and Q3, relative comparison of UCH-L1 partitioning is nearly similar. In the frontal cortex, there was 26% or 29% decrease of total UCH-L1 in the AD group in comparison to control group based on Q1 or Q3, respectively. Both decreases are statistically significant with P-values from Student’s t test less than 0.001. The most interesting observation is that, in addition to lower total level, the partitioning of UCH-L1 between soluble and particulate forms was also changed in AD (Figure 4). There was 41% (based on Q1) or 46% (based on Q3) decrease in the mean value of soluble UCH-L1. Both decreases for soluble UCH-L1 are statistically significant with P-values less than 0.001. At the same time, mean value of particulate UCH-L1 increased nearly 14% based on both Q1 and Q3 (Figure 4). Because of high donor-to-donor variability, the increases in particulate UCH-L1 were not statistically significant (P-values are around 0.14), and bigger sample size is necessary to draw a confident conclusion. However, overall combination of decrease in soluble UCH-L1 with increase of particulate UCH-L1 concurs well with partitioning of the whole UCH-L1. In other words, UCH-L1 not only occurred at lower level in AD, but it seems that UCH-L1 distribution between supernatant and pellet has been changed in AD.
Table 1.
Quantification of UCH-L1 in the High-Speed Supernatant and Pellet
| donor ID no. | UCH-L1 protein (pmol/mg tissue protein)a
|
|||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| control
|
AD
|
|||||||||||||||||
| 1 | 5 | 9 | 12 | 13 | 14 | 15 | 18 | 2 | 3 | 6 | 7 | 8 | 10 | 11 | 16 | 17 | 19 | |
| Supernatant | ||||||||||||||||||
| Q1 consensus | 245 ± 10 | 185 ± 12 | 235 ± 3 | 253 ± 16 | 198 ± 8 224 ± 31 |
192 ± 5 | 214 ± 16 | 269 ± 18 | 181 ± 16 | 67 ± 13 | 211 ± 3 | 168 ± 2 | 83 ± 1 | 130 ± 7 131 ± 50 |
121 ± 19 | 121 ± 10 | 65 ± 6 | 165 ± 13 |
| Q3 consensus | 342 ± 23 | 277 ± 18 | 387 ± 16 | 335 ± 13 | 280 ± 16 311 ± 63 |
183 ± 7 | 328 ± 3 | 354 ± 20 | 138 ± 20 | 76 ± 15 | 199 ± 21 | 233 ± 18 | 178 ± 6 | 164 ± 8 167 ± 56 |
162 ± 6 | 213 ± 15 | 77 ± 4 | 225 ± 11 |
| Pellet | ||||||||||||||||||
| Q1 consensus | 87 ± 2 | 83 ± 18 | 125 ± 4 | 81 ± 19 | 72 ± 6 90 ± 22 |
103 ± 9 | 57 ± 6 | 112 ± 17 | 62 ± 11 | 85 ± 12 | 113 ± 3 | 108 ± 10 | 110 ± 1 | 81 ± 4 102 ± 26 |
109 ± 11 | 100 ± 10 | 160 ± 4 | 94 ± 5 |
| Q3 consensus | 117 ± 7 | 111 ± 12 | 105 ± 14 | 115 ± 12 | 140 ± 16 119 ± 28 |
175 ± 8 | 78 ± 8 | 113 ± 4 | 75 ± 7 | 118 ± 10 | 138 ± 18 | 141 ± 20 | 117 ± 5 | 154 ± 9 136 ± 36 |
183 ± 17 | 150 ± 14 | 191 ± 6 | 96 ± 10 |
Measurements were performed on frontal cortex high-speed supernatant and pellet from control and severe AD human donors (Supporting Information, Table S1). The concentration was calculated for three experimental replicates by monitoring three transitions per individual Q1 and Q3 peptides. For the consensus, the data based on Q1 or Q3 peptides were combined for each control and AD groups. All data are presented as mean ± SD. The monitored transitions are summarized in the Supporting Information, Table S2.
Figure 4.
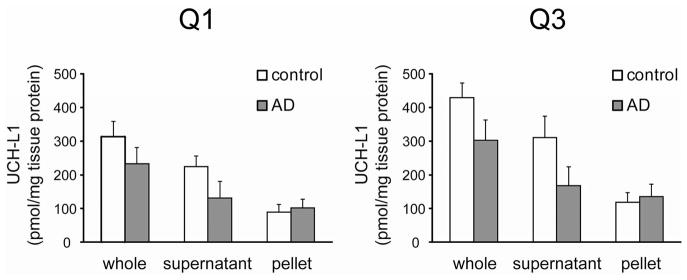
Quantification of UCH-L1 based on Q1 and Q3 peptides. Consensus values for supernatant and pellet from Table 1 are plotted as a bar graph. Values for whole homogenate were calculated as a sum of supernatant and pellet values in each group, control and AD. The data are presented as mean ± SD.
CONCLUSIONS
Partitioning of proteins between soluble and insoluble fractions changes the function(s) of proteins and is a major feature of neurodegenerative disorders, including AD. Oxidation and down-regulation of UCH-L1 in AD-affected brain suggest that UCH-L1 is involved in the pathogenesis of AD.6,8 We have composed a protocol for MRM quantification of UCH-L1 in the high-speed supernatant (soluble fraction) and high-speed pellet (particulate fraction) using 15N-labeled QconCAT as an internal standard. MRM performance of different UCH-L1 peptides and sources of error were carefully considered. Measurements on control and severe AD frontal cortexes have demonstrated that partitioning of UCH-L1 between soluble and particulate fractions is changed in AD.
Supplementary Material
Acknowledgments
This work was supported in part by the Washington University School of Medicine Alzheimer’s Disease Research Center Grant (P50 AG05681). Certain commercial materials, instruments, and equipment are identified in this manuscript in order to specify the experimental procedure as completely as possible. In no case does such identification imply a recommendation or endorsement by the National Institute of Standards and Technology nor does it imply that the materials, instruments, or equipment identified are necessarily the best available for the purpose.
Footnotes
Notes
The authors declare no competing financial interest.
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wilkinson KD, Lee K, Deshpande S, Duerksen-Hughes P, Boss JM, Pohl J. Science. 1989;246:670–673. doi: 10.1126/science.2530630. [DOI] [PubMed] [Google Scholar]
- 2.Liu Y, Fallon L, Lashuel HA, Liu Z, Lansbury PT., Jr Cell. 2002;111:209–218. doi: 10.1016/s0092-8674(02)01012-7. [DOI] [PubMed] [Google Scholar]
- 3.Osaka H, Wang YL, Takada K, Takizawa S, Setsuie R, Li H, Sato Y, Nishikawa K, Sun YJ, Sakurai M, Harada T, Hara Y, Kimura I, Chiba S, Namikawa K, Kiyama H, Noda M, Aoki S, Wada K. Hum Mol Genet. 2003;12:1945–1958. doi: 10.1093/hmg/ddg211. [DOI] [PubMed] [Google Scholar]
- 4.Walters BJ, Campbell SL, Chen PC, Taylor AP, Schroeder DG, Dobrunz LE, Artavanis-Tsakonas K, Ploegh HL, Wilson JA, Cox GA, Wilson SM. Mol Cell Neurosci. 2008;39:539–548. doi: 10.1016/j.mcn.2008.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Setsuie R, Wada K. Neurochem Int. 2007;51:105–111. doi: 10.1016/j.neuint.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 6.Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Free Radical Biol Med. 2002;33:562–571. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- 7.Choi J, Levey AI, Weintraub ST, Rees HD, Gearing M, Chin LS, Li LJ. Biol Chem. 2004;279:13256–13264. doi: 10.1074/jbc.M314124200. [DOI] [PubMed] [Google Scholar]
- 8.Liu Z, Meray RK, Grammatopoulos TN, Fredenburg RA, Cookson MR, Liu Y, Logan T, Lansbury PT., Jr Proc Natl Acad Sci USA. 2009;106:4635–4640. doi: 10.1073/pnas.0806474106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beynon RJ, Doherty MK, Pratt JM, Gaskell SJ. Nat Methods. 2005;2:587–589. doi: 10.1038/nmeth774. [DOI] [PubMed] [Google Scholar]
- 10.Pratt JM, Simpson DM, Doherty MK, Rivers J, Gaskell SJ, Beynon RJ. Nat Protoc. 2006;1:1029–1043. doi: 10.1038/nprot.2006.129. [DOI] [PubMed] [Google Scholar]
- 11.Nanavati D, Gucek M, Milne JL, Subramaniam S, Markey SP. Mol Cell Proteomics. 2008;7:442–447. doi: 10.1074/mcp.M700345-MCP200. [DOI] [PubMed] [Google Scholar]
- 12.Liao WL, Turko IV. Anal Biochem. 2008;377:55–61. doi: 10.1016/j.ab.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 13.Liao WL, Heo GY, Dodder NG, Pikuleva IA, Turko IV. Anal Chem. 2010;82:5760–5767. doi: 10.1021/ac100811x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turro NJ, Yekta A. J Am Chem Soc. 1978;100:5951–5952. [Google Scholar]
- 15.Wang M, Heo GY, Omarova S, Pikuleva IA, Turko IV. Anal Chem. 2012;84:5186–5191. doi: 10.1021/ac300587v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H, Liu Q, Zimmerman LJ, Ham AJL, Slebos RJC, Rahman J, Kikuchi T, Massion PP, Carbone DP, Billheimer D, Liebler DC. Mol Cell Proteomics. 2011;10:M110.006593. doi: 10.1074/mcp.M110.006593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.