

Abstract
Inflammatory responses to infection and injury must be restrained and negatively regulated to minimize damage to host tissue. One proposed mechanism involves enzymatic inactivation of the pro-inflammatory mediator leukotriene B4, but it is difficult to dissect the roles of various metabolic enzymes and pathways. A primary candidate for a regulatory pathway is omega oxidation of leukotriene B4 in neutrophils, presumptively by CYP4F3A in humans and CYP4F18 in mice. This pathway generates ω, ω-1, and ω-2 hydroxylated products of leukotriene B4, depending on species. We created mouse models targeting exons 8 and 9 of the Cyp4f18 allele that allows both conventional and conditional knockout of Cyp4f18. Neutrophils from wild-type mice convert leukotriene B4 to 19-hydroxy leukotriene B4, and to a lesser extent 18-hydroxy leukotriene B4, whereas these products were not detected in neutrophils from conventional Cyp4f18 knockouts. A mouse model of renal ischemia-reperfusion injury was used to investigate the consequences of loss of CYP4F18 in vivo. There were no significant changes in infiltration of neutrophils and other leukocytes into kidney tissue as determined by flow cytometry and immunohistochemistry, or renal injury as assessed by histological scoring and measurement of blood urea nitrogen. It is concluded that CYP4F18 is necessary for omega oxidation of leukotriene B4 in neutrophils, and is not compensated by other CYP enzymes, but loss of this metabolic pathway is not sufficient to impact inflammation and injury following renal ischemia-reperfusion in mice.
Keywords: Cytochrome P450, neutrophil, leukotriene B4, ω-hydroxylase, inflammation
1. Introduction
The cytochrome P450 (CYP) superfamily of enzymes catalyze oxidation of a diverse range of lipophilic substrates that include xenobiotics and endogenous compounds [1]. They play a role in generating and inactivating bioactive lipids that regulate physiological processes such as inflammation [2]. CYP4F subfamily members were first identified as enzymes that inactivate leukotriene B4 (LTB4), a potent pro-inflammatory lipid mediator [3,4]. LTB4 mobilizes neutrophils and other leukocytes to the site of an infection, then activates leukocyte secretion and phagocytosis to promote killing and removal of pathogens [5,6]. Mice deficient in LTB4 activity were developed by knocking out the genes for 5-lipoxygenase and leukotriene A4 hydrolase (enzymes that synthesize LTB4 from arachidonic acid), and the high affinity G protein-coupled receptor BLT1 [7,8,9,10,11]. These knockout mice have been used in many studies to confirm the role of LTB4 in cell recruitment during inflammation, and to implicate it as a pathological mediator in diverse disorders such as renal ischemia-reperfusion injury, arthritis, and asthma [6,12].
Less is known about mechanisms that negatively regulate LTB4, but it is inactivated by several metabolic pathways [13]. These include CYP-dependent omega oxidation [14], metabolism by LTB4 12-hydroxydehydrogenase that generates 12-oxo LTB4 and other products [15,16], and beta oxidation [17]. CYP-dependent omega oxidation was shown to be the major degradative pathway of LTB4 in neutrophils [14,18,19], and various studies indicate that it leads to loss of chemotactic activity and other responses of neutrophils to LTB4 [20,21]. The omega oxidation pathway is constitutively active in circulating neutrophils, and is present both before and during inflammatory recruitment. It is hypothesized that CYPs prevent excessive inflammation by dampening the LTB4 response and restraining neutrophil infiltration into tissues, but this has been difficult to test definitively in vivo without a knockout background. There are multiple members of the human and mouse CYP4F subfamilies (six and nine, respectively). Not all have been characterized, but several are described as LTB4 omega hydroxylases [2]. The possibility of functional compensation by related CYPs is a challenge for knockout studies. However, there are considerable differences in activity and tissue distribution of individual enzymes. The human CYP4F3 gene is alternatively spliced and generates two distinct isoforms [22]. CYP4F3A has high activity for LTB4, and is constitutively expressed at high levels in neutrophils as they develop in the bone marrow [4,22,23]. It is a candidate for a CYP enzyme that regulates LTB4-dependent inflammatory responses. The other isoform, CYP4F3B, is expressed in liver; it has activity for arachidonic acid as a substrate [22] and contributes to 20-hydroxyeicosatetraenoic acid (20-HETE) production in human liver cells [24, 25].
We identified CYP4F18 as the mouse homologue of CYP4F3A [21]. The Cyp4f18 gene is not alternatively spliced and generates a single translation product that is expressed in neutrophils. CYP-dependent omega oxidation of LTB4 results in hydroxylation at the ω-, ω-1, and ω-2 positions depending on species. 20-hydroxy LTB4 is produced by human neutrophils, and further converted to 20-carboxy LTB4 [18], whereas the principal product in rodents is 19-hydroxy LTB4 [21,26]. A targeted deletion in the Cyp4f18 gene described in this report results in loss of all LTB4 omega oxidation products in mouse neutrophils, but the deficiency does not significantly increase inflammatory cell infiltration or injury following renal ischemia-reperfusion. This is a first step in dissecting the complex interplay between different CYPs and other metabolic enzymes. Cyp4f18 knockout mice will be useful to explore the contribution of CYP4F18 in different physiological settings, to investigate other LTB4 inactivation pathways that operate in conjunction with omega oxidation, and to identify alternative substrates of CYP4F18.
2. Materials and methods
2.1. Mice
Generation of founder Cyp4f18 knockout mice was performed at the Gene Targeting and Transgenic Facility, University of Connecticut Health Center (GTTF UCHC). All procedures received prior approval by the Animal Care Committee at UCHC. Subsequent breeding and experimentation of mice was performed at the Massachusetts General Hospital in accordance with the guidelines of the Massachusetts General Hospital/Partners Committee on Research Animal Care. Mice of 6–12 weeks of age were used for experiments.
2.2. Construction of the Cyp4f18 targeting vector
The recombineering method [27] was used to construct a Cyp4f18 targeting vector. A bacterial artificial chromosome (BAC) containing the Cyp4f18 target region was obtained from the BACPAC Resources Center (BPRC), Children's Hospital Oakland Research Institute, Oakland, CA. A 12 kb DNA fragment spanning exon 5 through exon 13 of the Cyp4f18 gene was subcloned into pBluescript by gap repair. This method uses a bacterial strain (EL350) that can be induced to express λ phage Red proteins to mediate homologous recombination. A small mini-targeting vector was used to introduce a Neo selection cassette PL452 (loxP-PGK-EM7-NeobpA-loxP) in intron 7. The selection cassette was then excised by cre induction in EL350 cells, leaving a single loxP site upstream of exon 8. A second mini-targeting vector was used to introduce a Neo selection cassette PL451 (FRT-PGK-EM7-NeobpA-FRT-loxP) in intron 9.
2.3. Generation of Cyp4f18 fl/− and +/− mice
The Cyp4f18 targeting vector was linearized and electroporated into the mouse embryonic stem (ES) cell line D1, derived from (129S6/SvEvTac × C57BL/6J)F1 cells. Transfected cells were selected based on their resistance to G418 and ganciclovir, and surviving colonies were screened by PCR for homologous recombination of the target region. Selected ES cells were injected into blastocysts and implanted into foster mothers, resulting in chimeric offspring with germline transmission of the modified allele. To delete the Neo gene, mice were mated with a Rosa26FLP strain from Jackson Laboratories that express FLP recombinase. This generates mice with a “floxed” Cyp4f18 allele: the selection cassette flanked by FRT sites is deleted leaving exons 8 and 9 flanked by two loxP sites. The floxed allele (fl) was transferred to a C57BL/6 background by repeated backcrossing, and can be used to generate conditional (cell-specific) or conventional (ubiquitious) knockouts of exons 8 and 9. To generate the conventional knockout, male Cyp4f18 fl/+ mice were mated with female mice that have ubiquitous expression of cre recombinase (C57BL/6J-Hprt1 cre deleter strain provided by GTTF UCHC). Cyp4f18 +/− heterozygous offspring were identified by PCR genotyping, and mated to generate Cyp4f18 −/− homozygous knockouts and Cyp4f18 +/+ wild-type littermates. The mice used in this study were backcrossed to C57BL/6 background for at least 8 generations. Homozygous Cyp4f18 knockout (−/−) and floxed (fl/fl) mice were deposited and are available at the Mutant Mouse Regional Resource Center (MMRRC) with the following designations. Cyp4f18 knockout: B6.129S4(Cg)-Cyp4f18tm1.1Pchr, MMRRC 036956; Cyp4f18 floxed: B6.Cg-Cyp4f18tm1Pchr, MMRRC 036957.
2.4. Genotyping and PCR
Mouse genomic DNA was isolated from tail snips using a DNeasy Tissue Kit (Qiagen). Genotyping was performed by PCR using Thermo-Start Taq DNA polymerase (Fisher) and primer pair WTF-WTR (238 bp product) to detect the wild-type Cyp4f18 allele, or primer pair KOF-KOR (415 bp product) to detect the knockout allele. The sequences of primers are shown in Table 1 (0.5 μM of each primer was used in PCR reactions). Primer pair WTF-WTR flanks exon 9, a region that is deleted in the knockout. Primer pair KOF-KOR does not generate a product from the wild-type allele where the primers are separated by 2455 bp (KOF also has an 8 bp overlap with a junction marker that is not present in wild type). The PCR conditions were 95°C for 15 min followed by 30 cycles of: 95°C for 30 sec, 62°C for 1 min, 72°C for 1 min. PCR products were analyzed on a 3% agarose gel. To identify the floxed Cyp4f18 allele, PCR was performed with primer pair FLF-FLR that flanks an FRT-loxP sequence (393 bp product for floxed allele, 289 bp product for wild type allele which lacks the FRT-loxP sequence). The same PCR conditions were used except for an annealing temperature of 57°C. RNA transcripts in cells from wild-type and Cyp4f18 −/− mice were distinguished by RT-PCR, using primer pair exon7F-exon10R for PCR with an annealing temperature of 60°C.
Table 1.
Summary of primers.
| PCR primers | |
|---|---|
| WTF | GTGCTGTCCATTTGTTGGTG |
| WTR | GAGCAGGAAAGGAGCTCAGA |
| KOF | GTGGTTGCCTGTGAATTCCT |
| KOR | AAGCCCATGCCTACAATGAC |
| FLF | CCACAAATGACACACCTCCA |
| FLR | TAGGCAGAATCGATGGGAAA |
| FLF2 | AGGAACTTCATCAGTCAGGTACA |
| FLR2 | CTCTCTGGGCCAAGCATATT |
| Exon7F | CTCCTTGATCAGGGTGGTGT |
| Exon10R | GGGAGCACAATGTCCTGAGT |
| Taqman primers for real time PCR (Applied Biosystems) | |
|---|---|
| Cyp4f13 | Mm00504576_m1 |
| Cyp4f14 | Mm00491623_m1 |
| Cyp4f15 | Mm00506542_m1 |
| Cyp4f16 | Mm00775893_m1 |
| Cyp4f17 | Mm01345625_m1 |
| Cyp4f18 | Mm00499348_m1 |
| Cyp4f39 | Mm00624134_m1 |
| Cyp4f40 | Mm01342246_m1 |
| Custom Taqman primers | |
|---|---|
| Cyp4f18 | (exons 8/9) |
| Forward: | AGTGGACTTTCCTGGATCCTGTAC |
| Reverse: | GGCAGCGCTCCTGGTATTC |
| Probe: | ACCTGGCAAGACACC |
| Cyp4f37 | |
| Forward: | TCCCGCCTCAGATGTTTCC |
| Reverse: | CCCAAGTGACCCAAAAACCA |
| Probe: | TCAGCCTCCTAAAAGA |
Sequences are shown in 5' to 3' direction.
2.5. RNA isolation and real time PCR
Total RNA was isolated from cells and tissues using the RNeasy Plus mini kit with QIAshredders (Qiagen). Reverse transcription was performed with a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The cDNA was analyzed for target gene expression using Taqman primer sets and a StepOnePlus real time PCR machine from Applied Biosystems. A standard reaction protocol was followed (50°C for 2 min, 95°C for 10 min, 40 cycles of: 95°C for 15 sec, 60°C for 1 min). Relative quantification of gene expression in knockout samples compared to wild-type was performed by the ΔΔCt method using mouse GAPDH as endogenous control. Each sample was run in triplicate to determine ΔCt values, 2−ΔCt values, or fold-differences in expression (2−ΔΔCt). Values from four experiments were expressed as mean ± SEM. The Taqman primer-probe sets were purchased from Applied Biosystems as listed in Table 1.
2.6. Western blot analysis
Membrane proteins were extracted from bone marrow neutrophils using M-PER Reagent (Thermo Scientific), and the protein content of extracts was determined using Bio-Rad Reagent. Samples were fractionated by SDS-PAGE (10% gel), electrophoretically transferred to PVDF membranes (Millipore), and the membranes were processed using the SNAP i.d. Protein Detection System (Millipore). Tris buffered saline (TBS) containing 0.5% Tween 20 and 0.5% non-fat dried milk was used for blocking and all subsequent incubations and washes. After initial blocking, the membranes were incubated for 10 min with rabbit polyclonal anti-human CYP4F3[354–371] (Themo-Scientific PA1-41365) diluted 1:500. This antibody cross-reacts with mouse CYP4F18[353–370]. The membranes were washed 3 times and incubated for 10 min with peroxidase-conjugated AffiniPure goat anti-rabbit IgG (Jackson ImmunoResearch) diluted 1:3000. The membranes were washed as before, and immunoreactive protein bands were visualized using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific).
2.7. Isolation of bone marrow neutrophils
Mouse bone marrow cells were isolated from femurs and tibias by perfusion with PBS. The cells were filtered through a 40 μm cell strainer, washed in PBS, and layered on top of a discontinuous two-layer gradient of Histopaque 1077/1119 (Sigma). After centrifugation at 700 × g for 30 min at RT, neutrophils were separated from other cells including erythrocytes and recovered at the interface of the 1077 and 1119 fractions. Purity of the isolated polymorphonuclear cells was confirmed (>90%) by staining nuclei with DAPI and examination under a Nikon Eclipse Ti microscope with confocal imaging. Viability of the cells (>95%) was assessed by trypan blue exclusion.
2.8. Analysis of LTB4 and arachidonic acid metabolites by liquid chromatography-mass spectrometry (LC-MS/MS)
Bone marrow neutrophils were isolated from Cyp4f18 +/+ and −/− mice and incubated with LTB4 (1.0 μM/10 million cells/0.5 ml PBS) for 10 min at 37°C. The cells were then extracted and analyzed by LC-MS/MS at the Wayne State University Lipidomics Core Facility. Samples were spiked with LTB4-d4 as an internal standard, and extracted for LTB4 metabolites using C18 extraction columns as described previously [28]. The eluate was dried under nitrogen and redissolved in 50 μl methanol-25 mM aqueous ammonium acetate (1:1). HPLC was performed on a Prominence XR system (Shimadzu) using Luna C18 (3μ, 2.1×150 mm) column. The column was eluted with a gradient between A: methanol-water-acetonitrile (10:85:5 v/v) and B: methanol-water-acetonitrile (90:5:5 v/v), both containing 0.1% ammonium acetate. The gradient program with respect to the composition of B was as follows: 0–1 min, 50%; 1–8 min, 50–80%; 8–15 min, 80–95%; and 15–17 min, 95%. The flow rate was 0.2 ml/min. The HPLC eluate was directly introduced to ESI source of QTRAP5500 mass analyzer (ABSCIEX) in the negative ion mode with following conditions: curtain gas: 35 psi, GS1: 35 psi, GS2: 65 psi, temperature: 600 °C, ion spray voltage: −1500 V, collision gas: low, declustering potential: −60 V, and entrance potential: −7 V. The eluate was monitored by the Multiple Reaction Monitoring method for LTB4-d4 (m/z 339 to 197, Rt: 11.56 min), LTB4 (m/z 335 to 195, Rt: 11.56 min), LTB5 (m/z 333 to 195, Rt: 10.46 min), 12-oxo LTB4 (m/z 333 to 179, Rt: 11.35 min), 18-hydroxy LTB4 (m/z 351 to 195, Rt: 6.08 min), 19-hydroxy LTB4 (m/z 351 to 195, Rt: 5.5 min), and 20-HETE (m/z 319 to 257, Rt: 13.65 min) with 25 msec dwell time for each transition. Optimized collisional energies (18 – 35 eV) and collision cell exit potentials (7 – 10 V) were used for each MRM transition. The data was collected using Analyst 1.5.2 software and the MRM transition chromatograms were quantitated by MultiQuant software (both from ABSCIEX). The internal standard (LTB4-d4) signal in each chromatogram was used for normalization for recovery as well as relative quantitation of each analyte.
CYP4F18 microsomes were prepared as described previously [21]. Briefly, recombinant CYP4F18 was coexpressed with NADPH cytochrome P450 reductase in Sf9 cells using a baculovirus expression system, and microsomes were prepared from the Sf9 cells. CYP4F3A microsomes (Supersomes™) were from BD Biosciences. Reaction mixtures of 0.1 ml containing 20 μM LTB4, 5 pmol P450 (CYP4F18 or CYP4F3A microsomes), 1 mM NADPH, and 100 mM potassium phosphate buffer, pH 7.4, were incubated at 37°C for 5 min. Reactions with 20 μM arachidonic instead of LTB4 were performed under identical conditions. At the end of the incubation, 0.4 ml of 20% methanol and 10 ng LTB4-d4 internal standard was added, and the reaction mixtures were extracted with StrataX cartridges and analyzed by LC-MS/MS. Triplicate reactions were analyzed for quantification. Single reactions of each microsome preparation with 20 μM LTB4-d4 were used to assist product determination.
2.9. Renal ischemia-reperfusion injury (IRI) model, and preparation of a kidney cell suspension
An established mouse model of renal IRI was used [29]. Age-matched C57BL/6 male mice (8–12 weeks) were anesthetized using sodium pentobarbital (0.04 mg/g i.p.), and placed on a heating pad to keep them at constant temperature (37°C). The renal pedicles were occluded bilaterally with a non-traumatic vascular clamp (Robotz Surgical Instruments, RS-5426) to induce ischemia, and after reperfusion the kidneys were perfused with Ringer's solution in situ, then removed for analysis. Experiments were performed with 25 min ischemia and 4, 12, or 24 hours of reperfusion. Different times of ischemia (20 min, 30 min) and reperfusion (up to 72 hours) were also investigated, but data from these alternative conditions are not included in this report. Sham surgery was performed in a similar way except that the renal pedicles were not clamped. For assessment of renal injury, blood urea nitrogen (BUN) was measured using a diagnostic kit (Mega Diagnostics).
To prepare a cell suspension, kidneys were decapsulated, minced, and digested in Dulbecco's modified Eagle's medium (DMEM) containing 0.14 units/ml liberase blendzyme 3 (Roche Applied Science) and 200 μg/ml DNase I (Roche Applied Science) for 45 min at 37°C. The cell suspension was filtered through a 70 μm cell strainer, centrifuged at 500 × g for 5 min, resuspended in ACK lysing buffer (Biowhittaker) to lyse red blood cells, then centrifuged as before. The cell pellet was resuspended in PBS, pH 7.2, containing 0.5% BSA and 2 mM EDTA.
2.10. Flow cytometry and antibodies
Cell suspensions in PBS + 0.5% BSA were pre-incubated with Mouse Fc Block (BD Biosciences) for 5 min at 4°C (0.5 μg/106 cells/100 μl), then incubated with fluorophore-conjugated anti-mouse monoclonal antibodies for 30 min at 4°C (dilutions determined by titration). The cells were fixed in BD stabilizing fixative (BD Biosciences). Monoclonal antibodies specific for mouse hematopoietic surface lineage markers, or isotype-matched control antibodies, were used with the fluorophores V450 (eFluor450), PE, PE-Cy7, PerCP, FITC, AF647, and APC-eFluor 780 for seven color-labelling of cells. Anti-CD45-PerCP (clone 30-F11), anti-Ly6G-FITC (1A8), anti-Ly6C-PE or –PECy7 (AL-21), anti-CD11b-PECy7 (M1/70), anti-Siglec-F-PE (E50-2440), and anti-CD3e-V450 (500A2) were from BD Biosciences. Anti-CD11b-APCeFluor780 (M1/70), anti-F4/80-APCeFluor780 (BM8), anti-MHCII-APCeFluor780 (M5/114.15.2), anti-CD11c-AF647 or -eFluor450 (N418), anti-CD4-PECy7 or -AF647 (GK1.5) and anti-CD8a-PE (53-6.7) were from eBioscience. Appropriate conjugates of rat IgG2b, rat IgG2a, rat IgM, and Armenian hamster IgG (eBio299Arm) were used as isotype controls (eBioscience). Labeled cells were analyzed at the Flow Cytometry Core Facility, Massachusetts General Hospital, using a BD SORP 7 Laser LSRII and FlowJo software [30].
2.11. Histology
Kidneys were harvested for histology following renal ischemia-reperfusion and were fixed in 2% paraformaldehyde. The tissue was dehydrated, embedded in paraffin, and 3 μm sections were stained with hematoxylin and eosin (H&E) or Periodic acid Schiff stain (PAS). Tissue injury was scored on a 1–6 scale by adding parameters for severity and extent of tubular injury, similar to previously published protocols [31,32,33,34]. Our method differed from these approaches in that it accounts for both severity and extent of injury in a single score, and it gives a composite score for the extent of injury instead of randomly sampling a number of injured tubules (which could be subject to systematic error due to the location and the angle of the section). The scores were assigned as follows. Severity of tubular injury: 0, no injury; 1, mild injury with mild attenuation of the epithelium and a loss of brush border on PAS stain; 2, moderate injury with marked attenuation of the epithelium but without frank denudation of basement membrane; 3, severe injury with denudation of basement membrane. Extent of tubular injury: 0, no injury; 1, small isolated foci of injured epithelium; 2, confluent areas of injured epithelium but without uniform confluent involvement of cortico-medullary junction; 3, diffuse injury involving the entire cortico-medullary junction. Intermediate scores were assigned when appropriate, e.g. an isolated tubule with very mild injury would get a score of 0.5 + 0.5 = 1. The tissues were scored by a pathologist in a blind fashion. Immunohistochemical staining for myeloperoxidase (MPO) was performed according to the manufacturer's protocol using a rabbit polyclonal antibody (ab45977, Abcam). MPO-stained cells were examined under the x40 objective, and 10 fields were counted to derive a mean number of cells per high power field (hpf, 0.22 mm2). Images were taken with a Canon EOS Rebel T2i digital camera, mounted with a custom optical adapter onto a Olympus BX50 microscope.
2.12. Measurement of LTB4 in kidney tissue
LTB4 was assayed using an ELISA kit (Neogen). Kidney tissue was homogenized in methanol and centrifuged at 3000 rpm, 4°C, for 15 min to obtain supernatant and remove any cellular and tissue material. The protein concentration in the supernatant was determined using Bio-Rad Reagent. LTB4 was isolated from the supernatant using C18 Sep-Pak light columns, and aliquots of the samples were dried with nitrogen gas. The residue was dissolved in extraction buffer for the ELISA assay, and LTB4 concentration was measured according to the ELISA kit instructions. The results were expressed as ng LTB4 per mg of protein.
2.13. Statistical analysis
The results are expressed as the mean ± SEM. A t-test was used for comparisons of paired data, and multi-group data were analyzed by ANOVA. A P value of less than 0.05 was considered significant.
3. Results
3.1. Targeted disruption of the Cyp4f18 gene
A schematic of the Cyp4f18 gene and cDNA is shown (Fig. 1A). To generate a loss-of-function (knockout) mouse model, a targeted mutation was introduced to delete exons 8 and 9. These exons code for conserved core structures that are essential for enzymatic activity in the CYP superfamily. Deletion of these regions renders CYPs functionally inactive, and is frequently seen in human CYP mutations expressing pathological phenotypes [35]. The strategy for introducing the targeted deletion is summarized in Fig. 1B. A targeting vector was constructed with a loxP sequence in intron 7, and a neo cassette in intron 9 (the cassette has a neo gene flanked by a pair of FRT sequences, and a 3' loxP sequence). The targeting vector was electroporated into ES cells, and the cells were selected for neo expression based on their resistance to G418 and ganciclovir. Three positive clones were identified that contained homologous recombination of the target region spanning the loxP site in intron 7 and the neo cassette in intron 9. One of the clones was used to generate founder mice that carried the modified Cyp4f18 allele. Offspring were mated with mice that express FLP recombinase to delete the region flanked by FRT sites, including the neo gene. This generated the floxed (fl) Cyp4f18 allele (exons 8 and 9 flanked by two loxP sequences). Subsequent offspring were mated with mice that express cre recombinase to delete the region flanked by loxP sites, including exons 8 and 9. This generated the knockout (−) Cyp4f18 allele. Heterozygous (+/−) crosses generated homozygous (−/−) mice at the expected Mendelian ratios. The homozygous Cyp4f18 −/− mice were viable and did not exhibit any gross defects in development or breeding. Separate colonies of floxed and knockout mice were maintained on a C57BL/6 background, and different genotypes were distinguished by PCR of genomic DNA (Fig. 1C, D). The mice have been deposited and are available at the MMRRC as described in section 2.3.
Fig. 1.

Generation of Cyp4f18 −/− mice.
(A) Schematic drawings of the Cyp4f18 gene and cDNA. The cDNA locations of untranslated regions (UTR), and predicted locations of representative conserved core structures, are shown. Arrows indicate locations of primers exon7F and exon10R used for detection of mRNA by RT-PCR. (B) Strategy for deletion of exons 8 and 9. The insertion positions of a neo gene, the loxP sites (closed triangles), and FRT sites (open triangles) is shown in the targeting vector, the floxed (fl) allele, and the knockout (−) allele. The locations of primer pairs FLF-FLR (to detect floxed allele), KOF-KOR (to detect knockout allele), and WTF-WTR (a region deleted in the knockout allele) are indicated. (C) Genotyping results of DNA from Cyp4f18 +/+, +/fl, and fl/fl mice using primer pair FLF-FLR. (D) Genotyping results of DNA from Cyp4f18 +/+, +/−, and −/− mice using primer pairs WTF-WTR and KOF-KOR. The sizes of the PCR products are indicated.
3.2. Expression of CYP4F18 and analysis of enzymatic activity in wild-type and knockout mice
To confirm disrupted expression of Cyp4f18, RNA from mouse bone marrow neutrophils was analyzed by RT-PCR. The primer pair exon7F-exon10R amplifies a 395 bp region between exons 7 and 10 in cDNA from Cyp4f18 +/+ mice, but generates a PCR product of only 198 bp from Cyp4f18 −/− mice due to deletion of exons 8 and 9 (Fig. 2A). A real time PCR assay designed to detect exons 8 and 9 shows that expression of these exons is abolished in Cyp4f18 −/− mice (Fig. 2B). The mutant RNA transcript codes for a truncated translation product that lacks the C-terminal 219 amino acids, because deletion of exons 8 and 9 results in a frameshift mutation and introduces a premature UAA stop codon 40 bp downstream of the start of exon 10. Western blot analysis with an antibody that reacts with amino acids 353–370 confirms loss of expression of this critical C-terminal region in Cyp4f18 −/− mice (Fig. 2C).
Fig. 2.
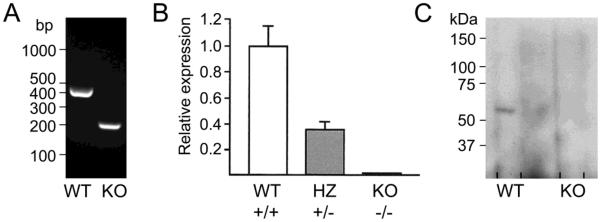
Expression of Cyp4f18 in bone marrow neutrophils from Cyp4f18 +/+ (WT), +/− (HZ), and −/− (KO) mice.
(A) Total RNA was extracted from neutrophils and used as a substrate for reverse transcription. The cDNA was analyzed by PCR using primer pair exon7F-exon10R.
(B) Real time PCR analysis of neutrophil cDNA using a primer-probe set that is specific for Cyp4f18 transcripts containing exons 8 and 9.
(C) Extracts of membrane proteins (20 μg) from neutrophils were analyzed by western blot. CYP4F18 protein appears as a 60 kDa band in WT extracts.
To confirm loss of enzymatic activity of CYP4F18, bone marrow neutrophils were incubated with LTB4 and metabolic products were analyzed by LC-MS/MS (Fig. 3). Experiments with recombinant CYP4F18 and CYP4F3A microsomes, which generate known products of LTB4, were performed in parallel as controls. We previously demonstrated that CYP4F18 microsomes convert LTB4 to 19-hydroxy LTB4, and to a lesser extent 18-hydroxy LTB4 (21). CYP4F3A microsomes convert LTB4 to 20-hydroxy LTB4; a small amount of 19-hydroxy LTB4 is also produced but represents < 5% of total product. The retention times of 19-, 20-, and 18-hydroxy LTB4 were 5.5, 5.84, and 6.08 min, respectively (Fig. 3A–C). The retention time of 20-hydroxy LTB4 was confirmed with a synthetic standard from Cayman (not shown). Neutrophils from wild-type (WT) mice exhibit the same profile of terminally hydroxylated products of LTB4 compared to CYP4F18 microsomes: 19-, and 18-hydroxy LTB4. Significantly, none of these products were detected in neutrophils from Cyp4f18 −/− (KO) mice. 12-Oxo LTB4, a product of an independent metabolic pathway involving LTB4 12-hydroxydehydrogenase, was produced by both wild-type and Cyp4f18 −/− neutrophils (retention time = 11.35 min). The identity of 12-oxo LTB4 was confirmed by an m/z transition of 333 to 179 Da (Fig. 3G). Hydroxylated products of LTB4 have similar fragmentation patterns, and MS/MS spectra show a characteristic peak at m/z 195 Da (Fig. 3D–F). In addition, unique peaks were observed for 18-, and 20-hydroxy LTB4 at m/z 275 and 189 Da, respectively; the structures of these fragments are shown (Fig. 4). The products of LTB4–d4 were analyzed after incubation with CYP4F18 and CYP4F3A microsomes, to assist fragment identification. For example, the fragments at m/z 275 and 189 Da retain all four deuterium atoms and are shifted to 279 and 193 Da, respectively (not shown).
Fig. 3.

LC-MS/MS analysis of LTB4-treated neutrophils.
Bone marrow neutrophils from wild-type mice (A), or Cyp4f18−/− (KO) mice (B), were incubated with LTB4 and extracted and analyzed by LC-MS/MS. Recombinant CYP4F18 and CYP4F3A microsomes, which generate known products of LTB4, were used as controls (C). HPLC chromatogram traces show detection of LTB4 (non-metabolized), 12-oxo LTB4 (product of 12-hydroxydehydrogenase metabolism), and 18-, 19, and 20-hydroxy LTB4 (products of CYP-dependent metabolism). LTB4-d4 was used as an internal standard. MS/MS spectra are shown for the metabolites with retention times of 6.08 min (D, 18-hydroxy LTB4), 5.5 min (E, 19, hydroxyl LTB4), 5.84 min (F, 20-hydroxy LTB4), and 11.35 min (G, 12-oxo LTB4). Characteristic fragments are observed at m/z 195 for all hydroxy LTB4 metabolites, and at m/z 179 for 12-oxo LTB4. Unique fragments are observed for 18-hydroxy LTB4 (m/z 275) and 20-hydroxy LTB4 (m/z 189), as indicated.
Fig. 4.
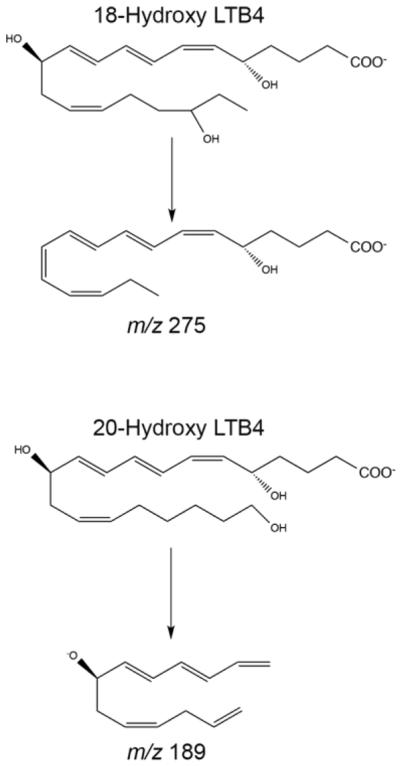
Characteristic fragments of 18- and 20-hydroxy LTB4.
The results of quantitation of LTB4 products in neutrophils are summarized in Table 2. Wild-type neutrophils generate 19-hydroxy LTB4 and 18-hydroxy LTB4 (~ 24%), whereas neither of these products were detected in neutrophils from Cyp4f18 −/− mice. There were no significant differences in 12-oxo LTB4 production in Cyp4f18 −/− neutrophils compared to wild-type (P < 0.05). Higher baseline levels of LTB4 were observed in Cyp4f18 −/− neutrophils in individual experiments, but the difference was not statistically significant (P = 0.13, n = 4).
Table 2.
Products of LTB4 metabolism in neutrophils
| picogram/million neutrophils | ||||
|---|---|---|---|---|
|
| ||||
| LTB4 | 12-oxo LTB4 | 18-hydroxy LTB4 | 19-hydroxy LTB4 | |
| Cyp4f18+/+ (WT) | 4620 ± 320 | 217 ± 8.3 | 54.5 ± 7.1 | 169 ± 23.8 |
| Cyp4f18−/− (KO) | 4903 ± 317 | 203 ± 11.5 | not detected | not detected |
|
| ||||
| Baseline WT | 74.8 ± 12.6 | 7.3 ± 1.1 | not detected | not detected |
| Baseline KO | 115.3 ± 19.3 | 10.8 ± 1.6 | not detected | not detected |
Cells were incubated with 1 μM LTB4 for 15 min at 37°C.
Baseline values are for cells that were incubated without LTB4.
Values indicate mean ± SEM, n = 4. Limit of detection = 5 picogram.
Mouse Cyp4f18 is a homologue of the human CYP4F3 gene, but is not alternatively spliced. There are two splice forms of CYP4F3 that have different activities: CYP4F3A metabolizes LTB4 and has little activity for arachidonic acid, whereas CYP4F3B has less activity for LTB4 and converts arachidonic acid to 20-HETE [22, 24, 25]. We compared the activity of recombinant CYP4F18 and CYP4F3A microsomes in parallel, under identical conditions (Table 3). Both have comparable overall activity for LTB4, although the site of terminal hydroxylation differs as previously reported [21]. CYP4F18 has even less activity for arachidonic acid than CYP4F3A, and the levels of 20-HETE product were barely above limits of detection. No other products of arachidonic acid were detected. Therefore, CYP4F18 does not combine the activities of the two CYP4F3 splice forms; it is not a significant 20-HETE producing enzyme and in this respect it is distinct from CYP4F3B.
Table 3.
Activity of CYP4F18 for LTB4 and arachidonic acid
| LTB4 substrate pmol product/(min × pmol P450) | Arachidonic acid substrate pmol product/(min × pmol P450) | |||
|---|---|---|---|---|
|
| ||||
| 20-OH LTB4 | 19-OH LTB4 | I8-OH LTB4 | 20-HETE | |
| CYP4F3A | 29.8 ± 5.2 | 1.45 ± 0.47 | not detected | 0.025 ± 0.003 |
| CYP4F18 | not detected | 20.8 ± 3.7 | 4.7 ± 0.8 | 0.01 ± 0.002 |
Values indicate mean ± SEM, n = 4.
3.3. Investigation of loss of function of CYP4F18 in vivo using a mouse model of renal ischemia-reperfusion injury (IRI)
Initially, we used flow cytometry analysis to investigate different conditions of IRI (Fig. 5). Single cell suspensions of kidney tissue were stained with antibodies to cell lineage-specific markers. A resident population of CD45+ cells was detected in sham-operated control kidneys, but the number of cells increased following renal ischemia and reperfusion, indicating infiltration of CD45+ leukocytes into kidney tissue (Fig. 5B). To identify neutrophils, the CD45+ cells were further analyzed for Ly6G (preferentially expressed in neutrophils) and CD11b (more broadly expressed in activated neutrophils and monocytes). Previous reports have demonstrated that renal CD11c+ dendritic cell subsets express a variety of proteins traditionally associated with other cell types, including Gr-1 (Ly6G + Ly6C) and CD11b [36]. We determined that CD11c+ cells comprise more than 50% of the resident CD45+ cell population in sham controls, but infiltrating populations of non-dendritic Ly6G+ CD11c- and CD11b+ CD11c- cells were clearly distinguished following ischemia-reperfusion (Fig. 5C). Quantitative analysis of these cell populations is summarized in Table 4. Ly6G+ CD11c- cells peaked at 12 hours post-ischemia (hpi), consistent with an early infiltration of neutrophils, whereas CD11b+ CD11c- cells continued to increase at 24 hpi. A comparison of wild-type and Cyp4f18 −/− mice showed no significant difference (P > 0.05) in the magnitude or time course of infiltration for either of these cell populations. Seven color flow cytometry permitted the use of multiple combinations of lineage–specific markers for neutrophils (Ly6G), monocytes (Ly6C), macrophages and dendritic cells (CD11c-, F4/80, MHC class II), eosinophils (Siglec F), and T cells (CD3, CD4, and CD8), under different conditions of ischemia (20, 25, and 30 minutes) and reperfusion (up to 72 hours), but did not detect any differences in leukocyte infiltration in Cyp4f18 −/− mice compared to wild-type (data not shown).
Fig. 5.

Analysis of CD45+ cells in mouse kidney tissue following renal ischemia-reperfusion. Peripheral blood was washed from kidneys by perfusion in situ before dissection and dissociation of kidney tissue. Kidney cell suspensions were labeled with anti-CD45-PerCP and other cell-specific markers and analyzed by flow cytometry.
(A) Total kidney cells were gated on forward versus side scatter (left panel), then for CD45 expression (middle panel), then reanalyzed for light scatter (right panel). A representative example is shown.
(B) Quantitative analysis of CD45+ cells in kidney tissue, expressed as a percentage of total kidney cells (mean ± SEM, n = 5), after 25 minutes ischemia and different times of reperfusion in wild-type (WT) and Cyp4f18 −/− knockout (KO) mice. Time 0 represents normal kidneys that were not subject to ischemia.
(C) Analysis of CD45+ cells in control (sham) and ischemic kidneys using antibodies to Ly6G (neutrophil marker), CD11b (myeloid marker), and CD11c (dendritic cell marker). Representative plots from individual WT mice are shown (25 min ischemia, 4 h reperfusion). An infiltrating population of Ly6G+ CD11c− cells and CD11b+ CD11c− cells is clearly distinguished in ischemic kidneys compared to sham controls. Numbers in each quadrant indicate the proportion of cells as a percentage of CD45+ cells.
Table 4.
Quantitative analysis of CD45+ cell subpopulations in mouse kidney tissue.
| Percent of total kidney cells | |||||
|---|---|---|---|---|---|
| Sham | 4 h | 12 h | 24 h | ||
| CD45+ Ly6G+ CD11c− | WT | 0.04 ± 0.008 | 1.5 ± 0.2 | 3.2 ± 0.3 | 2.9 ± 0.5 |
| KO | 0.05 ± 0.01 | 1.7 ± 0.3 | 3.4 ± 0.5 | 2.5 ± 0.8 | |
|
| |||||
| CD45+ CD11b+ CD11c− | WT | 0.37 ± 0.2 | 1.8 ± 0.2 | 3.75 ± 0.3 | 5.5 ± 0.7 |
| KO | 0.29 ± 0.1 | 2.0 ± 0.4 | 3.6 ± 0.2 | 5.8 ± 0.9 | |
Kidneys from wild-type (WT) and Cyp4f18 −/− (KO) mice were analyzed by flow cytometry following 25 min ischemia and 4, 12, or 24 h reperfusion (values indicate mean ± SEM, n = 5). Sham controls were performed for each time of reperfusion, and a combined value is shown.
Microscopic evaluation of ischemic kidney tissue revealed a progressive increase in the degree and extent of kidney injury as evidenced by loss of brush border, epithelial swelling, presence of luminal cell debris, epithelial attenuation with increased inter-nuclear distance and frank denudation of the basement membrane in the most severe cases (Fig. 6). The composite scores of epithelial injury increased from 2.6 ± 0.5 at 4 hpi to 5.5 ± 0.29 at 24 hpi in wild type controls, and 3.4 ± 0.5 at 4 hpi to 5.25 ± 0.25 at 24 hpi in Cyp4f18−/− mice (Table 5). There was no significant difference (P > 0.05) in total injury score between wild-type and Cyp4f18−/− mice between 4 and 24 hpi, or inflammatory cell infiltration into ischemic kidney tissue as assessed by immunohistochemistry for MPO (Table 5, Fig. 7). The number of MPO+ cells increased from 12 to 24 hpi in both wild-type and Cyp4f18−/− mice; this was consistent with the time course of CD11b+ cell infiltration observed in flow cytometry experiments, although Ly6G+ cells peaked at 12 hpi cells in the flow studies. Previous reports indicate that MPO is expressed in monocytes in addition to neutrophils [37], whereas Ly6G may be more specific for neutrophils. Blood urea nitrogen (BUN) levels at 24 hpi were 146.7 ± 14.8 mg/dl for wild-type mice and 150.2 ± 14.8 mg/dl for Cyp4f18−/− mice, P > 0.05, affirming no significant difference in renal injury (Table 5). LTB4 levels in ischemic kidney tissue were measured using an ELISA. At 12 hours post-ischemia, the levels were 2.25 ± 0.18 pg LTB4 per mg protein in wild-type, and 2.45 ± 0.2 pg LTB4 per mg protein in Cyp4f18−/− kidneys (P > 0.05, n = 4). In sham-operated mice, the levels were 1.0 ± 0.09 pg LTB4 per mg protein in wild-type, and 1.2 ± 0.06 pg LTB4 per mg protein in Cyp4f18−/− kidneys (P > 0.05, n = 4). We considered the possibility of compensatory increases in expression of other members of the CYP4F gene family in Cyp4f18−/− mice that might account for the comparable LTB4 levels, but no differences were detected by real time PCR (Fig. 8). A plot of 2−ΔCt values shows that CYP4F18 is the predominant transcript in bone marrow neutrophils in wild-type mice (CYP4F13 and −16 were detected at low levels), but only a minor transcript in normal kidney tissue (Fig. 8). Relative expression (2−ΔΔCt) was calculated, and the values indicate no change in expression of CYPs other than CYP4F18 in neutrophils or kidney. For example, the relative expression of CYP4F13 and −16 in kidney tissue from knockout mice compared to wild-type was 1.03 ± 0.08 and 0.93 ± 0.04, respectively (n = 4). We performed the real time PCR assays routinely during renal ischemia-reperfusion experiments, and did not observe any significant changes in the profile of CYP4F expression in kidney tissue at different times post-ischemia (not shown).
Fig. 6.

Microscopic morphology of kidney tissue after acute injury.
Hematoxylin and eosin stained kidney sections from wild-type mice (A, C, E, G) and Cyp4f18 −/− mice (B, D, F, H) at 4 h (C, D), 12 h (E, F), and 24 h (G, H) post-ischemia. Sham-operated mice (A, B) were not subject to renal ischemia. The histology shows correlation between the amount of injury and hours post-ischemia (hpi). At 4 hpi (C, D) both wild-type and Cyp4f18 −/− mice show mild to focally moderate epithelial injury evidenced by presence of luminal debris, loss of brush border (not shown), and variable epithelial attenuation with wide spaced nuclei. At 12 hpi (E, F) the injury is moderate to severe with focal epithelial denudation, but without a uniform continuous involvement of the cortico-medullary junction. The injury progresses to a uniform moderate to severe pattern by 24 hpi (G, H).
Table 5.
Quantitative analysis of inflammation and injury following renal ischemia-reperfusion.
| Time of Reperfusion | Cyp4f18 +/+ (WT) | Cyp4f18 −/−(KO) | Sham WT | Sham KO | |
|---|---|---|---|---|---|
| Histological score of injury (1–6 scale) | 4 h | 2.6 ± 0.5 | 3.4 ± 0.5 | 1 | 1 |
| 12 h | 4.7 ± 0.4 | 4.7 ± 0.4 | 1 | 1 | |
| 24 h | 5.5 ± 0.29 | 5.25 ± 0.25 | 1 | 1 | |
|
| |||||
| Cell infiltration: MPO+ cell number per hpf (0.22 mm2) | 12 h | 16.4 ± 5.1 | 18.7 ± 5.0 | 1 | 0.5 |
| 24 h | 25.2 ± 5.6 | 24.8 ± 5.4 | 2 | 2 | |
|
| |||||
| BUN level (mg/dl) | 24 h | 146.7 ± 14.8 | 150.2 ± 14.8 | 22.2 | 17.8 |
Results are expressed as mean ± SEM, n = 5.
Fig. 7.
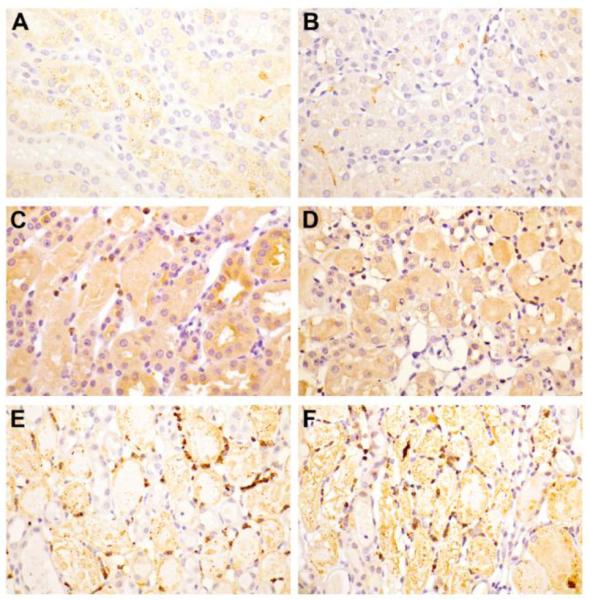
Myeloperoxidase immunohistochemistry.
Kidney sections from wild-type (A, C, E) and Cyp4f18 −/− mice (B, D, F) at 12 h (C, D) and 24 h (E, F) post-ischemia. Minimal background staining was observed in sham-operated mice (A, B). By 12 hpi there was an appreciable increase in the number of MPO positive cells. This number further increased by 24 hpi (E, F) in both the wild-type and Cyp4f18 −/− mice.
Fig. 8.

Expression of CYP4F family members.
Reverse transcription and real time PCR was used to measure CYP4F expression in bone marrow neutrophils (A), and normal kidney tissue (B). ΔCt values were determined for each CYP4F using GAPDH as endogenous control, and 2−ΔCt values were plotted for wild type (WT) and Cyp4f18−/− (KO) samples (n = 4). Relative quantitation (2−ΔΔCt) determined that there was no significant change in expression of any CYP4F in knockout samples compared to wild-type, except for the loss of CYP4F18.
4. Discussion
The activation and amplification of an immune response is critical to defend against microbial infection [38,39]. However, negative regulation of inflammation has emerged as an important mechanism to restrain the response within appropriate limits and minimize damage to host tissue [40,41,42,43]. Elucidating the relative contributions of inhibitory cytokines, receptor desensitization, and enzymatic inactivation of inflammatory mediators is a complex problem, but an important part of understanding and combating immune pathologies. Knockout mice have been generated to investigate loss of function of candidate regulatory molecules, and we created mice deficient in CYP418 for this purpose.
LTB4 is a potent inflammatory mediator implicated in a variety of immune pathologies, and multiple studies have demonstrated that a major pathway for enzymatic inactivation of LTB4 in neutrophils involves CYP-dependent omega oxidation [14,18,19]. CYP4F3A is the predominant CYP enzyme in human neutrophils [4,22,23], and the mouse homologue is CYP4F18 [21]. Low-level expression of other CYPs has been detected in neutrophils, including CYP4F12 in humans [44], and CYP4F13 and 16 in mice (Fig. 8). However, neutrophils from CYP4F18-deficient mice exhibit a knockout phenotype for LTB4 omega oxidation, and no products of this metabolic pathway were detected (Fig. 3; Table 2). This confirms a unique activity for CYP4F18 that is not compensated by other CYPs.
CYP4F18 is constitutively expressed in neutrophils, and it is predicted to play a role in restraining LTB4-dependent recruitment of the cells during inflammation. Previous studies using chemical inhibitors and in vitro systems have suggested that omega oxidation down-regulates LTB4 responses [20,21]. The mouse model of renal ischemia-reperfusion injury has a number of advantages for testing the role of CYP4F18 in vivo. It has been demonstrated that inhibition of LTB4 synthesis and BLT1 is accompanied by reduced leukocyte infiltration into renal tissue and reduced renal injury in this model [45,46,47]. A pivotal role for neutrophils in renal injury has been established [48], and there are established methods for measuring inflammatory cell infiltration into kidney tissue. However, we determined that there are no differences in cell infiltration or injury in Cyp4f18 knockout mice compared to wild-type. This was supported by experiments that varied the times of ischemia and reperfusion to adjust the extent of inflammation, and by the use of flow cytometry to complement traditional histological methods of evaluation (Figs. 5–7).
Flow cytometry allows rapid and sensitive detection of specific cell types in kidney tissue [48,49,50], and quantitation is representative of the entire tissue because single cell suspensions are prepared from whole kidneys. We used Ly6G in combination with CD45 and CD11c to identify neutrophils in ischemic kidneys, following perfusion in situ to remove circulating blood cells. Ly6G is a component of the myeloid marker Gr-1 that is preferentially expressed in neutrophils. The use of CD11c discriminates dendritic cells that co-express Gr-1. A population of infiltrating CD45+ Ly6G+ CD11c− cells was clearly distinguished in ischemic tissue compared to sham-operated controls (Fig. 5). The highest levels of this cell population were detected 12 hours post-ischemia, and levels declined by 24 hours, indicating an early infiltration of neutrophils. An identical time course was observed in wild-type and Cyp4f18 knockout mice.
Various myeloid and lymphoid cells infiltrate kidney tissue over different time courses following ischemia-reperfusion [51,52], and we extended our analysis to include these other cell types. Multiple combinations of lineage-specific markers were used in flow cytometry experiments, in conjunction with immunohistochemical staining with MPO. We did not detect any differences in leukocyte infiltration into kidneys in the Cyp4f18 knockout mice, but little is known about the expression patterns and potential roles of CYP4F18 in cells other than neutrophils. Human CYP4F3A is expressed in eosinophils and can be induced in macrophages [23,44,53], and there is evidence that CYP4F enzyme expression changes temporally in other cells such as hepatocytes and endothelia to facilitate clearance of LTB4 and resolution of inflammation in some species [2]. The functional contribution of CYP4F18 probably varies in different physiological settings, and analysis of different experimental models remains an important avenue of investigation. We created mice with a floxed Cyp4f18 allele that will permit generation of conditional knockouts in specific cells or tissues to facilitate research strategies.
The studies in this report represent a step in dissecting the role of enzymatic inactivation of LTB4. It appears that loss of omega oxidation is insufficient to affect neutrophil infiltration and injury following renal ischemia-reperfusion in mice. However, LTB4 is converted to 12-oxo LTB4 (a product of 12-hydroxydehydrogenase) in addition to 19-hydroxy LTB4 (the principal product of CYP4F18) in neutrophils from wild-type mice, and 12-hydroxydehydrogenase activity is unaffected in Cyp4f18 knockouts. We did not detect increased levels of 12-oxo LTB4 in knockout mice, but it is still possible that there is increased flux through the 12-hydroxydehydrogenase pathway that preserves overall rates of LTB4 metabolism. The development of double knockouts for CYP4F18 and LTB4 12-hydroxydehydrogenase would be needed to determine the effect of eliminating both pathways. Potential differences between humans and mice might arise from differential use of these pathways, or from the generation of different omega oxidation products.
In addition, it is possible that CYP4F18 has other functions besides LTB4 metabolism. CYP4F18 is a homologue of human CYP4F3, but is not alternatively spliced. The two splice forms of CYP4F3 preferentially use either LTB4 (4F3A) or arachidonic acid (4F3B) as substrates [22]. We determined that CYP4F18 has no significant activity for arachidonic acid and is not a significant 20-HETE producing enzyme; so it does not combine the activities of the two CYP4F3 splice forms and is more specifically a homologue of CYP4F3A. CYPs typically have broad substrate specificity, and it is a challenge to identify new biological functions for individual enzymes. A comparison of lipid metabolism in neutrophils from wild-type and knockout mice will provide an experimental system to search for other physiologically relevant substrates of CYP4F18.
Highlights
We created Cyp4f18 knockout mice and made them available at the MMRRC.
Neutrophils from knockout mice lack LTB4 omega oxidation.
Other CYP enzymes do not compensate for loss of CYP4F18 activity in neutrophils.
Neutrophil infiltration and tissue injury is not increased following renal IRI.
Knockout neutrophils retain production of 12-oxo LTB4 by 12-hydroxydehydrogenase.
Acknowledgements
This project was supported by National Institutes of Health Grant R01DK074821 (to P. Christmas). LC-MS/MS analysis was performed at the Wayne State University Lipidomics Core Facility and was supported in part by National Center for Research Resources, National Institutes of Health Grant S10RR027926; we thank Dr. Krishnarao Maddipati for help with the analysis.
Abbreviations
- AA
arachidonic acid
- BUN
blood urea nitrogen
- CYP
cytochrome P450
- 20-HETE
20-hydroxyeicosatetraenoic acid
- HPLC
high performance liquid chromatography
- IRI
ischemia-reperfusion injury
- LTB4
leukotriene B4
- LC-MS
liquid chromatography-mass spectrometry
- MPO
myeloperoxidase
- RT-PCR
reverse transcription-polymerase chain reaction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Nelson DR, Zeldin DC, Hoffman SMG, Maltais LJ, Wain HM, Nebert DW. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes pseudogenes and alternative-splice variants. Pharmacogenetics. 2004;14:1–18. doi: 10.1097/00008571-200401000-00001. [DOI] [PubMed] [Google Scholar]
- [2].Kalsotra A, Strobel HW. Cytochrome P450 4F subfamily: at the crossroads of eicosanoid and drug metabolism. Pharmacol. Therapeut. 2006;112:589–611. doi: 10.1016/j.pharmthera.2006.03.008. [DOI] [PubMed] [Google Scholar]
- [3].Chen L, Hardwick JP. Identification of a new P450 subfamily, CYP4F1, expressed in rat hepatic tumors. Arch. Biochem. Biophys. 1993;300:18–23. doi: 10.1006/abbi.1993.1003. [DOI] [PubMed] [Google Scholar]
- [4].Kikuta Y, Kusunose E, Endo K, Yamamoto S, Sogawa K, Fujii-Kuriyama Y, Kusunose M. A novel form of cytochrome P-450 family 4 in human polymorphonuclear leukocytes. cDNA cloning and expression of leukotriene B4 ω-hydroxylase. J. Biol. Chem. 1993;268:9376–9380. [PubMed] [Google Scholar]
- [5].Peters-Golden M, Henderson WR. Leukotrienes. N. Engl. J. Med. 2007;357:1841–1854. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- [6].Flamand N, Mancuso P, Serezani CHC, Brock TG. Leukotrienes: mediators that have been typecast as villains. Cell. Mol. Life Sci. 2007;64:2657–2670. doi: 10.1007/s00018-007-7228-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chen X-S, Sheller JR, Johnson EN, Funk CD. Role of leukotrienes revealed by targeted disruption of the 5-lipoxygenase gene. Nature. 1994;372:179–182. doi: 10.1038/372179a0. [DOI] [PubMed] [Google Scholar]
- [8].Goulet JL, Snouwaert JN, Latour AM, Coffman TM, Koller BH. Altered inflammatory responses in leukotriene-deficient mice. Proc. Natl. Acad. Sci. USA. 1994;91:12852–12856. doi: 10.1073/pnas.91.26.12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Byrum RS, Goulet JL, Snouwaert JN, Griffiths RJ, Koller BH. Determination of the contribution of cysteinyl leukotrienes and leukotriene B4 in acute inflammatory responses using 5-lipoxygenase- and leukotriene A4 hydrolase-deficient mice. J. Immunol. 1999;163:6810–6819. [PubMed] [Google Scholar]
- [10].Haribabu B, Verghese MW, Steeber DA, Sellars DD, Bock CB, Snyderman R. Targeted disruption of the leukotriene B4 receptor in mice reveals its role in inflammation and platelet-activating factor-induced anaphylaxis. J. Exp. Med. 2000;192:433–438. doi: 10.1084/jem.192.3.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tager AM, Dufour JH, Goodarzi K, Bercury SD, von Andrian UH, Luster AD. BLTR mediates leukotriene B4-induced chemotaxis and adhesion and plays a dominant role in eosinophil accumulation in a murine model of peritonitis. J. Exp. Med. 2000;192:439–446. doi: 10.1084/jem.192.3.439. 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Haeggstrom JZ, Funk CD. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem. Rev. 2011;111:5866–5898. doi: 10.1021/cr200246d. [DOI] [PubMed] [Google Scholar]
- [13].Murphy RC, Gijon MA. Biosynthesis and metabolism of leukotrienes. Biochem. J. 2007;405:379–395. doi: 10.1042/BJ20070289. [DOI] [PubMed] [Google Scholar]
- [14].Kikuta Y, Kusunose E, Kusunose M. Prostaglandin and leukotriene omega-hydroxylases. Prostaglandins Other Lipid Mediat. 2002;68–69:345–362. doi: 10.1016/s0090-6980(02)00039-4. [DOI] [PubMed] [Google Scholar]
- [15].Wainwright SL, Powell WS. Mechanism for the formation of dihydro metabolites of 12-hydroxyeicosanoids. Conversion of leukotriene B4 and 12-hydroxy-5,8,10,14-eicosatetraenoic acid to 12-oxo intermediates. J. Biol. Chem. 1991;266:20899–20906. [PubMed] [Google Scholar]
- [16].Yokomizo T, Ogawa Y, Uozumi N, Kume K, Izumi T, Shimizu T. cDNA cloning, expression, and mutagenesis study of leukotriene B4 12-hydroxydehydrogenase. J. Biol. Chem. 1996;271:2844–2850. doi: 10.1074/jbc.271.5.2844. [DOI] [PubMed] [Google Scholar]
- [17].Shirley MA, Murphy RC. Metabolism of leukotriene B4 in isolated rat hepatocytes. Involvement of 2,4-dienoyl-coenzyme A reductase in leukotriene B4 metabolism. J. Biol. Chem. 1990;265:16288–16295. [PubMed] [Google Scholar]
- [18].Shak S, Goldstein IM. Omega oxidation is the major pathway for the catabolism of LTB4 in human polymorphonuclear leukocytes. J. Biol. Chem. 1984;259:10181–10187. [PubMed] [Google Scholar]
- [19].Soberman RJ, Harper TW, Murphy RC, Austen KF. Identification and functional characterization of leukotriene B4 20-hydroxylase of human polymorphonuclear leukocytes. Proc. Natl. Acad. Sci. USA. 1985;82:2292–2295. doi: 10.1073/pnas.82.8.2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pettipher ER, Salter ED, Breslow R, Raycroft L, Showel HJ. Specific inhibition of leukotriene B4-induced neutrophil emigration by 20-hydroxy LTB4: implications for the regulation of inflammatory responses. Br. J. Pharmacol. 1993;110:423–427. doi: 10.1111/j.1476-5381.1993.tb13827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Christmas P, Tolentino K, Primo V, Zemski Berry K, Murphy RC, Chen M, Lee DM, Soberman RJ. Cytochrome P-450 4F18 is the leukotriene B4 ω-1/ω-2 hydroxylase in mouse polymorphonuclear leukocytes. Identification as the functional orthologue of human polymorphonuclear leukocyte CYP4F3A in the down-regulation of responses to LTB4. J. Biol. Chem. 2006;281:7189–7196. doi: 10.1074/jbc.M513101200. [DOI] [PubMed] [Google Scholar]
- [22].Christmas P, Jones JP, Patten CJ, Rock DA, Zheng Y, Cheng S-M, Weber BM, Carlesso N, Scadden DT, Rettie AE, Soberman RJ. Alternative splicing determines the function of CYP4F3 by switching substrate specificity. J. Biol. Chem. 2001;276:38166–38172. doi: 10.1074/jbc.M104818200. [DOI] [PubMed] [Google Scholar]
- [23].Christmas P, Carlesso N, Shang H, Cheng S-M, Weber BM, Preffer FI, Scadden DT, Soberman RJ. Myeloid expression of cytochrome P450 4F3 is determined by a lineage-specific alternative promoter. J. Biol. Chem. 2003;278:25133–25142. doi: 10.1074/jbc.M302106200. [DOI] [PubMed] [Google Scholar]
- [24].Antoun J, Goulitquer S, Amet Y, Dreano Y, Salaun J-P, Corcos L, Plee-Gautier E. CYP4F3B is induced by PGA1 in human liver cells: a regulation of the 20-HETE synthesis. J. Lipid Res. 2008;49:2135–2141. doi: 10.1194/jlr.M800043-JLR200. [DOI] [PubMed] [Google Scholar]
- [25].Plee-Gautier E, Antoun J, Goulitquer S, Le Jossic-Corcos C, Simon B, Amet Y, Salaun J-P, Corcos L. Statins increase cytochrome P450 4F3-mediated eicosanoids production in human liver cells: a PXR dependent mechanism. Biochem. Pharmacol. 2012;84:571–579. doi: 10.1016/j.bcp.2012.05.012. [DOI] [PubMed] [Google Scholar]
- [26].Powell WS, Gravelle F. Metabolism of arachidonic acid by peripheral and elicited rat polymorphonuclear leukocytes. Formation of 18- and 19-oxygenated dihydro metabolites of leukotriene B4. J. Biol. Chem. 1990;265:9131–9139. [PubMed] [Google Scholar]
- [27].Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Maddipati KR, Zhou SL. Stability and analysis of eicosanoids and docosanoids in tissue culture media. Prostaglandins Other Lipid Mediat. 2011;94:59–72. doi: 10.1016/j.prostaglandins.2011.01.003. [DOI] [PubMed] [Google Scholar]
- [29].Kelly KJ, Williams WW, Colvin RB, Meehan SM, Springer TA, Gutierrez-Ramos JC, Bonventre JV. Intercellular adhesion molecule-1-deficient mice are protected against ischemic renal injury. J. Clin. Invest. 1996;97:1056–1063. doi: 10.1172/JCI118498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Preffer F, Dombkowski D. Advances in complex multiparameter flow cytometry technology: applications in stem cell research. Cytometry Part B. 2009;76B:295–314. doi: 10.1002/cyto.b.20480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tögel F, Isaac J, Westenfelder C. Hematopoietic stem cell mobilization-associated granulocytosis severely worsens acute renal failure. J. Am. Soc. Nephrol. 2004;15:1261–1267. doi: 10.1097/01.asn.0000123692.01237.0a. [DOI] [PubMed] [Google Scholar]
- [32].Sharples EJ, Patel N, Brown P, Stewart K, Mota-Philipe H, Sheaff M, Kieswich J, Allen D, Harwood S, Raftery M, Thiemermann C, Yaqoob MM. Erythropoietin protects the kidney against the injury and dysfunction caused by ischemia-reperfusion. J. Am. Soc. Nephrol. 2004;15:2115–2124. doi: 10.1097/01.ASN.0000135059.67385.5D. [DOI] [PubMed] [Google Scholar]
- [33].Sadis C, Teske G, Stokman G, Kubjak C, Claessen N, Moore F, Loi P, Diallo B, Barvais L, Goldman M, Florquin S, Le Moine A. Nicotine protects kidney from renal ischemia/reperfusion injury through the cholinergic anti-inflammatory pathway. PLoS One. 2007;2:e469. doi: 10.1371/journal.pone.0000469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wever KE, Wagener FA, Frielink C, Boerman OC, Scheffer GJ, Allison A, Masereeuw R, Rongen GA. Diannexin protects against renal ischemia reperfusion injury and targets phosphatidylserines in ischemic tissue. PLoS One. 2011;6:e24276. doi: 10.1371/journal.pone.0024276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Stoilov I, Akarsu AN, Alozie I, Child A, Barsoum-Homsy M, Turacli ME, Or M, Lewis RA, Ozdemir N, Brice G, Aktan SG, Chevrette L, Coca-Prados M, Sarfarazi M. Sequence analysis and homology modeling suggest that primary congenital glaucoma on 2p21 results from mutations disrupting either the hinge region or the conserved core structures of cytochrome P4501B1. Am. J. Hum. Genet. 1998;62:573–584. doi: 10.1086/301764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hochheiser K, Tittel A, Christian K. Kidney dendritic cells in acute and chronic renal disease. Int. J. Exp. Path. 2011;92:193–201. doi: 10.1111/j.1365-2613.2010.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ysebaert DK, De Greef KE, Vercauteren SR, Ghielli M, Verpooten GA, Eyskens EJ, De Broe ME. Identification and kinetics of leukocytes after severe ischaemia/reperfusion renal injury. Nephrol. Dial. Transplant. 2000;15:1562–1574. doi: 10.1093/ndt/15.10.1562. [DOI] [PubMed] [Google Scholar]
- [38].Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- [39].Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- [41].Kobayashi KS, Flavell RA. Shielding the double-edged sword: negative regulation of the innate immune system. J. Leukoc. Biol. 2004;75:428–433. doi: 10.1189/jlb.0703321. [DOI] [PubMed] [Google Scholar]
- [42].Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell. 2010;140:771–776. doi: 10.1016/j.cell.2010.03.006. [DOI] [PubMed] [Google Scholar]
- [43].Scrivo R, Vasile M, Bartosiewicz I, Valesini G. Inflammation as “common soil” of the multifactorial diseases. Autoimmun. Rev. 2011;10:369–374. doi: 10.1016/j.autrev.2010.12.006. [DOI] [PubMed] [Google Scholar]
- [44].Kikuta Y, Mizomoto J, Strobel HW, Ohkawa H. Expression and physiological function of CYP4F subfamily in human eosinophils. Biochim. Biophys. Acta. 2007;1771:1439–1445. doi: 10.1016/j.bbalip.2007.10.004. [DOI] [PubMed] [Google Scholar]
- [45].Klausner JM, Paterson IS, Goldman G, Kobzik L, Rodzen C, Lawrence R, Valeri CR, Shepro D, Hechtman HB. Postischemic renal injury is mediated by neutrophils and leukotrienes. Am. J. Physiol. 1989;256:F794–F802. doi: 10.1152/ajprenal.1989.256.5.F794. [DOI] [PubMed] [Google Scholar]
- [46].Noiri E, Yokomizo T, Nakao A A, Izumi T, Fujita T, Kimura S, Shimizu T. An in vivo approach showing the chemotactic activity of leukotriene B4 in acute renal ischemic-reperfusion injury. Proc. Natl. Acad. Sci. USA. 2000;97:823–828. doi: 10.1073/pnas.97.2.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Patel NSA, Cuzzocrea S, Chatterjee PK, Di Paola R, Sautebin L, Britti D, Thiemermann C. Reduction of renal ischemia-reperfusion injury in 5-lipoxygenase knockout mice and by the 5-lipoxygenase inhibitor zileuton. Mol. Pharmacol. 2004;66:220–227. doi: 10.1124/mol.66.2.220. [DOI] [PubMed] [Google Scholar]
- [48].Awad AS, Rouse M, Huang L, Vergis AL, Reutershan J, Cathro HP, Linden J, Okusa MD. Compartmentalization of neutrophils in the kidney and lung following acute ischemic kidney injury. Kidney Int. 2009;75:689–698. doi: 10.1038/ki.2008.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Dong X, Swaminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney Int. 2007;71:619–628. doi: 10.1038/sj.ki.5002132. [DOI] [PubMed] [Google Scholar]
- [50].Furuichi K, Gao J-L, Horuk R, Wada T, Kaneko S, Murphy PM. Chemokine receptor CCR1 regulates inflammatory cell infiltration after renal ischemia-reperfusion injury. J. Immunol. 2008;181:8670–8676. doi: 10.4049/jimmunol.181.12.8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Jang HR, Rabb H. The innate immune response in ischemic acute kidney injury. Clin. Immunol. 2009;130:41–50. doi: 10.1016/j.clim.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Li L, Okusa MD. Macrophages, dendritic cells, and kidney ischemia-reperfusion injury. Semin. Nephrol. 2010;30:268–277. doi: 10.1016/j.semnephrol.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kikuta Y, Yamashita Y, Kashiwagi S, Tani K, Okada K, Nakata K. Expression and induction of CYP4F subfamily in human leukocytes and HL60 cells. Biochim. Biophys. Acta. 2004;1683:7–15. doi: 10.1016/j.bbalip.2004.03.007. [DOI] [PubMed] [Google Scholar]