

Abstract
Despite compelling genetic evidence indicating that cerebral amyloidosis can be, at least sometimes, the primary cause of Alzheimer's disease (AD), clinical trials for symptomatic AD with amyloid-reducing agents have succeeded at target engagement but failed to cause clinical benefit. In a landmark shift, the U.S. Food and Drug Administration now proposes to approve prophylaxis that alters the trajectory of what is now believed to be typical AD biomarker evolution. The first prevention trials are now beginning in patients with genetic guarantees for or high genetic risks for AD. The expectation is that clues to their outcomes will begin to emerge from these trials in approximately 2018. In the meantime, new strategies point to nonneuronal cells and to system pathology. A review of the current state of the art of AD science follows herein.
Keywords: Amyloid plaques, amyloidosis, microglia, neurodegeneration, systems biology, tauopathy
Over the last 30 years, Alzheimer's disease (AD) research has exploded, such that it is now possible to demonstrate unequivocally that some amyloid-reduction treatments reduce the burden of cerebral amyloidosis (1). The current challenge is converting that “target engagement” into meaningful clinical benefit, something that was not seen by Rinne et al. (1). Interventions in AD vary from vaccines to antibody infusions to orally active, small molecules (i.e., pills). Among these, pills are far and away the most desirable, because they are less expensive, less likely to cause side effects, and do not require refrigeration. The current status of development for amyloid-β (Aβ)-reducing pills focuses on two enzymatic targets known as β- and γ-secretases; these are complex aspartyl proteinases that specify Aβ generation [for review, see Gandy and DeKosky (2)]. The discovery that pathogenic mutations in the transmembrane amyloid precursor protein (APP) and in presenilins 1 and 2 underlie familial AD brought much attention to the biology and pharmacology of γ-secretase, the enzyme responsible for specifying the Aβ carboxy terminus. The specification of the Aβ carboxy terminus is critical for pathogenesis, leading to the initial popularity of γ-secretase inhibitors (GSIs) and γ-secretase modulators (GSMs) as therapeutic strategies. Side effects—perhaps due to mechanism-based, off-target inhibition of Notch processing—have limited GSIs. One such compound, known as semagacestat, unexpectedly even caused an acceleration of cognitive decline in a trial that was halted in 2011. The GSMs are now the major focus of γ-secretase pharmacotherapies.
Since its discovery in 1999, the aspartyl proteinase that marks the committed step toward Aβ generation, known as β-APP site cleaving enzyme-1 (BACE1), has been a popular target for therapeutic reduction of Aβ generation. In 2012, the popularity of BACE1 as a target soared on the discovery in Iceland of protective APP mutations located near the scissile bond where BACE1 cuts and releases the amino terminus of Aβ. The Icelandic mutations reduce Aβ generation and prevent AD, even in 25 subjects with two copies of the high-risk APOE ε4 allele. The BACE1 seems to have only a handful of substrates, leading to the notion that BACE inhibitors might be less toxic than GSIs. Still, this enzyme is critical in myelin formation, so some possibility of central nervous system toxicity is present. Together, β- and γ-secretases represent the most popular therapeutic opportunities for drug discovery, and current hopes are pinned on emerging clinical trials of GSMs and BACE inhibitors as the “next wave” of orally active, Aβ-reducing small molecules.
Asymptomatic cerebral amyloidosis can now be established by examination of cerebrospinal fluid for reduction of Aβ42 concentration or by positron emission tomography imaging with a 11C-or 18F-labelled ligand. Asymptomatic cerebral amyloidosis seems to be a frequent occurrence, because one third of asymptomatic individuals 65 years of age have positive amyloid brain scans (3). Current conventional wisdom holds that presymptomatic intervention and prophylaxis with an Aβ-reducing compound holds the most promise for arresting the progression of the amyloidosis and the eventual clinical presentation of cognitive decline and dementia. A key challenge lies in determining how early is early enough? Midlife surveillance amyloid imaging has been proposed as the best strategy for identifying candidates for Aβ-reducing secondary prevention. However, we cannot exclude the possibility that even a trace of amyloidosis will set into motion a series of intra- and intercellular signaling cascades and aggregated protein transfers that propagate in an Aβ-independent manner and culminate in clinical dementia even in the face of therapeutic reduction of Aβ burden. There might also be an Aβ-dependent disease initiation phase that is followed by Aβ-independent disease propagation and evolution. If either of these scenarios is true, then even timing intervention as early as the asymptomatic cerebral amyloidosis stage might still be too late to arrest progression. Of note, severe traumatic brain injury (TBI) is a well-known risk for the eventual development of AD. Acute Aβ deposition accompanies severe TBI—apropos of the question of how early to intervene with Aβ-reducing therapies—and there is new interest in the prospect of initiating Aβ-reducing therapies at the time of the acute TBI to neutralize the AD risk caused by that TBI (4). Several initiatives around this strategy are in development, and the results of those initiatives might inform the answer to the “How-early-is-early-enough?” question.
What this means is that we would like to have interventions that clear away existing fibrillar and oligomeric Aβ to give the best shot at stabilization of brain function at an early or preclinical stage. In addition to the small molecule approach, the advent of biologic drugs has led to the study of monoclonal anti-Aβ antibody therapy and, more recently, to the study of intravenous immunoglobulin (IVIg) containing naturally occurring anti-Aβ antibodies (5). These antibodies are presumed to bind circulating and/or interstitial Aβ and promote its catabolism by microglia (6). Thus, these compounds offer some of the first hope for therapeutic promotion of Aβ clearance. Bapineuzumab and solanezumab, anti-Aβ monoclonal antibodies, have been demonstrated to engage the target of cerebral fibrillar Aβ, and in the case of the former, bapineuzumab has been shown to arrest the progression of amyloidosis. However, when these were tested in patients with mild-to-moderate AD, only one of the two antibodies (solanezumab) offered a hint of benefit on neuropsychological testing, although this was not associated with any meaningful functional benefit. Furthermore, this subclinical benefit was detected among those with mild dementia but not those with moderate dementia, reinforcing the notion that, if Aβ-reduction is to succeed, we must get drugs on board as early as possible in the course of AD. Still, the solanezumab results stand as a milestone in that this is the first intervention to have any statistical benefit on neuropsychological measures. This is especially significant, because the U.S. Food and Drug Administration (FDA) has recently proposed that “moving the needle” on neuropsychological examination might be sufficient for registering a drug for AD prevention (7), although meaningful clinical benefit would remain the standard for approving a drug for treatment of symptomatic AD.
Trial results have recently revealed that infusions of naturally occurring anti-Aβ antibodies as they exist in current commercial preparations of immunoglobulin might slow progression in APOE ε4 carriers with moderate-stage AD. In the best possible scenario, if dementia prevention with solanezumab or some other biologic drug provides sufficient benefit on neuropsychological testing so as to be approvable by the FDA or its equivalent elsewhere in the world, we will be instantly faced with new problems: namely, affordability and availability. The annual per-person cost of a typical biologic drug is roughly equivalent to the annual per-person cost for custodial dementia care in nursing homes, but infusion of the biologic drug would begin 10 to 20 years sooner than custodial care. As a result, a successful biologic drug would not reduce the cost of dementia care but would double or triple a cost that is already unaffordable. Furthermore, in the case of IVIg, it is not clear how close the blood products industry could come to meeting the worldwide volume demand that would be required for AD prevention. Never has the adage “be careful what you wish for” been more apt.
The recent discovery that the microglial molecule CR1 is linked to AD and that CR1 genotype apparently modulates the impact of the APOE ε4 allele on brain fibrillar amyloid burden establishes that microglial dysfunction can be genetically linked to AD (8). The molecular pathological explanation for CR1-associated AD remains to be revealed (9), but this linkage dovetails well with recent evidence that Aβ can directly mediate dysfunction of microglia (Figure 1), resulting in their failure to clear Aβ burden in transgenic AD mice, a phenomenon previously described as microglial senescence (10). Two other recent articles linked a known microglial Aβ-clearing molecule, TREM2, to AD (11,12). More interestingly, Zhang et al. (13) have recently shown that a “multiscale” genetics/genomics approach links sporadic AD to TYROBP, another microglial molecule. Thus, evidence converges from several directions that microglia are key players in AD pathogenesis (Figure 2).
Figure 1.
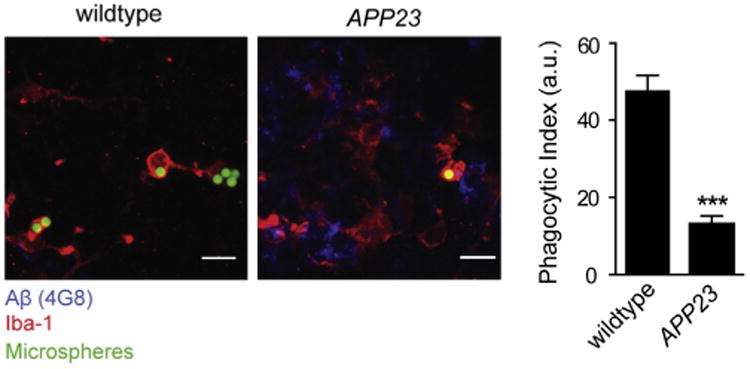
Phagocytic capacity of cortical microglia is impaired in mouse models of cerebral amyloidosis. Representative images (left) and microglial phagocytic index of 20 month old amyloid precursor protein (APP)23 and age-matched control mice (3 mice/genotype, p < .001, right) are shown. Data are mean ± SEM; ***p < .001. Scale bars: 10 μm. Aβ, amyloid beta; a.u., arbitrary units; Iba-1: ionized calcium-binding adapter molecule 1. Reprinted, with permission, from Krabbe et al. (24).
Figure 2.
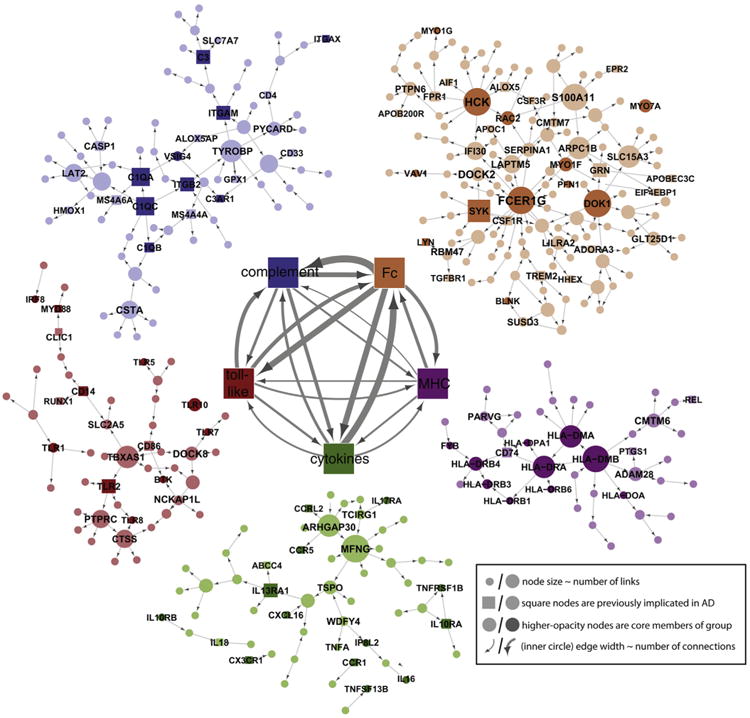
The Bayesian Brain Immune and Microglia Module. A module that correlates with multiple late-onset Alzheimer's disease (LOAD) clinical covariates and is enriched for immune functions and pathways related to microglia activity (prefrontal cortex module shown). (Inner networks) The prefrontal cortex module is enriched in genes that can be classified as members of the complement cascade (complement), toll-like receptor signaling (toll-like), chemokines/cytokines (cytokines), the major histocompatibility complex (MHC), or Fc-receptor system (Fc). The direction and strength of interactions between these pathways are collected across all gene–gene causal relationships that span different pathways. The minimum line width corresponds to a single interaction (MHC–toll-like) and scales linearly to a maximum of 17 interactions (Fc–complement). (Outer networks) Each color-coded group of genes consists of the core members of the different families, and genes that are causally related to a given family. Core family members of each pathway are shaded darkly, whereas square nodes in any family denote literature-supported nodes (at least two PubMed [http://www.ncbi.nlm.nih.gov/pubmed.] abstracts implicating the gene or final protein complex in LOAD or a model of LOAD). Labeled nodes are either highly connected in the original network, literature-implicated LOAD genes, or core members of one of the five immune families. Node size is proportional to connectivity in the module. Reprinted from Zhang et al. (13) with permission from Elsevier, copyright 2013.
On its surface, this growing evidence of the importance of microglia seems to conflict with the data of Grathwohl et al. (14), who showed that microglia were apparently dispensable in cerebral amyloidogenesis in a mouse model. However, recent articles suggest that the role(s) of microglia is (are) not black and white. Inhibition of interleukins (IL)-12 and IL-23 (15) or dysfunction of CD45 (16), of CD33 (17-19), or of NALP3—a component of the inflammasome (20)—can lead to a substantial reduction of AD pathology. It is becoming clear that microglia, having long been assumed to be invariably beneficial, might be helpful or harmful either alternately or even simultaneously. After migrating through the interstitial space of the brain to reach a nascent Aβ deposit, the balance between beneficial and harmful effects is unpredictable and might vary according to individual factors. Once the microglial cell has been poisoned by Aβ and/or once the Aβ deposit exceeds a size that would permit its engulfment by microglia, microglia might facilitate or instigate neuronal dysfunction and disease progression. This view does not exclude the possibility that Aβ per se is neurotoxic; the two pathways (i.e., intrinsic Aβ neurotoxicity and Aβ-driven microglial dysfunction) are not mutually exclusive but might rather act in concert to exacerbate AD pathology.
Perhaps the most exciting recent development from the therapeutic point of view is the recent success by vom Berg et al. (15) in modulating microglia and AD pathology via immunotherapeutic inhibition of IL-12/23. That article provides clear evidence that microglia or, more precisely, certain microglial pathways can be viable therapeutic targets (Figure 3). The network approach might also point us toward novel targets. Zhang et al. (13) has suggested that networks and subnetworks form multi-molecular modules controlled by drivers and that superior drugs might act on these drivers. Indeed, this prediction has already proven true in a non–central nervous system inflammatory disease (21).
Figure 3.
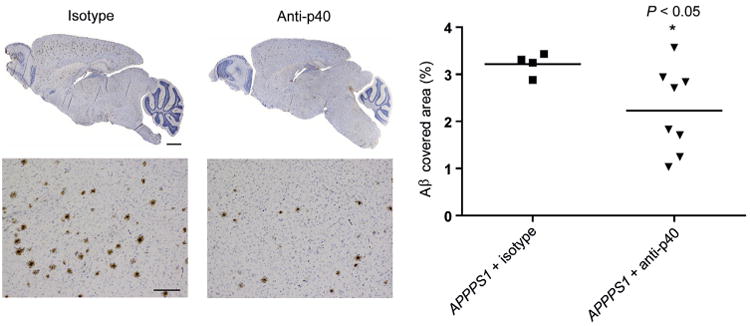
Peripheral p40-antibody (Anti-p40) treatment reduces amyloid-β (Aβ) plaque load in APPPS1 mice. Results from APPPS1 mice treated with p40-specific antibodies (C17.8, n = 8) or isotype control antibodies (2A3, n = 4) twice/week starting at 4 weeks of age and lasting until 120 days of age. (Left) Whole-brain, sagittal sections and high-magnification images of plaque burden within representative cortical areas in antibody-treated mice (4G8 staining). Scale bars, low-magnification images, 500 μm; high-magnification images, 200 μm. (Right) Morphometric analysis of Aβ-covered area in antibody-treated mice. Each symbol represents the mean of the morphometrically assessed Aβ plaque load or the number of Congo red–positive plaques of one mouse. For statistical analyses, one-way analysis of variance with Dunnet's post hoc analysis or a mixed-effects analysis of variance model with fixed effects was used. Reprinted by permission from Macmillan Publishers Ltd: Nat Med (15), copyright (2012).
Thus, as we contemplate the state of the translational science of AD circa 2014, there is evidence for both progress and promise at the basic and clinical levels. Basic AD science, long a bastion of discrimination toward neuroscience and against glial science, finally embraces the obvious fact that the brain is composed of multiple cell types. Genetics, multiscale genomics, cell biology, and experimental therapeutics all converge on microglia as the next frontier for drug discovery. In the clinic, solanezumab and IVIg show glimmers of benefit at particular stages (i.e., mild or presymptomatic AD) or in particular populations (i.e., APOE ε4–positive AD) on the basis of flickers of signals on neuropsychological testing. Although meaningful clinical benefit might not be at hand, the FDA has endorsed the hope that neuropsychological test benefit will be the harbinger of eventual meaningful clinical benefit, the holy grail of translational AD research. There is good reason to believe that success might be at hand, although “at hand” in this case means at least another 5 years before the first registration, and even that would be the most optimistic prediction; 10 years is a much more realistic estimate.
The real downside of the current status of translational AD research is that the focus on moving earlier and earlier in intervention threatens to leave behind those already diagnosed with disease. This presents an important opportunity for symptomatic drug discovery or perhaps the identification of a strategy that combines both symptomatic benefit with disease modification. New data suggest that metabotropic antagonists might, through a combination of stem cell stimulation and reduction of Aβ42 generation, represent such agents (22,23). Further basic and clinical work is required to establish whether the apparent promise of this combination of novel and unexpected bioactivities can be realized as an effective AD drug. If the stem cell stimulation is critical for the benefit of this drug, that too will represent an important milestone that opens yet another new frontier in drug discovery. More importantly, this strategy might offer hope for the 36 million persons already diagnosed with AD worldwide.
Acknowledgments
SG acknowledges the support of National Institute of Neurological Disorders and Stroke, National Institute on Aging, the Veterans Administration, the American Health Assistance Foundation, and the Cure Alzheimer's Fund.
SG is a member of a Data Management Safety Board for a Pfizer/Johnson and Johnson Alzheimer Immunotherapy Alliance clinical trial and a member of the Science Advisory Board of Cerora. Within the past 5 years, SG has held research grants from Amicus Therapeutics and from Baxter Pharmaceuticals.
References
- 1.Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, Klunk WE, et al. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer's disease treated with bapineuzumab: A phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;9:363–372. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- 2.Gandy S, DeKosky ST. Toward the treatment and prevention of Alzheimer's disease: Rational strategies and recent progress. Annu Rev Med. 2013;64:367–383. doi: 10.1146/annurev-med-092611-084441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aizenstein HJ, Nebes RD, Saxton JA, Price JC, Mathis CA, Tsopelas ND, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. 2008;65:1509–1517. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gandy S, Ikonomovic MD, Mitsis E, Elder G, Ahlers ST, Barth J, et al. Chronic traumatic encephalopathy. Molecular Neurodegen. doi: 10.1186/1750-1326-9-37. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Szabo P, Relkin N, Weksler ME. Natural human antibodies to amyloid beta peptide. Autoimmun Rev. 2008;7:415–420. doi: 10.1016/j.autrev.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 6.Gold M, Mengel D, Röskam S, Dodel R, Bach JP. Mechanisms of action of naturally occurring antibodies against β-amyloid on microglia. J Neuroinflammation. 2013;10:5. doi: 10.1186/1742-2094-10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kozauer N, Katz R. Regulatory innovation and drug development for early-stage Alzheimer's disease. N Engl J Med. 2013;368:1169–1171. doi: 10.1056/NEJMp1302513. [DOI] [PubMed] [Google Scholar]
- 8.Thambisetty M, An Y, Nalls M, Sojkova J, Swaminathan S, Zhou Y, et al. Effect of complement CR1 on brain amyloid burden during aging and its modification by APOE genotype. Biol Psychiatry. 2013;73:422–428. doi: 10.1016/j.biopsych.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gandy S, Haroutunian V, DeKosky ST, Sano M, Schadt EE. CR1 and the “vanishing amyloid” hypothesis of Alzheimer's disease. Biol Psychiatry. 2013;73:393–395. doi: 10.1016/j.biopsych.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller KR, Streit WJ. The effects of aging, injury and disease on microglial function: A case for cellular senescence. Neuron Glia Biol. 2007;3:245–253. doi: 10.1017/S1740925X08000136. [DOI] [PubMed] [Google Scholar]
- 11.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell. 2013;153:707–720. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grathwohl SA, Kälin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, et al. Formation and maintenance of Alzheimer's disease beta-amyloid plaques in the absence of microglia. Nat Neurosci. 2009;12:1361–1363. doi: 10.1038/nn.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.vom Berg J, Prokop S, Miller KR, Obst J, Kälin RE, Lopategui-Cabezas I, et al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer's disease-like pathology and cognitive decline. Nat Med. 2012;18:1812–1819. doi: 10.1038/nm.2965. [DOI] [PubMed] [Google Scholar]
- 16.Zhu Y, Hou H, Rezai-Zadeh K, Giunta B, Ruscin A, Gemma C, et al. CD45 deficiency drives amyloid-β peptide oligomers and neuronal loss in Alzheimer's disease mice. J Neurosci. 2011;31:1355–1365. doi: 10.1523/JNEUROSCI.3268-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bertram L, Lange C, Mullin K, Parkinson M, Hsiao M, Hogan MF, et al. Genome-wide association analysis reveals putative Alzheimer's disease susceptibility loci in addition to APOE. Am J Hum Genet. 2008;83:623–632. doi: 10.1016/j.ajhg.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, et al. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78:631–643. doi: 10.1016/j.neuron.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–678. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dudley JT, Sirota M, Shenoy M, Pai RK, Roedder S, Chiang AP, et al. Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci Transl Med. 2011;3:96ra76. doi: 10.1126/scitranslmed.3002648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim SH, Fraser PE, Westaway D, St George-Hyslop PH, Ehrlich ME, Gandy S. Group II metabotropic glutamate receptor stimulation triggers production and release of Alzheimer's amyloid(beta)42 from isolated intact nerve terminals. J Neurosci. 2010;30:3870–3875. doi: 10.1523/JNEUROSCI.4717-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gandy S, Steele JW, Lee SW, Clemenson D, Gadient R, Wedel P, et al. WIP1104. Behavioral deficits in APP transgenic mice reversed by mGluR inhibitor with pro-neurogenic, Aβ-reducing, and anxiolytic properties. Ann Neurol. 2012;72:S126. abstract. [Google Scholar]
- 24.Krabbe G, Halle A, Matyash V, Rinnenthal JL, Eom GD, Bernhardt U, et al. Functional impairment of microglia coincides with beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS One. 2013;8:e60921. doi: 10.1371/journal.pone.0060921. [DOI] [PMC free article] [PubMed] [Google Scholar]