

Abstract
Context:
Recombinant factor VIIa (rFVIIa) has been used as an adjunctive therapy for acute post-traumatic hemorrhage and reversal of iatrogenic coagulopathy in trauma patients in the hospital setting. However, investigations regarding its potential use in pre-hospital management of traumatic brain injury (TBI) have not been conducted extensively.
Aims:
In the present study, we investigated the physiology, hematology and histology effects of a single pre-hospital bolus injection of rFVIIa compared to current clinical practice of no pre-hospital intervention in a swine model of moderate fluid percussion TBI.
Materials and Methods:
Animals were randomized to receive either a bolus of rFVIIa (90 μg/kg) or nothing 15 minutes (T15) post-injury. Hospital arrival was simulated at T60, and animals were euthanized at experimental endpoint (T360).
Results:
Survival was 100% in both groups; baseline physiology parameters were similar, vital signs were comparable. Animals that received rFVIIa demonstrated less hemorrhage in subarachnoid space (P = 0.0037) and less neuronal degeneration in left hippocampus, pons, and cerebellum (P = 0.00009, P = 0.00008, and P = 0.251, respectively). Immunohistochemical staining of brain sections showed less overall loss of microtubule-associated protein 2 (MAP2) and less Flouro-Jade B positive cells in rFVIIa-treated animals.
Conclusions:
Early pre-hospital administration of rFVIIa in this swine TBI model reduced neuronal necrosis and intracranial hemorrhage (ICH). These results merit further investigation of this approach in pre-hospital trauma care.
Keywords: Coagulopathy, necrosis, pre-hospital, rFVIIa, swine, traumatic brain injury
INTRODUCTION
Traumatic brain injury (TBI) is the primary cause of death and severe disability following head trauma.[1] In the U.S. alone, approximately 1.4 million people sustain TBI annually and casualties account for 53,000 deaths and 235,000 hospitalizations.[2] TBI is also of growing concern for the U.S. military, including recent conflicts in Iraq and Afghanistan, in which the widespread use of improvised explosive devices (IEDs) has exposed soldiers to increased incidents that can cause TBI.[3] Patients who suffer TBI often develop coagulopathy.[4] Several studies have shown increased mortality in coagulopathic TBI patients compared to non-coagulopathic TBI patients.[5,6,7] Dilutional coagulopathy may occur following concomitant hemorrhage from injuries followed by aggressive crystalloid resuscitation or packed red blood cell (RBC) transfusion that dilute clotting factors and cause platelet dysfunction.[8] Coagulopathy can also develop as the direct result of the brain injury itself and secondary to release of tissue thromboplastin, which activates local clotting factors and promotes cerebral microthrombosis or disseminated intravascular coagulation (DIC), resulting in increased risk of cerebral ischemia.[4,5,9,10,11,12,13] Regardless of its origin, TBI-associated coagulopathy may stimulate bleeding diathesis and preclude rapid neurosurgical intervention and, if left untreated, may exacerbate the primary injury.
Reversal of coagulopathy is necessary to perform safe neurosurgical procedures. Coagulopathy reversal begins as the patient arrives at the hospital and is commonly achieved with fresh frozen plasma (FFP) and vitamin K. FFP, however, must be thawed at the blood bank prior to administration and may take hours to normalize the patient coagulation profile (International Normalized Ratio, INR), creating costly delays in appropriate neurosurgical interventions.[14] Also, the volume of plasma required to correct coagulopathy is unpredictable, resulting in a risk of pulmonary complications due to massive volume over-load.[8]
Optimal management of TBI would be improved by availability of pre-hospital treatments, which in turn may reduce the severity of morbidity of survivors and improve patients’ functional outcome. However, there are no currently approved therapies for arresting intracranial hemorrhage (ICH) or reversing TBI-associated coagulopathy prior to hospital arrival, thus presenting severe TBI patients with substantially increased risk of morbidity and mortality.
Recombinant factor VIIa (rFVIIa) is a Food and Drug Administration (FDA)-approved drug for prevention and treatment of bleeding in patients with hemophilia A and B.[15] rFVIIa has been used “off-label” as a last resort to reverse trauma-associated coagulopathy.[16,17,18,19] The use of rFVIIa in trauma patients has been claimed to be beneficial; however, the supporting data comes largely from anecdotal reports and case scenarios.[20,21,22] In phase II clinical trials, rFVIIa administration for hemorrhage control in blunt and penetrating trauma resulted in significantly reduced red blood cell (RBC) transfusion requirements and a trend towards reduction in mortality and critical complications.[16,23] However, data from the subsequent phase III trial (CONTROL trial) confirmed reduction in blood product use, but did not show reduction in mortality.[24] Additionally, whereas rFVIIa for ICH in phase II clinical trials was shown to be promising, the suggested benefits could not be confirmed in the phase III FAST (The Factor Seven for Acute Hemorrhagic Stroke) trials.[25,26,27] However, rFVIIa administration in these patients did not occur until after the patients had been transported to the hospital.
The early pre-hospital use of rFVIIa in trauma patients with brain injury has not been well studied so far. We hypothesize that early administration of rFVIIa before the development of irreversible brain injury and subsequent coagulopathy will reduce ICH and mitigate secondary brain injury, thus potentially improving neurological outcome compared to current clinical practice of no pre-hospital intervention in a swine model of fluid percussion TBI.
MATERIALS AND METHODS
The experiments reported herein were conducted according to the principles set forth in the “Guide for the Care and Use of Laboratory Animals”, Institute of Laboratory Animals Resources, National Research Council, National Academy Press, 1996. The study was approved by the Naval Medical Research Center/Walter Reed Army Institute of Research Institutional Animal Care and Use Committee (IACUC) and all procedures were performed in an animal facility approved by the Association for Assessment and Accreditation for Laboratory Animal Care International (AAALAC).
Animal preparation and catheter instrumentation
Twenty farm bred male and female immature Yorkshire swine (Animal Biotech Industries, Doylestown, PA) weighing 28.40 ± 0.64 kilogram (kg) (n = 10/group) were fasted for approximately 12 hours prior to the surgical procedure with water provided ad libitum. Anesthesia was induced with an intramuscular injection of ketamine hydrochloride (33 mg/kg) and atropine sulfate (0.05 mg/kg) followed by mask ventilation with isoflourane inhalant (1-5%) delivered in 100% O2 (1.0 FiO2) to facilitate endotracheal intubation. After intubation, the isoflourane concentration was lowered to 2-2.5% to maintain a surgical plane of anesthesia (Instrumentarium Corp., Stockholm, Sweden). Animals were then placed in dorsal recumbence on the operating table, and end-tidal carbon dioxide (ETCO2) was monitored continuously. Animals were ventilated if needed for apnea or ETCO2 >50 milimeters of mercury (mmHg) with a tidal volume of 5-10 mililiter (mL) per kg at a rate of 12-15 breaths per min adjusted to maintain ETCO2 between 35 and 40 mmHg. An intramuscular (IM) injection of 1mL buprenorphine was given 10 minutes before incision for pre-emptive analgesia. A Foley catheter was placed into the bladder via the urethra (female) or directly inserted into the bladder via cut down (male) to monitor urine production. The right external jugular vein and carotid artery were isolated. A 9 French (Fr) introducer sheath was placed in the external jugular vein and a 7.5 Fr pulmonary artery catheter (PAC, Edwards Life Sciences, Irvine, CA) was inserted for continuous hemodynamic monitoring, and sampling of mixed venous blood. An 18-gauge (Ga) angiocatheter (Johnson and Johnson, Somerville, NJ) was placed in the carotid artery for blood pressure monitoring and blood sampling. The right femoral vein was isolated and an 18-Ga angiocatheter was placed in the femoral vein for fluid and drug infusion.
Brain instrumentation
The animal was then placed in prone position. The scalp was incised with electrocautery and the cranium exposed. A 16-mm diameter craniotomy was performed in the right parietal region adjacent to the sagittal suture and anterior to the coronal suture. A T-shaped bolt was screwed into the craniotomy so it abuts the intact dura. The lateral end of the bolt was connected to a high-pressure transducer (Sensym, Sunnyvale, CA), and the transducer was attached to an oscilloscope (EZ Digital Co.LTD, South Korea) for recording force of injury. The top of the bolt was connected to the fluid percussion injury (FPI) device. A second craniotomy was performed in the left frontoparietal region through which a neonatal intraventricular catheter (Phoenix Biomedical Corp.) was placed into the left lateral ventricle and connected to a monitor for measuring intracranial pressure (ICP). A third craniotomy site was prepared just anterior to the anion, for placement of a sagittal sinus catheter for cerebral venous blood sampling. Any cortical bleeding that occurred from this process was controlled with cotton applicators and bone wax (Ethicon, Inc., NJ). All craniotomy sites were sealed with dental cement (3M ESPE, St Paul, MN).
Experimental design
Fluid-percussion TBI was inflicted in standard fashion by pulling back the pendulum and weighted mallet portion of the device at a pre-set level, releasing it, and allowing it to strike the fluid-filled column (start of experiment [T0]). Immediately before infliction of fluid percussion TBI at time 0 (T0) a baseline reading of all parameters was taken. During the next fifteen minutes, animals were monitored and received no treatment. At T15, animals were randomized to receive either a bolus of rFVIIa (90 ug/kg; NovoSeven, Novo Nordisk, Bagsvaerd, Denmark) or no treatment (NON) [Table 1]. Infusion was administered via the catheterized femoral vein. FiO2 was increased from 0.21 to 1.0 between T15 and T30 and then titrated to maintain transcutaneous O2 saturation (SaO2) above 92%. Normal saline (NS) was administered as a maintenance fluid at the rate of 1.5-2 ml/kg/hour by using an infusion pump (Abbott Laboratory, North Chicago, IL) [Table 1].
Table 1.
Experimental timeline

Hospital emergency room arrival and definitive operating room (OR) medical stabilization were simulated at T60 and T75, respectively. Upon simulated hospital arrival (T60), care consistent with emergency department/intensive care unit was implemented as clinically indicated: initial intravenous injection of vecuronium (0.1 mg/kg) was performed at T60 to assist in mechanical ventilation, and additional doses (0.01 mg/kg) were administered every 30 minutes. A maximum of 2 doses of mannitol (1 g/kg) was provided to control intracranial hypertension [ICP >20 mmHg (only if MAP was >40 mmHg)] between T60 and T330. At T360, euthanasia was performed with pentobarbital and phenytoin (Euthasol, 100 mg/kg, IV, Virbac Carros-Cedex, France), and brain tissues were collected upon necropsy.
Data collection
Standard hemodynamic parameters were measured by both invasive and non-invasive monitoring for 360 minutes. Heart rate, respiratory rate, ETCO2, FiO2, SaO2, arterial blood pressures, pulmonary blood pressures, central venous pressure, cardiac output (CO), and ICP were recorded every 5 minutes until hospital arrival (T60) and every 15 minutes, thereafter. Pulmonary artery wedge pressure (PAWP) and urine output (UO) were measured every 15 minutes. CO was measured continuously and cerebral perfusion pressure (CPP) calculated by subtracting ICP from mean arterial pressure (MAP). Blood gas content (arterial, mixed venous and sagittal sinus) was analyzed by using an automatic analyzer (ABL 705, Radiometer, Copenhagen, Denmark) every 15 minutes until T60, then every 60 minutes, thereafter. Blood samples were also taken at T0, T15, T60, and T360 for in vitro assessments. Complete CBC (Pentra 60C+ cell counter, HORIBA ABX Diagnostics, Irvine, CA), thromboelastography (TEG) (TEG 5000 Hemostasis Analyzer, Haemoscope Corp., Niles, IL), coagulation parameters (Stat Compact, Diagnostica Stago, Parsippany, NJ), and thrombin-antithrombin (TAT) (Enzygnost, Dade Behring, Stuart, FL) were determined. The level of rFVIIa in blood samples was determined using a factor VIIa kit (Sta-FVIIa, Diagnostica Stago).
Histopathology
Brain was harvested and sections were immersion fixed in 10% buffered formalin. After a minimum of 48 hours fixation time, blocks of tissue from the injured right parietal cortex, left parietal cortex, cerebellum, hippocampus, pons, and right thalamus were processed for routine paraffin embedding, sectioned and mounted on slides for light microscopic evaluation. Brain sections were evaluated via light microscopy by a board-certified veterinary pathologist blinded to treatment group. Tissue changes were identified and semiquantitatively graded. Semi-quantitative assessment of neutrophil infiltration, hemorrhage, and neuronal necrosis was made. Neutrophil infiltration and hemorrhage were scored 0 to 4 considering distribution (confined to meninges or neutropil involvement) and severity. Neuronal necrosis was scored 0 to 4 by assessing the number of fields that contained 5 or more necrotic neurons per high-powered field [Table 2].
Table 2.
Brain histologic semiquantitative scoring

To assess for the presence of microthrombi, sections of the right and left cerebrum at the injury site and the right hippocampus were routinely stained with phosphotungstic acid and hematooxylin (PTAH) for the presence of fibrin. Fibrin formation within vessels was scored 0 to 4 with the range of 1 assigned for 1-2 microthrombi and 4 assigned for greater than 10 microthrombi per section [Table 2].
Immuno-histochemistry
Microtubule-associated protein (MAP)2 localization
Brain sections were formalin fixed, paraffin embedded, and mounted on negatively charged microscope slides and then deparaffinized using xylene followed by graded baths of ethanol. A DAKO Autostainer Plus Universal Staining System (DAKO, Carpenteria, CA) was used for a portion of the immunohistochemical staining process.
Immunohistochemical detection of MAP2 was performed on sections of formalin fixed, paraffin embedded blocks of swine cerebrum and cerebellum. Antigen retrieval was performed using citra (Biogenex, San Ramon, CA) in a pressure cooker for approximately 40 minutes. Mouse monoclonal MAP2 antibody, Ab7756, (Abcam, Cambridge, MA) was used at a dilution of 1:250 and incubated overnight at 4°C. Labeled streptavidin-biotin 2 (LSAB2) link-label goat anti-mouse was applied as secondary antibody per manufacturer's instruction for 30 minutes at room temperature. The chromogen applied was 3,3′ diaminobenzidine (DAKO, Carpenteria, CA) for 10 minutes. The sections were counterstained with hematoxylin and then cover-slipped. Similar brain sections between the treatment groups were immunostained according to the method described. Areas of neuronal loss were outlined and measured with Image Pro Plus (Media Cybernetics, Bethesda, MD) software. Total areas of loss were determined for right cerebrum, left cerebrum, and right hippocampus. Negative controls were available from a tissue library and were used to identify non-specific background staining.
Flouro-Jade B localization
Formalin fixed, paraffin embedded brain sections were cut 4 μm thick, mounted with distilled water onto gelatin coated slides and dried on a slide warmer at 45°C. Tissues were then fully hydrated. Slides were transferred to a solution of 0.06% potassium permanganate for 15 minutes and gently shaken on a rotating platform. Under reduced lighting conditions, slides were rinsed for 1 minute in distilled water and were then transferred to the Flouro-Jade B at a 0.0004% concentration. After staining, sections were rinsed thrice with 1-minute changes of distilled water. Excess water was drained and slides were rapidly dried on a slide warmer. When dry, slides were immersed in xylene and then cover-slipped with distyrene plasticizer and xylene (DPX) mounting media, (VWR International Ltd, Poole, UK). Sections were examined with an epifluorescence microscope using a filter system suitable for visualizing fluorescein (Excitation range: 465-495 nm) (Nikon Eclipse 800).
Statistical analysis
Pre-study power analysis for mixed models indicated a sample size of 10 per group would provide sufficient power (i.e., 80%) to detect significant group differences of 50% or greater (alpha = 0.05). T-test was used to compare differences between the rFVIIa and NON-groups at specific time points and between certain time intervals with respect to hemodynamic and other measurements that were continuous in nature. When the data distribution did not appear normal, the Mann-Whitney U-test was used. Chi-square tests were used to compare between group differences in histopathologic measures. P values of less than or equal to 0.05 (two-sided) were considered statistically significant.
RESULTS
Hemodynamics
There were no significant group differences in the baseline physiological parameters. Survival rate at 6 hours was 100% in both groups. CO, PAWP and UO were similar between the groups. As a response to TBI, ICP rapidly increased in both groups and remained elevated above the baseline during the entire experiment [Figure 1a]. Following the brain injury, overall decline in systemic blood pressure was continuous and persistent in both groups without any significant difference (P = 0.056) [Figure 1b]. The rFVIIa group demonstrated less decline in MAP between T60 and T180 (P = 0.0001) compared to the NON-group [Figure 1b]. There was no statistically significant difference in CPP over time between the groups (P = 0.2589) [Figure 1c]. There was no evidence of pulmonary hypertension in either group as shown by consistent mean pulmonary arterial pressure (MPAP) with minimal deviation from the baseline and similarity in both groups [Figure 1d].
Figure 1.
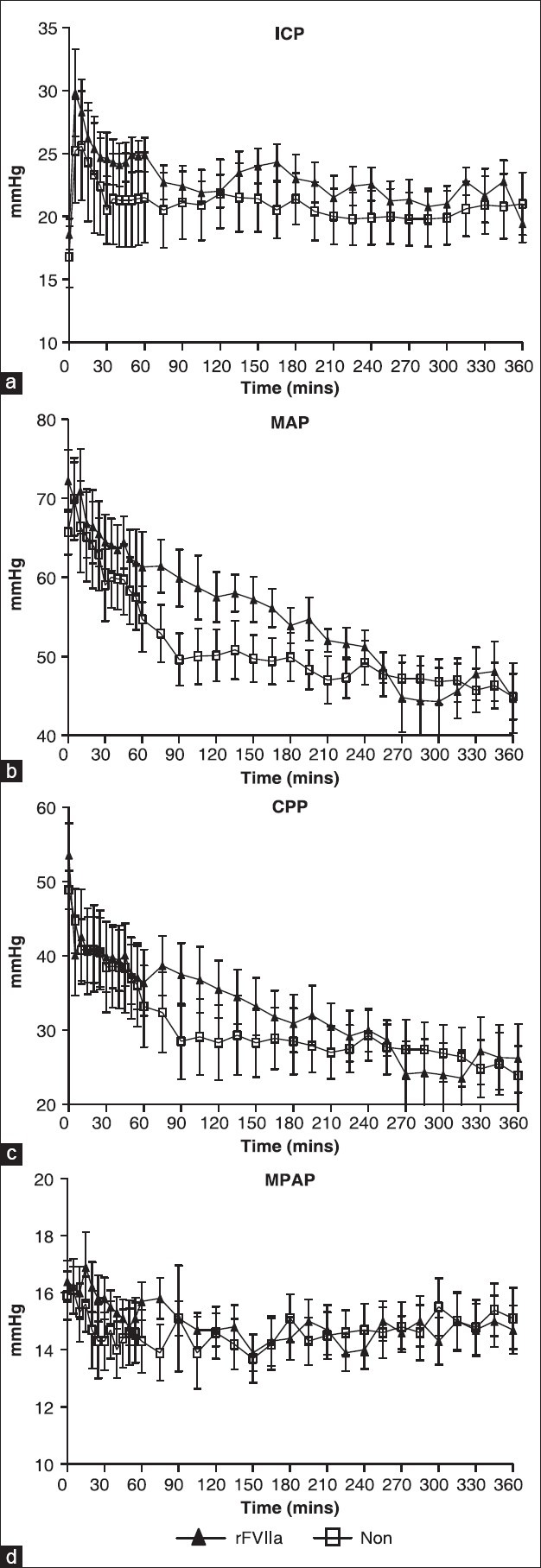
Hemodynamics. Comparison between rFVIIa and controls (NON). (a) Intracranial pressure (ICP), (b) mean arterial pressure (MAP), (c) cerebral perfusion pressure (CCP), (d) mean pulmonary arterial pressure (MPAP). Note differing scales on Y-axes
Hematology and blood sample analysis
There were no statistically significant group differences in measurements of blood gas level and electrolyte concentration in arterial, mixed venous, or sagittal venous blood [Table 3]. However, the rFVIIa group maintained base excess, bicarbonate, and pH at slightly higher levels than the NON-group [Table 3]. There was an initial reduction in sagittal venous blood pH at T15, but it was soon normalized and tended to be higher in rFVIIa group. The lactate level did not differ between the groups [Table 3]. The intravascular rFVIIa level peaked shortly after injection; at T60, levels were 58.6 ± 11.6 μg/ml, which was significantly higher than T0 value (P = 3.23 × 10−10). rFVIIa was barely detectable after 6 hours (4.8 ± 2.7 μg/ml,) due to its short half-life of rFVIIa. This correlates with the significant reduction of prothrombin time (PT) at T60 compared to T0 in the rFVIIa group (P = 2.44 × 10−10) [Figure 2a]. There were no differences in PT at different time points in the NON-group but their T60 and T360 PT values are significantly different than T60 and T360 PT values of the rFVIIa group (P = 1.34 × 10−9 for T60, P = 3.12 × 10−8 for T360). There were no time or group differences for platelets [Figure 2b] and Thromboelastography reaction time (TEG-R) [Figure 2c]. There was a significant increase in TAT value at T15 compared to T0 in both the groups (P = 0.0004678) and TAT value remained elevated in both the groups throughout the experiment [Figure 2d].
Table 3.
Blood gas analysis
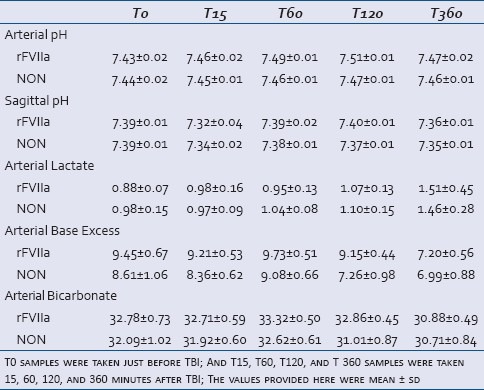
Figure 2.
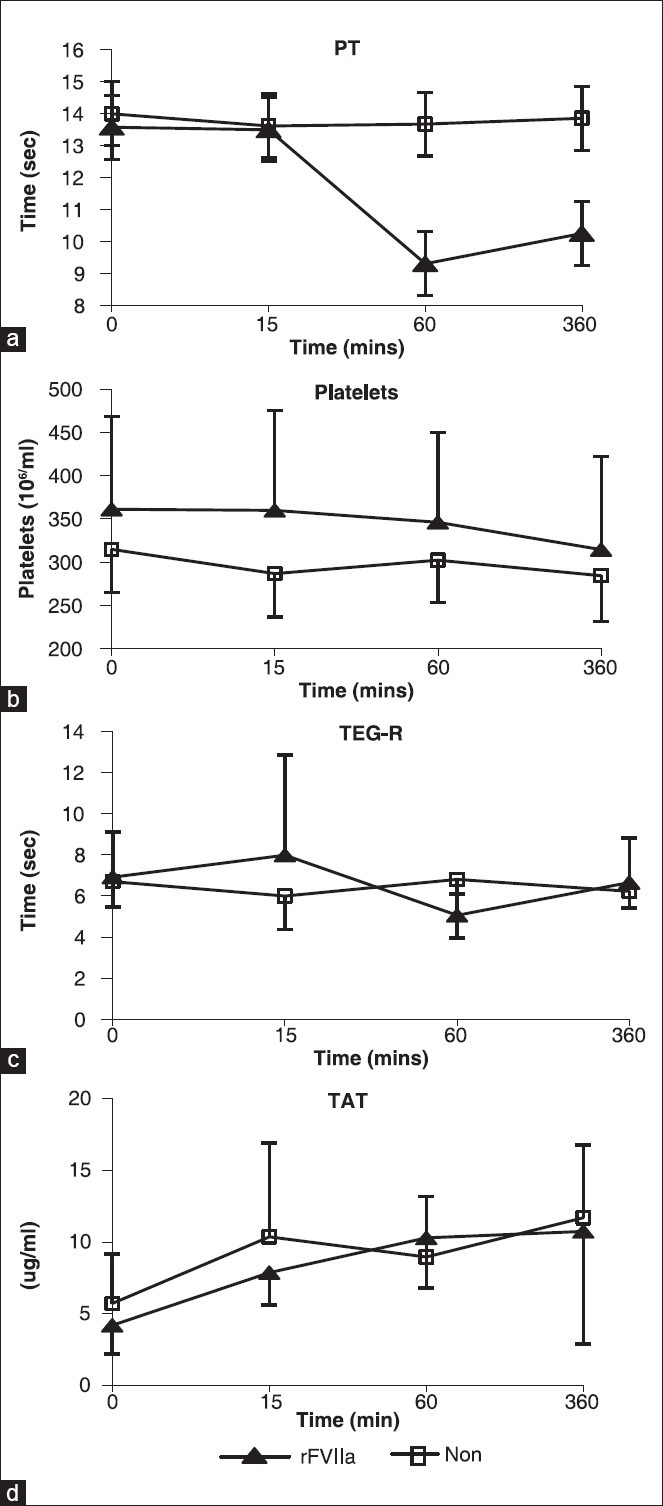
Coagulation parameters. (a) Prothrombin time (PT), (b) platelet count, (c) thromboelastography reaction time (TEG-R), (d) thrombin-antithrombin (TAT)
Histopathology
In the right frontoparietal cortex, the site of brain injury, the severity of hemorrhage in the subarachnoid space was significantly lower in the rFVIIa group (P = 0.0037) [Figure 3a]. There were no statistically significant differences in subarachnoid hemorrhage scores in the left cortex, pons, or cerebellum. The severity of hemorrhage in both groups was equally greater in cerebellum and pons than in contralateral cortex [Figure 3a]. There was neutrophil (polymorphonuclear leukocytes, (PMN)) infiltration in both right and left cortex, and there were no differences in perivascular or parenchymal neutrophil infiltration between the treatment groups [Figure 3b]. There was no difference in the severity scores of microscopic cerebral hemorrhage in the right cortex, right thalamus, pons, and cerebellum between the treatment groups; however, examination of the left cortex showed a statistically significant reduction in cerebral hemorrhage scores in the rFVIIa group (P = 0.03) [Figure 3c]. Varying degrees of neuro-degeneration and necrosis were observed across different regions of the brain with right cortex and right thalamus presenting the highest severity score without any group difference [Figure 4a]. Although mild in severity, left hippocampal necrosis was detected in the NON-group, whereas in the rFVIIa group, no necrosis was found in this area (P = 0.00009). Necrosis in pons and cerebellum were found in both groups; however, the rFVIIa group demonstrated less neuro-degeneration than the NON-group (P = 0.00008 and P = 0.0251, respectively). Intravascular microthrombosis was observed in both groups; however, only five animals were affected. In each of these animals, only one section was affected and none had a severity greater than two. Additionally, the presence of microthrombi was not significantly different between the groups (P = 0.71).
Figure 3.
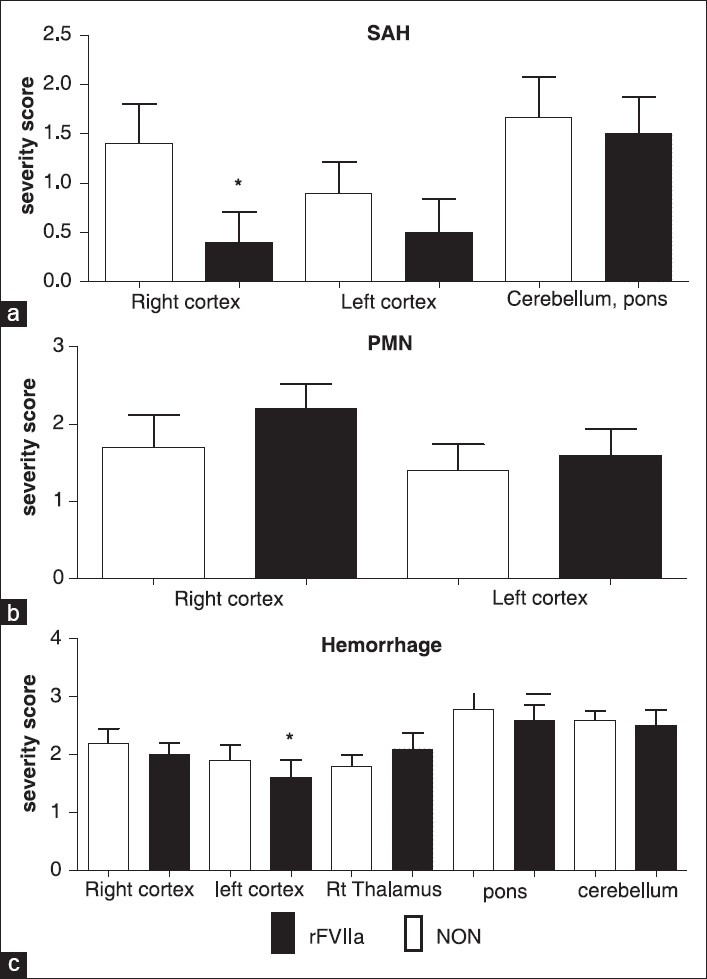
Cerebral Hemorrhage. (a) Subarachnoid hemorrhage severity scores (SAH), (b) polymorphonuclear neutrophil infiltration severity scores (PMN), (c) meningeal/neuropil hemorrhage severity scores (hemorrhage). See Table 2 for definitions of score values. Hemorrhage scores (2.c.) were subdivided by brain regions. * indicates the value was significantly different between the groups (P < 0.05)
Figure 4.
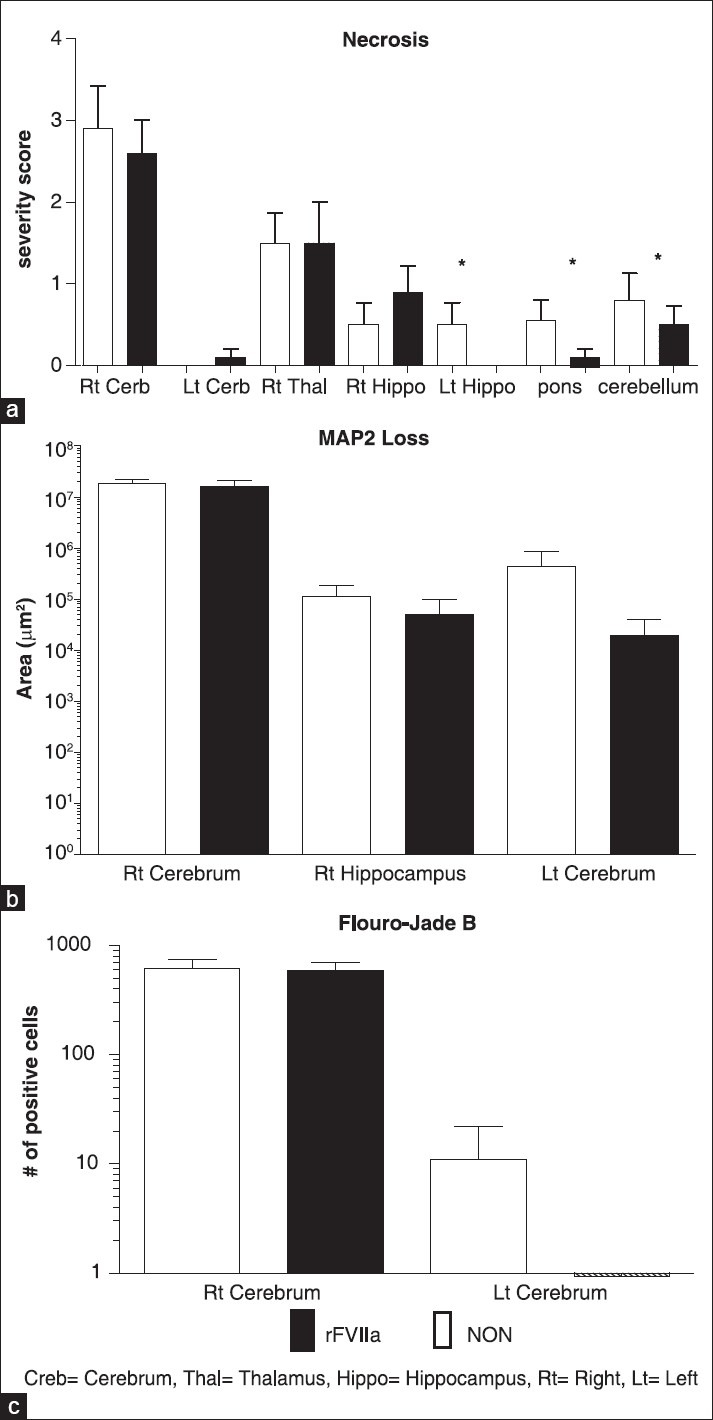
Pathologic evidence of cell damage by brain region. (a) severity score of neuronal necrosis, (b) MAP2 loss, (c) Fluoro- Jade B positive cell count. Creb = Cerebrum, Thal = Thalamus, Hippo = Hippocampus, Rt = Right, Lt = Left. * Indicates the value was significantly different between the groups (P < 0.05)
MAP2 reactivity
The data from immunohistochemical staining of porcine cerebrum demonstrated evidence of MAP2 immunoreactivity in all gray matter areas (cortex, thalamus, and hippocampus), but absence in white matter areas with the exception of neuronal cells in crossover areas. In gray matter areas, varying degrees of intracytoplasmic reactivity in neuronal cells and their axons was observed. Although, MAP2 loss showed no statistically significant regional differences (P = 0.66 right cerebrum, P = 0.49 right hippocampus, P = 0.32 left cerebrum), overall MAP2 loss was less in the rFVIIa group (157,761,876 μm2 for rFVIIa and 188,347,887 μm2 for NON) [Figure 4b].
Fluoro-jade B localization
Fluoro-Jade B positive cells were generally localized in the region of MAP2 loss. As with MAP2 loss, the total number of fluoro-Jade B positive cells was greater in the NON-group than in the rFVIIa group (6,359 for NON and 1,435 for FVIIa). Of the two examined regions, the right cerebrum showed the greatest number of fluoro-Jade B positive cells, followed by the left cerebrum. There were no positive cells in the left cerebrum of the rFVIIa group [Figure 4c].
DISCUSSION
In this study, we tested the efficacy of rFVIIa as a pre-hospital treatment in a swine moderate TBI model. While there is no equivalent to human TBI severity scales, it has been shown by multiple investigators[28,29] that a fluid percussion injury force of 2.5-3 atmospheres in swine reliably reproduces an injury consisting of physiological responses (apnea, transient hypertension, elevated ICP, etc.), histological evidence of significant injury (hemorrhage, axonal damage, necrosis, etc.) while not compromising short-term survival. The injury in this study produced a trend towards hypotension in both experimental groups. Hypotension can eventually be associated with poor outcome in patients with closed head injuries.[30] The underlying mechanisms which may explain this phenomenon have been explored by several investigators.[31] In one model, it is suggested that fluid percussion TBI may have extended to medulla and hypothalamus which very much contributed to the loss of blood pressure regulation.[32] Another study indicates an elevated level of catecholamines that is proportional to the severity of the brain injury.[33] Thus, it is hypothesized that sustained level of catecholamines may cause hypotension which is mainly cardiogenic in nature by decreased myocardial contractility.
In this study, the NON-group tended to be more hypotensive than the rFVIIa group. This difference was most evident between T60 (early hospital phase) and T180 and then tapered off towards the end of the experiment. The transient improvement in MAP due to the administration of rFVIIa did not affect survivability, and the mechanism by which rFVIIa causes transient improvement in MAP in this TBI model remains uncertain. However, rFVIIa had a pronounced effect on the severity of subarachnoid hemorrhage: the lower severity of subarachnoid hemorrhage in the right cortex despite similar ICP and CPP was significant. The dose used in this study (90 μg/kg) rFVIIa had a hemostatic effect as measured by PT, but there were no differences in thrombotic complications observed between the groups. This aligns with previously reported studies in hemorrhagic shock.[34] Clinically significant intravascular coagulation events usually arise concomitantly with TBI and are serious contributing factors to increased mortality and morbidity. This has been shown in several animal studies.[9,10,13] However, although it is a common belief that intravascular coagulation may follow non-fatal TBI, our pathological finding indicated negligible microthrombotic incidence and severity which may not be clinically relevant in this 6 hours of experimental observation period. This may be due to the specific method of creating mild/moderate fluid percussion injury that may not have been severe enough to cause significant formation of microclots. More importantly, despite the use of the pro-coagulant rFVIIa, the treated animals did not manifest a more severe level of microthrombosis (vs. NON). In a similar study, the use of rFVIIa at 720 μg/kg, 8 times the dosage used in our study, in a swine model of diffuse axonal TBI did not show significant difference in thromboembolic events between the control vehicle and the rFVIIa group.[35]
One of the problems neurosurgeons encounter in management of TBI is reversal of coagulopathy to surgically optimize patients. Infusion of rFVIIa has been tried as alternative or adjunct to infusion of FFP, the standard method of reversing coagulopathy. The use of rFVIIa in severe TBI patients resulted in substantial reduction in time to neurosurgical intervention and amount of FFP used (114 vs. 446 minutes, P = 0.0003 and 2 vs. 6 units of FFP, P = 0.0006, respectively).[8] More rapid reversal of coagulopathy directly translates into faster neurosurgical intervention which can make profound differences in patients’ functional and neurologic outcome. There are also a number of reports of successful use of rFVIIa as a coagulopathy reversal agent without any adverse involvement of major thrombotic complications such as deep vein thrombosis and thromboembolism.[8,36,37,38]
These findings suggest that the early use of rFVIIa in the pre-hospital setting may be beneficial to TBI patients with coagulopathy because it effectively and rapidly lowers INR without eliciting adverse clinical thrombotic complications. This may provide survival benefits to TBI patients who are in immediate need of surgical evacuation of an expanding hematoma by enabling faster time to emergent craniotomy.
Besides its hemostatic properties that halt active bleeding and rapidly reverse coagulopathy, rFVIIa has also been shown to have neuroprotective effects. In a clinically relevant pig double injury model Zhang et al., evaluated the safety and efficacy of rFVIIa for traumatic brain injury (cerebral contusion and diffuse axonal injury) and subsequent coagulopathy. The administration of rFVIIa five minutes post-injury resulted in reduced expansion of contusion volume and slower progression of hippocampal neuronal and axonal necrosis, but without exacerbating the severity of microthrombosis.[35] This report is consistent with our histopathological finding that the rFVIIa group animals showed a substantial reduction in neuronal necrosis in the left hippocampus, pons, and cerebellum. The hippocampus is an area of the brain that is selectively vulnerable to hypoxia,[39] and damage to hippocampus due to necrosis or hypoxia often results in memory loss and disorientation.[40] We observed that overall MAP2 loss and fluoro-Jade B positive cells in the right and left cortices and hippocampus were reduced in the animals receiving rFVIIa. MAP2, a principle cross-linking protein component of microtubules that is predominantly confined to the somatodendritic region, is associated with a number of neuronal functions including neuronal morphogenesis and neuronal structure.[41] Therefore, a significant decrease in MAP2 level may indicate neuronal damage. Fluoro-Jade stain selectively labels degenerating neurons. A few studies have observed decreased levels of MAP2 following TBI-induced global cerebral ischemia in rodents,[42,43,44] and in one study, a decreased level of hippocampal MAP2 was reported immediately following TBI.[45] The degree of MAP2 loss in our experiment was most severe in the right cortex near the injury site which is consistent with the data reported in the literature.[46] However, MAP2 loss in other areas of the brain was minimal, indicating that MAP2 loss is confined to the injury area. Although, the mechanism of rFVIIa in mitigating MAP2 loss near the site of injury is unknown, it may play an important neuroprotective role in addition to its conventionally known hemostatic role. Overall, the exact neuroprotective role of rFVIIa is not identified; but, the findings suggest that the early use of rFVIIa may be beneficial to combat the progression of rapid neuronal pathogenesis.
CONCLUSION
In this study, we have explored the use of rFVIIa in the pre-hospital TBI setting and demonstrated a trend towards improvement in maintenance of MAP, significant reduction in subarachnoid hemorrhage (SAH) severity scores and a benefit to brain tissue integrity on histopathologic examination, but with no discernible survival benefit or improvements in ICP or CPP. Future recovery and behavioral studies including uncontrolled hemorrhage may show a long-term survival benefit.
DISCLAIMERS
The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, or the U.S. Government. I am a military service member (DF) or employee of the U.S. Government (TS, RMc). This work was prepared as part of my official duties. Title 17 U.S.C. §105 provides that ‘Copyright protection under this title is not available for any work of the United States Government.’ Title 17 U.S.C. §101 defines a U.S. Government work as a work prepared by a military service member or employee of the U.S. Government as part of that person's official duties.
ACKNOWLEDGMENTS
We thank our NeuroTrauma Department laboratory technicians (Noemy Carballo, Eileen Sagini, Rolli Pessu, and Michael Hammett) for their support and assistance during the study and the personnel at the Department of Veterinary Medicine for their help in facilitating the animal surgery.
Footnotes
Source of Support: This work was supported by BUMED Core Capability Funding Work Unit Number 602236N.M04426. B26.A0241.
Conflict of Interest: None declared.
REFERENCES
- 1.Shackford SR, Mackersie RC, Holbrook TL, Davis JW, Hollingsworth-Fridlund P, Hoyt DB, et al. The epidemiology of traumatic death. A population-based analysis. Arch Surg. 1993;128:571–5. doi: 10.1001/archsurg.1993.01420170107016. [DOI] [PubMed] [Google Scholar]
- 2.Langlois J, Rutland-Brown W, Thomas K. Traumatic brain injury in the united states: Emergency department visits, hospitalizations, and deaths. Centers for Disease Control and Prevention. 2004. p. 60. Available. http://www.cdc.gov/ncipc/pub-res/TBI_in_US_04/TBI-USA_Book-Oct1.pdf .
- 3.Galarneau MR, Woodruff SI, Dye JL, Mohrle CR, Wade AL. Traumatic brain injury during operation Iraqi freedom: Findings from the united states navy-marine corps combat trauma registry. J Neurosurg. 2008;108:950–7. doi: 10.3171/JNS/2008/108/5/0950. [DOI] [PubMed] [Google Scholar]
- 4.Cortiana M, Zagara G, Fava S, Seveso M. Coagulation abnormalities in patients with head injury. J Neurosurg Sci. 1986;30:133–8. [PubMed] [Google Scholar]
- 5.Hulka F, Mullins RJ, Frank EH. Blunt brain injury activates the coagulation process. Arch Surg. 1996;131:923–7. doi: 10.1001/archsurg.1996.01430210021004. [DOI] [PubMed] [Google Scholar]
- 6.Carrick MM, Tyroch AH, Youens CA, Handley T. Subsequent development of thrombocytopenia and coagulopathy in moderate and severe head injury: Support for serial laboratory examination. J Trauma. 2005;58:725–9. doi: 10.1097/01.ta.0000159249.68363.78. [DOI] [PubMed] [Google Scholar]
- 7.Zygun DA, Kortbeek JB, Fick GH, Laupland KB, Doig CJ. Non-neurologic organ dysfunction in severe traumatic brain injury. Crit Care Med. 2005;33:654–60. doi: 10.1097/01.ccm.0000155911.01844.54. [DOI] [PubMed] [Google Scholar]
- 8.Stein DM, Dutton RP, Kramer ME, Handley C, Scalea TM. Recombinant factor VIIa: Decreasing time to intervention in coagulopathic patients with severe traumatic brain injury. J Trauma. 2008;64:620–7. doi: 10.1097/TA.0b013e3181650fc7. [DOI] [PubMed] [Google Scholar]
- 9.Dietrich WD, Alonso O, Halley M. Early microvascular and neuronal consequences of traumatic brain injury: A light and electron microscopic study in rats. J Neurotrauma. 1994;11:289–301. doi: 10.1089/neu.1994.11.289. [DOI] [PubMed] [Google Scholar]
- 10.Stein SC, Chen XH, Sinson GP, Smith DH. Intravascular coagulation: A major secondary insult in nonfatal traumatic brain injury. J Neurosurg. 2002;97:1373–7. doi: 10.3171/jns.2002.97.6.1373. [DOI] [PubMed] [Google Scholar]
- 11.Stein SC, Smith DH. Coagulopathy in traumatic brain injury. Neurocrit Care. 2004;1:479–88. doi: 10.1385/NCC:1:4:479. [DOI] [PubMed] [Google Scholar]
- 12.Zehtabchi S, Soghoian S, Liu Y, Carmody K, Shah L, Whittaker B, et al. The association of coagulopathy and traumatic brain injury in patients with isolated head injury. Resuscitation. 2008;76:52–6. doi: 10.1016/j.resuscitation.2007.06.024. [DOI] [PubMed] [Google Scholar]
- 13.Schwarzmaier SM, Kim SW, Trabold R, Plesnila N. Temporal profile of thrombogenesis in the cerebral microcirculation after traumatic brain injury in mice. J Neurotrauma. 2010;27:121–30. doi: 10.1089/neu.2009.1114. [DOI] [PubMed] [Google Scholar]
- 14.Goldstein JN, Thomas SH, Frontiero V, Joseph A, Engel C, Snider R, et al. Timing of fresh frozen plasma administration and rapid correction of coagulopathy in warfarin-related intracerebral hemorrhage. Stroke. 2006;37:151–5. doi: 10.1161/01.STR.0000195047.21562.23. [DOI] [PubMed] [Google Scholar]
- 15.Weiskopf RB. Recombinant-activated coagulation factor VIIa (Novoseven): Current development. Vox Sang. 2007;92:281–8. doi: 10.1111/j.1423-0410.2006.00888.x. [DOI] [PubMed] [Google Scholar]
- 16.Boffard KD, Riou B, Warren B, Choong PI, Rizoli S, Rossaint R, et al. Recombinant factor VIIa as adjunctive therapy for bleeding control in severely injured trauma patients: Two parallel randomized, placebo-controlled, double-blind clinical trials. J Trauma. 2005;59:8–15. doi: 10.1097/01.ta.0000171453.37949.b7. [DOI] [PubMed] [Google Scholar]
- 17.Dutton RP, McCunn M, Hyder M, D’Angelo M, O’Connor J, Hess JR, et al. Factor VIIa for correction of traumatic coagulopathy. J Trauma. 2004;57:709–18. doi: 10.1097/01.ta.0000140646.66852.ab. [DOI] [PubMed] [Google Scholar]
- 18.Harrison TD, Laskosky J, Jazaeri O, Pasquale MD, Cipolle M. “Low-dose” recombinant activated factor VII results in less blood and blood product use in traumatic hemorrhage. J Trauma. 2005;59:150–4. doi: 10.1097/01.ta.0000171470.39742.8e. [DOI] [PubMed] [Google Scholar]
- 19.Rizoli SB, Nascimento B, Jr, Osman F, Netto FS, Kiss A, Callum J, et al. Recombinant activated coagulation factor VII and bleeding trauma patients. J Trauma. 2006;61:1419–25. doi: 10.1097/01.ta.0000243045.56579.74. [DOI] [PubMed] [Google Scholar]
- 20.O’Neill PA, Bluth M, Gloster ES, Wali D, Priovolos S, DiMaio TM, et al. Successful use of recombinant activated factor VII for trauma-associated hemorrhage in a patient without preexisting coagulopathy. J Trauma. 2002;52:400–5. doi: 10.1097/00005373-200202000-00034. [DOI] [PubMed] [Google Scholar]
- 21.Kenet G, Walden R, Eldad A, Martinowitz U. Treatment of traumatic bleeding with recombinant factor VIIa. Lancet. 1999;354:1879. doi: 10.1016/S0140-6736(99)05155-7. [DOI] [PubMed] [Google Scholar]
- 22.Shander A, Goodnough LT, Ratko T, Matuszewski KA, Cohn SM, Diringer M, et al. Consensus recommendations for the off-label use of recombinant human factor VIIa (novoseven®) therapy. Pharm Ther. 2005;30:644–58. [Google Scholar]
- 23.Geeraedts LM, Jr, Kamphuisen PW, Kaasjager HA, Verwiel JM, van Vugt AB, Frolke JP. The role of recombinant factor VIIa in the treatment of life-threatening haemorrhage in blunt trauma. Injury. 2005;36:495–500. doi: 10.1016/j.injury.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 24.Hauser CJ, Boffard K, Dutton R, Bernard GR, Croce MA, Holcomb JB, et al. Results of the control trial: Efficacy and safety of recombinant activated factor vii in the management of refractory traumatic hemorrhage. J Trauma. 2010;69:489–500. doi: 10.1097/TA.0b013e3181edf36e. [DOI] [PubMed] [Google Scholar]
- 25.Diringer MN, Skolnick BE, Mayer SA, Steiner T, Davis SM, Brun NC, et al. Risk of thromboembolic events in controlled trials of rFVIIa in spontaneous intracerebral hemorrhage. Stroke. 2008;39:850–6. doi: 10.1161/STROKEAHA.107.493601. [DOI] [PubMed] [Google Scholar]
- 26.Mayer SA, Brun NC, Begtrup K, Broderick J, Davis S, Diringer MN, et al. Efficacy and safety of recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med. 2008;358:2127–37. doi: 10.1056/NEJMoa0707534. [DOI] [PubMed] [Google Scholar]
- 27.Mayer SA, Brun NC, Broderick J, Davis SM, Diringer MN, Skolnick BE, et al. Recombinant activated factor VII for acute intracerebral hemorrhage: Us phase IIA trial. Neurocrit Care. 2006;4:206–14. doi: 10.1385/NCC:4:3:206. [DOI] [PubMed] [Google Scholar]
- 28.Stern S, Rice J, Philbin N, McGwin G, Arnaud F, Johnson T, et al. Resuscitation with the hemoglobin-based oxygen carrier, HBOC-201, in a swine model of severe uncontrolled hemorrhage and traumatic brain injury. Shock. 2009;31:64–79. doi: 10.1097/SHK.0b013e3181778dc3. [DOI] [PubMed] [Google Scholar]
- 29.King DR, Cohn SM, Proctor KG. Resuscitation with a hemoglobin-based oxygen carrier after traumatic brain injury. J Trauma. 2005;59:553–60. [PubMed] [Google Scholar]
- 30.Manley G, Knudson MM, Morabito D, Damron S, Erickson V, Pitts L. Hypotension, hypoxia, and head injury: Frequency, duration, and consequences. Arch Surg. 2001;136:1118–23. doi: 10.1001/archsurg.136.10.1118. [DOI] [PubMed] [Google Scholar]
- 31.Fuller G, Hasler R, Mealing N, Lawrence T, Woodford M, Juni P, et al. The association between admission systolic blood pressure and mortality in significant traumatic brain injury: A multi-centre cohort study. Injury. 2013 doi: 10.1016/j.injury.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 32.Fulton RL, Flynn WJ, Mancino M, Bowles D, Cryer HM. Brain injury causes loss of cardiovascular response to hemorrhagic shock. J Invest Surg. 1993;6:117–31. doi: 10.3109/08941939309141603. [DOI] [PubMed] [Google Scholar]
- 33.Rosner MJ, Newsome HH, Becker DP. Mechanical brain injury: The sympathoadrenal response. J Neurosurg. 1984;61:76–86. doi: 10.3171/jns.1984.61.1.0076. [DOI] [PubMed] [Google Scholar]
- 34.Arnaud F, Hammett M, Philbin N, Scultetus A, McCarron R, Freilich D. Hematologic effects of recombinant factor VIIa combined with hemoglobin-based oxygen carrier-201 for prehospital resuscitation of swine with severe uncontrolled hemorrhage due to liver injury. Blood Coagul Fibrinolysis. 2008;19:669–77. doi: 10.1097/MBC.0b013e3283089198. [DOI] [PubMed] [Google Scholar]
- 35.Zhang J, Groff RF, 4th, Chen XH, Browne KD, Huang J, Schwartz ED, et al. Hemostatic and neuroprotective effects of human recombinant activated factor VII therapy after traumatic brain injury in pigs. Exp Neurol. 2008;210:645–55. doi: 10.1016/j.expneurol.2007.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cohen MJ, Brohi K, Ganter MT, Manley GT, Mackersie RC, Pittet JF. Early coagulopathy after traumatic brain injury: The role of hypoperfusion and the protein c pathway. J Trauma. 2007;63:1254–61. doi: 10.1097/TA.0b013e318156ee4c. [DOI] [PubMed] [Google Scholar]
- 37.Roitberg B, Emechebe-Kennedy O, Amin-Hanjani S, Mucksavage J, Tesoro E. Human recombinant factor VII for emergency reversal of coagulopathy in neurosurgical patients: A retrospective comparative study. Neurosurgery. 2005;57:832–6. doi: 10.1227/01.neu.0000180816.80626.c2. [DOI] [PubMed] [Google Scholar]
- 38.Yusim Y, Perel A, Berkenstadt H, Attia M, Knoller N, Sidi A. The use of recombinant factor VIIa (novoseven) for treatment of active or impending bleeding in brain injury: Broadening the indications. J Clin Anesth. 2006;18:545–51. doi: 10.1016/j.jclinane.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 39.Collins RC. Selective vulnerability of brain: New insights from the excitatory synapse. Metab Brain Dis. 1986;1:231–40. doi: 10.1007/BF00999353. [DOI] [PubMed] [Google Scholar]
- 40.Luzzi S, Pucci E, Di Bella P, Piccirilli M. Topographical disorientation consequent to amnesia of spatial location in a patient with right parahippocampal damage. Cortex. 2000;36:427–34. doi: 10.1016/s0010-9452(08)70851-7. [DOI] [PubMed] [Google Scholar]
- 41.Tucker RP. The roles of microtubule-associated proteins in brain morphogenesis: A review. Brain Res Brain Res Rev. 1990;15:101–20. doi: 10.1016/0165-0173(90)90013-e. [DOI] [PubMed] [Google Scholar]
- 42.Inuzuka T, Tamura A, Sato S, Kirino T, Toyoshima I, Miyatake T. Suppressive effect of E-64c on ischemic degradation of cerebral proteins following occlusion of the middle cerebral artery in rats. Brain Res. 1990;526:177–9. doi: 10.1016/0006-8993(90)90269-h. [DOI] [PubMed] [Google Scholar]
- 43.Inuzuka T, Tamura A, Sato S, Kirino T, Yanagisawa K, Toyoshima I, et al. Changes in the concentrations of cerebral proteins following occlusion of the middle cerebral artery in rats. Stroke. 1990;21:917–22. doi: 10.1161/01.str.21.6.917. [DOI] [PubMed] [Google Scholar]
- 44.Kitagawa K, Matsumoto M, Niinobe M, Mikoshiba K, Hata R, Ueda H, et al. Microtubule-associated protein 2 as a sensitive marker for cerebral ischemic damage--immunohistochemical investigation of dendritic damage. Neuroscience. 1989;31:401–11. doi: 10.1016/0306-4522(89)90383-7. [DOI] [PubMed] [Google Scholar]
- 45.Taft WC, Yang K, Dixon CE, Hayes RL. Microtubule-associated protein 2 levels decrease in hippocampus following traumatic brain injury. J Neurotrauma. 1992;9:281–90. doi: 10.1089/neu.1992.9.281. [DOI] [PubMed] [Google Scholar]
- 46.Posmantur RM, Kampfl A, Taft WC, Bhattacharjee M, Dixon CE, Bao J, et al. Diminished microtubule-associated protein 2 (map2) immunoreactivity following cortical impact brain injury. J Neurotrauma. 1996;13:125–37. doi: 10.1089/neu.1996.13.125. [DOI] [PubMed] [Google Scholar]