

Abstract
Whole exome sequencing in a family with suspected dominant Kufs disease identified a novel Presenilin 1 mutation p.Leu(381)Phe in three brothers who, along with their father, developed progressive dementia and motor deficits in their early 30s. All affected relatives had unusually rapid disease progression (on average 3.6 years from disease onset to death). In silico analysis of mutation p.Leu(381)Phe predicted more detrimental effects when compared to the common Presenilin 1 mutation p.Glu(280)Ala. Electron microscopy study of peripheral fibroblast cells of the proband showed lysosomal inclusions typical for Kufs disease. However his brain autopsy demonstrated typical changes of Alzheimer disease.
Introduction
The Neuronal Ceroid Lipofuscinoses (NCL) are at least eleven genetic disorders, characterized by progressive neurodegeneration and early death. Adult-onset NCL, also referred to as Kufs disease has an autosomal recessive (MIM 204300) and an autosomal dominant (MIM162350) forms. We, and others, have recently identified mutations in the gene DNAJC5 associated with dominant Kufs disease [1,2,3,4]. About 20% of the individuals included in our study had mutations in DNAJC5 [3]. We have thus performed whole exome sequencing (WES) on probands of DNAJC5 - negative families in order to identify mutations associated with their neurodegenerative condition(s). One family tested positive for a novel significant sequence change in Presenilin 1 (PSEN1).
Materials and Methods
Studied individuals
The pedigree of the family studied is shown in Figure 1. The proband (Individual III-1) was in good health until his early 30s, except for a ruptured vertebral disk at the age of 31. At 32 he started developing progressive dementia and ataxia. His fine motor coordination became impaired, and he developed remote memory deficits. His speech became slurred and his writing was affected. He denied having visual or sensory changes. His mother described that he was unable to locate objects in the home where he had lived for 20 years. His neurological exam at age 33 revealed increased tone in his lower extremities and unsteady gait. His EEG was abnormal, with marked slowing throughout the recording with some delta activity at the patient’s highest state of alertness. No epileptiform discharges were noted. Enzyme activity for a panel of lysosomal storage disorders was normal. Electron microscopy study of skin biopsy showed lipofuscin containing phagocytic cells and distinct curvilinear lysosomal inclusion bodies. These findings were highly suggestive of NCL. Later in his life, he developed spastic paraparesis and died at the age of 36 from aspiration pneumonia and sepsis as a consequence of his neurological impairment. His neuropathological examination showed changes consistent with Alzheimer’s disease, including extensive replacement of neocortex, limbic structures, and cerebellar cortex by neuritic and amyloid containing plaques. The presence of Hirano bodies, neurofibrillary tangles, and granulovacuolar degeneration in the hippocampus were also reported.
Figure 1.
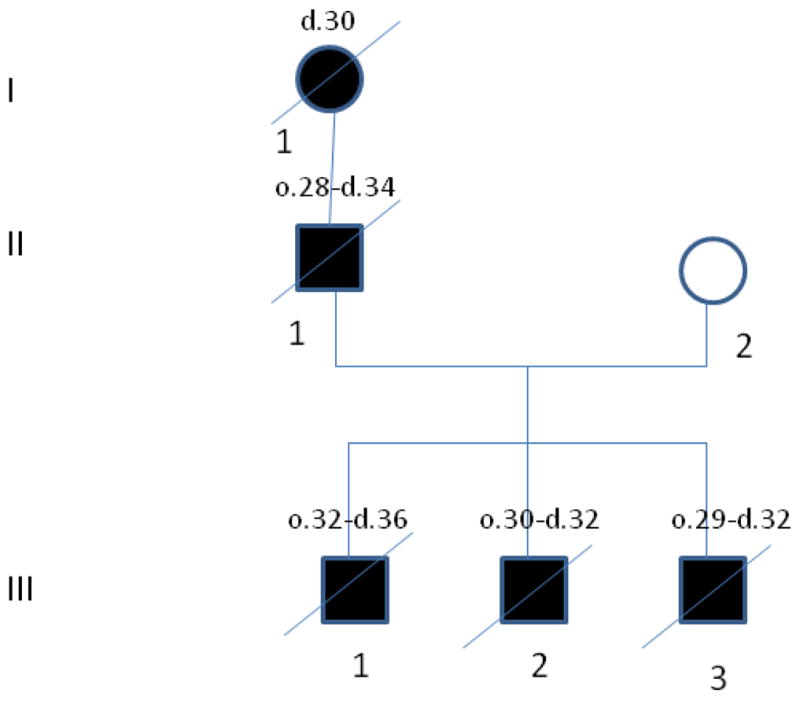
The family pedigree. The affected individuals are shown with black symbols. The age of disease onset (o.) and time of death (d.) are shown above every individual’s symbol in years.
One of this patient’s brothers (Individual III-2, Figure 1) developed very similar manifestations, first noticed at 30 years. He died at 32. Another brother (Individual III-3, Figure 1) had similar manifestations starting at 29. He died at 32. These brothers’ father had his first manifestations at age 28. His initial clinical manifestations consisted in weakness in his legs. He died at the age of 34. The paternal grandmother died at the age of 30 after progressive neurological disorder, but no information about the age of onset was available for her. Given the family history, autosomal dominant Kufs disease was considered as the most likely clinical diagnosis. Informed consent was obtained for all individuals and the Institutional Review Boards of the participating medical centers approved the study.
Methods
Whole exome sequencing
The proband (III-1) was found to be negative by Sanger sequencing for mutations in the gene DNAJC5, previously associated with autosomal dominant Kufs disease [1,2,3,4]. His genomic DNA was further studied with whole exome sequencing (WES). WES was performed as previously described [3]. Briefly, BWA and GATK software packages [5, 6, 7] were used to align sequence reads to the reference and call variant positions. All data were then annotated and imported into GEnomes Management Application (GEM.app), a web-based tool for next generation sequencing data analysis [8] (genomics.med.miami.edu). Variants were filtered for presence of non-synonymous heterozygous variants, frequency in public databases, conservation, protein predictions, and genotype quality (GATK quality index >100 and genotype quality GQ > 75)
Sanger sequencing was done for individuals III-1, III-2, III- 3, and II-2 in order to confirm the initial findings on WES and to assess the segregation of the identified sequence change. No specimen for study was available from individual II-1.
In silico protein analysis
The effects of the novel p.Leu381Phe amino acid change as compared to the well established p.Glu280Ala mutation [11] on the protein structure’s stability, flexibility and function was investigated in silico utilizing the experimental structure 2kr6.pdb available at Protein Data Bank [9] and a model created using the webserver I-Tasser (http://www.ncbi.nlm.nih.gov/pubmed/22238268) )[10]. The combined structure covers amino acids 260 – 467. Analyses of the effects of the mutations on the flexibility and stability of the protein were accomplished using Molecular Dynamic simulations and an in-house structure-based predictor (http://www.ncbi.nlm.nih.gov/pubmed/22238268)[10], along with third parties webservers. The positions of the mutations in PSEN1 were investigated in order to determine if they were located within highly conserved sequence regions.
Results
WES of individual III-1 identified 23 potentially significant changes in as many genes. Upon manual investigation to see if any of these genes or their pathways was associated with Kufs-like phenotype, we identified a heterozygous sequence change in Presenilin 1 (PSEN1). This change, c.1141C>T (NM_000021.3), resulted in a p.Leu381Phe amino acid substitution, was highly conserved (GERP score 5.69, PhastCons 1, phyloP 2.84) and was predicted to be damaging to the protein function by five in silico methods (Phylop: 2.84; Polyphen 2: 1; MutationAssessor: high; MutationTaster: damaging; LRT class: damaging; SIFT: 0.01). Further, this variant was not present in 6,500 exomes available through the EVS server and was unique amongst 1,901 exomes in GEM.app. However, a different change of the same amino acid residue (p.Leu381Val) has been shown previously to cause early onset Alzheimer disease with spastic paraplegia and extrapyramidal signs [12]. We confirmed the novel mutation by Sanger sequencing and showed co-segregation with the disease status in all available family members (III-2, III-3 and II-2). All affected brothers were heterozygous for p.Leu381Phe and their mother was negative for the change indicating that this mutation was inherited from the patients’ father.
In order to gather further confirmation of the pathogenicity of this novel change we performed comparative in silico analysis including a known PSEN1 mutation, p.Glu280Ala [11]. Both mutations, p.Glu280Ala and p.Leu381Phe, significantly affect the folding free energy and flexibility of the corresponding structural domains. However, the energy changes and changes in the flexibility are predicted to be greater for the novel p.Leu381Phe mutation as compared to p.Glu280Ala. Figure 2A illustrates part of the three dimensional structure of PSEN1 (amino acids 260 – 467), showing the position of the common mutation p.Glu280Ala indicated as E280 and the novel mutation p.Leu381Phe indicated as L381. The p.Leu381Phe mutation is very close to the putative active site X385 and is located within the structurally conserved large protein domain (Fig 2A), which is involved in interactions with beta and delta catenins. The catenins were previously shown to have important role in neuronal development, including formation of neuronal circuits and synaptic functioning [13,14]. In addition, p.Glu280Ala is located within a relatively polymorphic stretch of the PSEN1 protein, while p.Leu381Phe resides in a less polymorphic area (Fig 2B). Taking these findings all together, the novel p.Leu381Phe mutation is predicted to be more deleterious than the established p.Glu280Ala mutation (for additional information see supplement).
Figure 2.
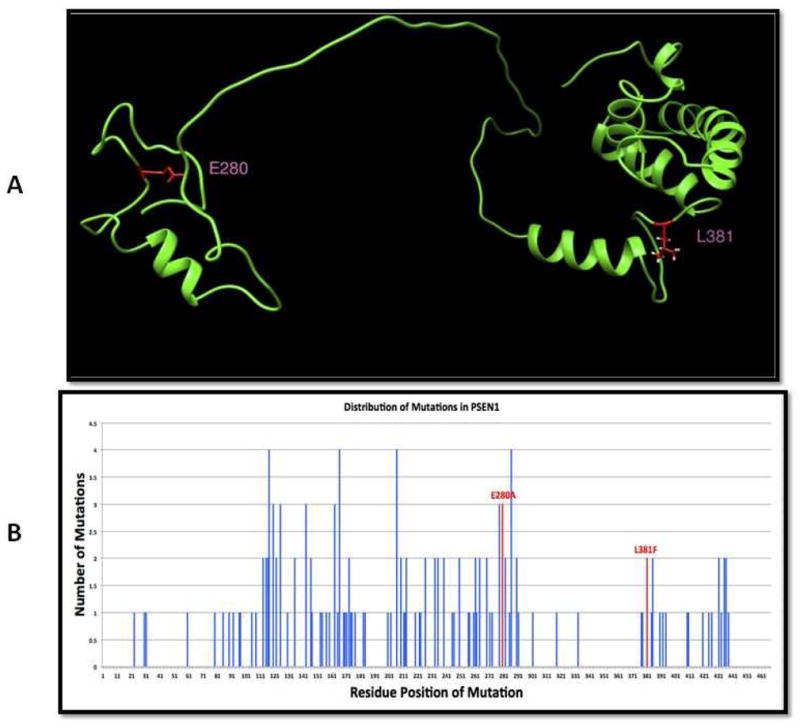
Figure 2A: A three dimensional structure model of Presenilin 1 is shown. This model was built by combining NMR structures and models built using I-Tasser, and it covers both the common mutation E280 and the novel mutation L381 (shown in red). L381 is located within a large, structurally conserved domain –on the figure shown on the right, and E280 is located within a smaller, more structurally flexible region-on the figure shown on the left.
Figure 2B: The distribution of mutations known to be associated with disease in Presenilin 1 is shown. The graph displays the numbers of known mutations for every amino acid position; there are three known mutations for E280 and two for L381 (shown in red).
Discussion
The identification of mutations in DNAJC5 associated with dominant Kufs disease helped better define this disorder in 20–25% of the individuals suspected to be affected. Prior to identifying these genetic mutations, the definite diagnosis was only possible by neuropathological studies. Lipofuscin storage material and characteristic submicroscopic lysosomal inclusions are noted in neuropathological studies of affected individuals [1,3]. Such submicroscopic inclusions are also variably found in peripheral fibroblasts or blood cells. Submicroscopic inclusions were inconsistently found in cases with DNAJC5 mutations. There is lack of consistency not only between different family members, but also when the same individual is studied at different ages or using different tissues [3]. However, once identified, such inclusions are considered highly suggestive of NCL. Conversely, this report demonstrates that such submicroscopic inclusions are not exclusive to NCL. Thus, ultimately, molecular studies for mutations in DNAJC5, PSEN1, and yet to be identified factors will provide a more accurate marker for disease classification.
PSEN1 mutations are the most common single gene cause of early-onset Alzheimer disease [11,15,16]. The first manifestations typically start in adults in their 40s. All studies involving larger number of patients show progression from first manifestation to death to be longer than 5 years, and most commonly over 10 years [11]. A proportion of PSEN1 mutations lead to very early disease onset (<35 years of age) [17]. Even in these most severe cases the disease course is on average more than 5 years. The reported novel PSEN 1 mutation consistently resulted in unusually fast progression in all affected individuals with mean disease duration of 3.6 years. Another missence mutation affecting the same codon, but changing Leucin to Valine was previously reported [12]. The proband with this mutation had disease onset at 32 and died at 37. Her affected mother’s disease onset was at 40 and she died at 48, while her affected maternal grandfather had his first symptoms at 65 and died at 69.
In conclusion, the reported novel PSEN1 mutation is associated with unusually severe disease course and lysosomal inclusions, previously believed to be specific for NCL.
Supplementary Material
Figure S1: (A) The superimposed 20 NMR structures of the large domain 1 are shown. The residue Leu(381) is displayed in red; (B) the model of the 3D structure of the small domain 2. The residue Leu(381) is shown in red.
References
- 1.Nosková L, Stránecký V, Hartmannová H, Přistoupilová A, Barešová V, Ivánek R, Hůlková H, Jahnová H, van der Zee J, Staropoli JF, Sims KB, Tyynelä J, Van Broeckhoven C, Nijssen PC, Mole SE, Elleder M, Kmoch S. Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. Am J Hum Genet. 2011;89:241–5. doi: 10.1016/j.ajhg.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benitez BA, Alvarado D, Cai Y, Mayo K, Chakraverty S, Norton J, Morris JC, Sands MS, Goate A, Cruchaga C. Exome-sequencing confirms DNAJC5 mutations as cause of adult neuronal ceroid-lipofuscinosis. PLoS One. 2011;6:e26741. doi: 10.1371/journal.pone.0026741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Velinov M, Dolzhanskaya N, Gonzalez M, Powell E, Konidari I, Hulme W, Staropoli JF, Xin W, Wen GY, Barone R, Coppel SH, Sims K, Brown WT, Züchner S. Mutations in the gene DNAJC5 cause autosomal dominant Kufs disease in a proportion of cases: study of the Parry family and 8 other families. PLoS One. 2012;7:e29729. doi: 10.1371/journal.pone.0029729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cadieux-Dion M, Andermann E, Lachance-Touchette P, Ansorge O, Meloche C, Barnabé A, Kuzniecky R, Andermann F, Faught E, Leonberg S, Damiano J, Berkovic S, Rouleau G, Cossette P. Recurrent mutations in DNAJC5 cause autosomal dominant Kufs disease. Clin Genet. 2013;83:571–5. doi: 10.1111/cge.12020. [DOI] [PubMed] [Google Scholar]
- 5.Margolin DH, Kousi M, Chan YM, Lim ET, Schmahmann JD, Hadjivassiliou M, Hall JE, Adam I, Dwyer A, Plummer L, Aldrin SV, O’Rourke J, Kirby A, Lage K, Milunsky A, Milunsky JM, Chan J, Hedley-Whyte ET, Daly MJ, Katsanis N, Seminara SB. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N Engl J Med. 2013;368:1992–2003. doi: 10.1056/NEJMoa1215993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gonzalez MA, Lebrigio RF, Van Booven D, Ulloa RH, Powell E, Speziani F, Tekin M, Schüle R, Züchner S. GEnomes Management Application (GEM. app): a new software tool for large-scale collaborative genome analysis. Hum Mutat. 2013;34:842–6. doi: 10.1002/humu.22305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rose PW, Bi C, Bluhm WF, Christie CH, Dimitropoulos D, Dutta S, Green RK, Goodsell DS, Prlic A, Quesada M, Quinn GB, Ramos AG, Westbrook JD, Young J, Zardecki C, Berman HM, Bourne PE. The RCSB Protein Data Bank: new resources for research and education. Nucleic Acids Res. 2013;41(Database issue):D475–82. doi: 10.1093/nar/gks1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Z, Wang L, Gao Y, Zhang J, Zhenirovskyy M, Alexov E. Predicting folding free energy changes upon single point mutations. Bioinformatics. 2012;28:664–71. doi: 10.1093/bioinformatics/bts005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sepulveda-Falla D, Glatzel M, Lopera F. Phenotypic profile of early-onset familial Alzheimer’s disease caused by presenilin-1 E280A mutation. J Alzheimers Dis. 2012;32:1–12. doi: 10.3233/JAD-2012-120907. [DOI] [PubMed] [Google Scholar]
- 12.Dintchov Traykov L, Mehrabian S, Van den Broeck M, Radoslavova Raycheva M, Cruts M, Kirilova Jordanova A, Van Broeckhoven C. Novel PSEN1 mutation in a Bulgarian patient with very early-onset Alzheimer’s disease, spastic paraparesis, and extrapyramidal signs. Am J Alzheimers Dis Other Demen. 2009;24:404–7. doi: 10.1177/1533317509341464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wexler EM, Rosen E, Lu D, Osborn GE, Martin E, Raybould H, Geschwind DH. Genome-wide analysis of a Wnt1-regulated transcriptional network implicates neurodegenerative pathways. Sci Signal. 2011;4:ra65. doi: 10.1126/scisignal.2002282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matter C, Pribadi M, Liu X, Trachtenberg JT. Delta-catenin is required for the maintenance of neural structure and function in mature cortex in vivo. Neuron. 2009;64:320–7. doi: 10.1016/j.neuron.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, Thomas-Anterion C, Michon A, Martin C, Charbonnier F, Raux G, Camuzat A, Penet C, Mesnage V, Martinez M, Clerget-Darpoux F, Brice A, Frebourg T. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999;65:664–70. doi: 10.1086/302553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larner AJ, Doran M. Genotype-phenotype relationships of presenilin-1 mutations in Alzheimer’s disease: an update. J Alzheimers Dis. 2009;17:259–65. doi: 10.3233/JAD-2009-1042. [DOI] [PubMed] [Google Scholar]
- 17.Filley CM, Rollins YD, Anderson CA, Arciniegas DB, Howard KL, Murrell JR, Boyer PJ, Kleinschmidt-DeMasters BK, Ghetti B. The genetics of very early onset Alzheimer disease. Cogn Behav Neurol. 2007;20:149–56. doi: 10.1097/WNN.0b013e318145a8c8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: (A) The superimposed 20 NMR structures of the large domain 1 are shown. The residue Leu(381) is displayed in red; (B) the model of the 3D structure of the small domain 2. The residue Leu(381) is shown in red.