

Abstract
Glia account for 90% of human brain cells and have a significant role in brain homeostasis. Thus, specific in vivo imaging markers of glial metabolism are potentially valuable. In the brain, 2-fluoroacetate is selectively taken up by glial cells and becomes metabolically trapped in the tricarboxylic acid cycle. Recent work in rodent brain injury models demonstrated elevated lesion uptake of 2-[18F]fluoroacetate ([18F]FACE), suggesting possible use for specifically imaging glial metabolism. To assess this hypothesis, we evaluated [18F]FACE kinetics in rodent models of cerebral hypoxia–ischemia at 3 and 24 hours post insult. Lesion uptake was significantly higher at 30 minutes post injection (P<0.05). An image-based method for input function estimation using cardiac blood was validated. Analysis of whole blood showed no significant metabolites and plasma activity concentrations of ∼50% that of whole blood. Kinetic models describing [18F]FACE uptake were developed and quantitatively compared. Elevated [18F]FACE uptake was found to be driven primarily by K1/k2 rather than k3, but changes in the latter were detectable. The two-tissue irreversible uptake model (2T3k) was found to be necessary and sufficient for modeling [18F]FACE uptake. We conclude that kinetic modeling of [18F]FACE uptake represents a potentially useful tool for interrogation of glial metabolism.
Keywords: cerebral hypoxia–ischemia, fluoroacetate, glial metabolism, kinetic modeling, MRI, PET
Introduction
Glia account for 90% of human brain cells and have a significant role in the response to neuropathological insults such as stroke and other neurodegenerations.1 Of particular interest is the activation and proliferation of astrocytes and microglia, and their resulting effects on glial–neuronal interactions. A detailed understanding of the response is vital for treatment development and in vivo methods to interrogate glial cell metabolism under abnormal conditions can shed light on such processes.
The use of 2-deoxy-2-[18F]fluoro-D-glucose for imaging cerebral metabolism has been well documented,2, 3 and while fluoro-D-glucose signal is sensitive to regional differences in glucose metabolism, it is not specific to glial metabolism.4 Radioligands of the 18-kDa translocator protein, particularly [11C]PK11195, have been used extensively for imaging upregulated expression of translocator protein in glia during neuroinflammation.5, 6 However, it does not directly measure glial metabolism.
In the brain, acetate is readily and specifically taken up in glial cells by different isoforms of the monocarboxylate transporter family of transmembrane proteins and then metabolized in the tricarboxylic acid cycle.4, 7 Mouse astrocytes express both MCT1 and MCT4 isoforms, the latter of which is thought to be primarily involved in efflux of lactate, and a subpopulation of rat astrocytes also express MCT2.8 Activated microglia in rats have been shown to upregulate MCT1 and MCT2 expression after ischemia.9 Radiotracers of glial acetate metabolism have been used in the past, primarily for autoradiography of naïve rodent brain, and include [3H] and [11C] labeled acetate.4, 10 [14C]acetate has been used in vitro in astrocyte cultures to measure uptake,11 and has also been used for autoradiography in a rat model of short-term middle cerebral artery occlusion (MCAo)—a reduction in uptake was attributed to transient depression of glial metabolism in the model.12 Similar studies have also examined the incorporation of radiolabeled carbon from [14C]acetate into metabolites to examine changes in astrocyte metabolism.13 The study of acetate metabolism in the brain has also been explored with nuclear magnetic resonance (see Supplementary Material).
A fluorinated analog of acetate, 2-fluoroacetate (FACE), behaves similarly to acetate but becomes metabolically trapped as fluorocitrate in the tricarboxylic acid cycle.14 In the past, this mechanism has been exploited by the use of sodium fluoroacetate as a potent rodenticide and also for the study of brain metabolism.15, 16 The use of [18F]fluoroacetate ([18F]FACE) as a positron emission tomography (PET) tracer was suggested as early as 1986 after the authors demonstrated glia-specific uptake of [3H]FACE with autoradiography.4 More recently, [18F]FACE imaging of CWR22 tumor-bearing mice showed increased tumor-to-background signal, and [18F]FACE was suggested as a potential alternative to [11C]acetate for tumor imaging.17 The value of [18F]FACE compared with [11C]acetate is still a subject of debate. Lindhe et al18 compared [18F]FACE and [11C]acetate and concluded that the former is not a functional analog of the latter, but only in normal physiology, as these studies did not evaluate [18F]FACE uptake in the brain under abnormal conditions.
The first known use of [18F]FACE in PET imaging of the brain demonstrated elevated lesion uptake of [18F]FACE in animal models of glioblastoma, focal cerebral ischemia, and cerebral hypoxia–ischemia (H–I). 2-[18F]fluoroacetate was suggested to be a potentially useful PET tracer of glial metabolism in the central nervous system and the need for further validation—including evaluation of the [18F]FACE kinetics—was emphasized.19, 20 These results suggested the intriguing possibility that the observed [18F]FACE uptake is metabolism-driven, thus suggesting the potential use of this tracer for imaging glia-specific metabolism in these animal models. In this study, we have investigated the extent to which [18F]FACE uptake in cerebral H–I is driven by metabolism or by other factors. To do so, we have conducted multi-modal PET and magnetic resonance imaging (MRI) studies to characterize [18F]FACE and analyze its kinetics in rodent models of cerebral H–I. Initial kinetic analysis with mouse data suggested potential improvement of sensitivity to parameters of interest with a longer scan duration and larger regions of interest. Thus, in the following discussion, we present data for both mouse and rat.
Materials and Methods
All animal handling and procedures described herein, and according to the Animal Research: Reporting In Vivo Experiments (ARRIVE) guidelines, were performed in accordance with the protocols approved by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) accredited Institutional Animal Care and Use Committee at Genentech.
Cerebral Hypoxia–Ischemia Model
The cerebral H–I model was originally developed in rats and subsequently modified and adapted to mice.21, 22 For these studies, we used both mice (C57BL/6, male, 8 to 12 weeks old) (Charles River Laboratories, Hollister, CA, USA) and rats (Sprague–Dawley, male 3 to 5 weeks old) (Harlan Sprague–Dawley, Livermore, CA, USA). Throughout surgery and hypoxic challenge, animals were anesthetized with isoflurane (1% to 2% for mouse, 1.5% to 2.5% for rat) and maintained at 37°C. The right common carotid artery was permanently ligated with 6-0 gauge silk suture. The duration of surgery was between 15 to 30 minutes. Animals were then allowed to recover until ambulatory and subsequently challenged in a hypoxic environment (8% oxygen and 92% nitrogen, with 0.8% isoflurane) for 30 minutes (mouse) or 30 to 40 minutes (rat). Animals were imaged either at 3 hours post insult (nmouse=3, nrat=4) or 24 hours post insult (nmouse=4, nrat=5). Assessment of neurological deficit score was performed after recovery from hypoxia, and where applicable, prior to imaging at 24 hours.
Magnetic Resonance Imaging
Animals were anesthetized with isoflurane, and body temperature monitored and maintained at 37°C with a rectal temperature probe (Small Animal Instruments, Stony Brook, NY, USA) and a proportional-integral-derivative controller implemented in LabVIEW (National Instruments, Austin, TX, USA). The animal's head was secured in place by ear bars. Imaging was performed in a 4.7T horizontal bore magnet using a DirectDrive console and VnmrJ software (Agilent Technologies, Santa Clara, CA, USA). For mouse imaging, a 40 mm (inner diameter) quadrature coil (Millipede, Agilent Technologies) was used. For rat imaging, a 40 mm quadrature coil (Morris Instruments, Ottawa, ON, Canada) was used. Diffusion weighted images were acquired in mice with a fast spin echo sequence with the following parameters: repetition time, 4000 milliseconds; echo time, 33 milliseconds (1st echo); echo train length, 8; echo spacing, 9 milliseconds; a target b-value of 965 s/mm2 was applied in four directions with three repetitions each: none, readout, phase encode, and slice encode. Parametric images of apparent diffusion coefficient (ADC) were then calculated from this data. For both mouse and rat, T2 parametric images were calculated from data acquired with a multispin echo sequence with the following parameters: repetition time, 3,000 milliseconds; echo time, 10.10 milliseconds (mouse) or 15 milliseconds (rat); number of echoes, 8; averages, 4. All mouse images were acquired with a 20 mm × 20 mm field of view, with 16 axial slices and 0.8 mm slice thickness. The field of view for rat images was 36 mm × 36 mm, with 20 axial slices and 1.0 mm slice thickness.
2-[18F]fluoroacetate Positron Emission Tomography
Following MRI, animals were allowed to recover from anesthesia and then imaged in a preclinical PET/computed tomography system (Inveon; Siemens Medical Solutions USA, Knoxville, TN, USA). 2-[18F]fluoroacetate was synthesized as described previously19 and delivered via tail vein infusion at an average activity concentration of 249±96 μCi (mouse) or 853±104 μCi (rat) in 100 μL saline during the first minute of a 30-minute (mouse) or 90-minute (rat) dynamic scan. A short infusion delivery was selected for reproducibility and peak identifiability given the chosen frame durations. For the 30-minute acquisition, data was framed as follows: 12 × 10 seconds, 6 × 60 seconds, 6 × 120 seconds, and 4 × 15 seconds. The 90-minute acquisition was framed as follows: 12 × 10 seconds, 6 × 60 seconds, 6 × 120 seconds, 6 × 180 seconds, 4 × 330 seconds, and 4 × 450 seconds. A computed tomography scan was acquired immediately following PET imaging for attenuation and scatter correction, and to facilitate image registration. Positron emission tomography reconstruction consisted of two iterations of three dimensional ordered subset expectation maximization followed by 18 maximum a posteriori iterations. The imaging matrix was set to 128 × 128 × 159, with corresponding voxel sizes of 0.4 mm × 0.4 mm × 0.769 mm.
Image Analysis
Co-registration was performed in PMOD (PMOD Technologies, Zurich, Switzerland). Computed tomorgraphy data were manually registered to MRI, with the resulting transformation applied to PET data in order to co-register the latter with MRI. The lesion region of interest (ROI) was manually segmented from the parametric T2 images with the assistance from the isocontour function (lower threshold 68 to 70 milliseconds, upper threshold 95 milliseconds) in PMOD. The lesion ROI was mirrored about the midline to form the contralateral ROI. To derive whole blood time-activity curves (TACs) from the PET imaging data, a cardiac blood ROI was manually determined from an average of the first 10 frames, assisted by the isocontour function in PMOD.
Kinetic Modeling and Input Function Determination
Fitting of kinetic models to the data was performed in PMOD. Lear and Ackermann10 suggested adapting Sokoloff's two-tissue irreversible models for fluoro-D-glucose to model FACE uptake. Here, this model (2T3k) as well as a simpler 1 tissue model (1T2k) and a modified irreversible uptake model based on slow but existent wash-out of metabolized tracer in the form of [18F]fluoride (denoted as 2T4k-k5, not to be confused with a two-tissue reversible uptake model, 2T4k) were fit to the [18F]FACE uptake data. The models are depicted in Figure 1 and are described by the following:
Figure 1.

Schematic of the compartment models used to model [18F]fluoroacetate kinetics.
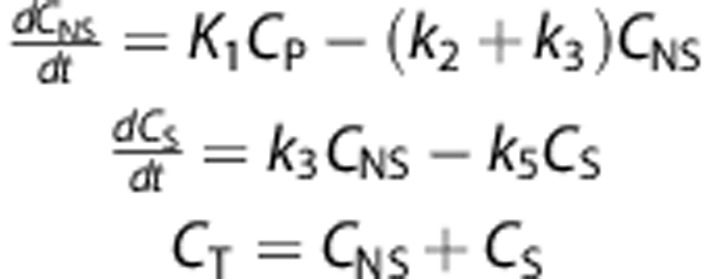 |
where CP, CNS, CS, and CT refer respectively to the concentration of radiotracer in the plasma, free and non-specific (parent), specific (metabolically trapped), and total tissue compartments.
The paired student's t-test was used to compare parameter group means in lesion versus contralateral. The unpaired student's t-test was used to compare parameter means between time point groups. Comparison of fits was performed with the F-test for nested models and the Akaike and Bayesian criteria.23, 24, 25 Parametric maps of kinetic parameters (2T3k) were generated using PMOD.
In previous [18F]FACE biodistribution studies of various species, it has been shown that myocardial TACs resemble blood curves.17, 18 On this basis, and with the assumption that partial volume effects should be negligible between tissues with similar TACs, we have investigated the validity of using an image-derived whole blood curve from which a plasma curve can be derived.
Three methods of whole blood activity measurement were compared: (1) manual sampling of cardiac blood at discrete time points, (2) real-time arterial blood sampling with concurrent PET imaging, and (3) image-derived sampling of cardiac blood. For the first method, naïve animals (n=14) were injected as described in the initial steps of the PET imaging protocol. ∼0.5 mL of blood was withdrawn by cardiac puncture and the animal was euthanized immediately afterwards. Whole blood samples were centrifuged at 1000 g for 10 minutes to separate plasma and blood cells. Measurements were taken at 1, 2, 5, 15, and 30 minutes post injection (n=4, 4, 2, 2, and 2, respectively).
Separately, for 2 animals at 30 minutes post injection, the supernatant was extracted, diluted 4:1 with methanol, vortexed for 30 to 60 seconds, and then incubated at −20°C for 10 minutes. The diluted samples were then centrifuged at 20,800 g for 10 minutes at 4°C to separate precipitated proteins and plasma. Plasma samples were vacuum concentrated (pressure: <1 Torr, temperature: 45°C, time: 1.5 hours) and diluted 2:1 with water + 0.1% formic acid for high performance liquid chromatography. Samples were analyzed via high performance liquid chromatography (Agilent Technologies) using a reverse phase column (250 × 4.6 mm, 5 μm pore size). The mobile phase consisted of (A) water + 0.1% formic acid and (B) acetonitrile + 0.1% formic acid. Samples were eluted with a gradient starting at 95% A for 2 minutes, changing to 5% in 4 minutes, and holding at 5% for 2 minutes.
Continuous, in vivo whole blood activity measurements were taken using a blood sampling system (Twilite; Swisstrace GmbH, Switzerland) recorded in PMOD. The carotid cannula in naïve mice (n=2, female FVB strain) was exteriorized to the blood sampling system with a jugular vein return via a peristaltic pump at 120 μL per minute. Animals then underwent [18F]FACE PET imaging as described while the activity in arterial blood was measured.
Histology
Brains were excised, sliced into 2 mm sections, and then incubated with 20 mg/mL triphenyltetrazolium chloride (Sigma-Aldrich, St Louis, MO, USA) in phosphate-buffered saline at 37°C for 10 minutes. Slices were transferred to transparent slides and imaged with a document scanner. For rat, slices were then fixed in 10% formalin at 4°C overnight and transferred to 70% ethanol for long-term storage. Further preparation and immunohistochemical staining for glial fibrillary acidic protein (GFAP) and ionized calcium-binding adapter molecule (IBA1) was performed using standard techniques (Cureline, South San Francisco, CA, USA).
Results
[18F]fluoroacetate Uptake in Rodent Cerebral Hypoxia–Ischemia Lesions
In both species and at both 3 and 24 hours post insult, lesions appeared with the characteristic elevated T2 values, lowered diffusion coefficient, and negative triphenyltetrazolium chloride staining. Mean T2 values and mean trace (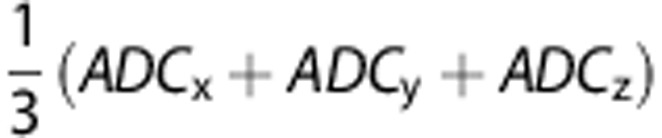 ) values in lesion were significantly higher and lower, respectively, than in contralateral tissue (Table 1). [18F]fluoroacetate uptake in lesion regions of interest was elevated (Figure 2) and, at 30 minutes post injection, consistently higher than contralateral uptake in both species at both 3 and 24 hours post insult (P<0.001, Table 1). 2-[18F]fluoroacetate was retained in the blood at notable activity concentrations after initial clearance (Figure 2), a behavior that has been observed by others in rodent, porcine, and non-human primate species.18, 26 This resulted in an input function with the appearance of a bolus plus constant infusion, making [18F]FACE a tracer amenable to steady-state analysis methods.27
) values in lesion were significantly higher and lower, respectively, than in contralateral tissue (Table 1). [18F]fluoroacetate uptake in lesion regions of interest was elevated (Figure 2) and, at 30 minutes post injection, consistently higher than contralateral uptake in both species at both 3 and 24 hours post insult (P<0.001, Table 1). 2-[18F]fluoroacetate was retained in the blood at notable activity concentrations after initial clearance (Figure 2), a behavior that has been observed by others in rodent, porcine, and non-human primate species.18, 26 This resulted in an input function with the appearance of a bolus plus constant infusion, making [18F]FACE a tracer amenable to steady-state analysis methods.27
Table 1. Summary of select parameters for mouse and rat cerebral hypoxia–ischemia.
| Mouse | Rat | |
|---|---|---|
| T2 (ms) | ||
| 3 hours | ||
| Lesion | 76±1 | 73±0.5 |
| Contralateral | 62±2 | 66±1 |
| 24 hours | ||
| Lesion | 78±8 | 81±2 |
| Contralateral | 58±6 | 64±1 |
| Trace (10−4 mm2/s) | ||
| 3 hours | ||
| Lesion | 5.3±0.4 | |
| Contralateral | 6.4±0.1 | |
| 24 hours | ||
| Lesion | 5.5±0.3 | |
| Contralateral | 6.5±0.2 | |
| [18F]FACE uptake, 30 minutes post injection (%ID/g) | ||
| 3 hours | ||
| Lesion | 10±0.8 | 1.9±0.3 |
| Contralateral | 7.0±0.6 | 1.0±0.1 |
| 24 hours | ||
| Lesion | 10±2 | 2.1±0.5 |
| Contralateral | 6.2±1 | 1.3±0.2 |
| Lesion volume (cc) | ||
| MRI (T2) | ||
| 3 hours | 0.0029±0.0005 | 0.30±0.05 |
| 24 hours | 0.048±0.1 | 0.16±0.09 |
| TTC | ||
| 3 hours | 0.019±0.01 | 0.075±0.03 |
| 24 hours | 0.15±0.1 | 0.16±0.09 |
| Neurological deficit score | ||
| 3 hours | 1.5, 2, 2 | 2.5, 3, 3.5, 4 |
| 24 hours | 0, 0, 2, 3 | 0, 1, 1.5, 2.5, 3 |
[18F]FACE, 2-[18F]fluoroacetate; MRI, magnetic resonance imaging; TTC, triphenyltetrazolium chloride.
Figure 2.

(A and B) Example multi-modal data for mouse (24 hours post insult) and rat (3 hours post insult). A z-axis apparent diffusion coefficient (ADCz) map was generated in one rat, shown here. (C and D) Averaged time-activity curves for whole blood, brain lesion, and brain contralateral regions of interest for 3 hours and (E and F) 24 hours time points. DWI, diffusion weighted image; GFAP, glial fibrillary acidic protein; H&E, hematoxylin and eosin; Iba1, ionized calcium-binding adapter molecule 1; PET, position emission tomography, TTC, triphenyltetrazolium chloride.
Immunohistochemical staining of brain tissues collected at 3 hours post insult from both species using anti-GFAP antibody specific for activated astrocytes showed lower GFAP reactivity in the necrotic area located in the striatum compared with the corresponding area in the contralateral hemisphere, and stronger diffuse staining was observed in the ipsilateral hippocampus in some samples (Figure 2B). Samples collected at 24 hours post insult showed diffuse GFAP reactivity in the lesion compared with corresponding areas in contralateral hemisphere (Figure 2A). Such patterns of GFAP reactivity are suggestive of early astrocytic death followed by infiltration of GFAP-reactive debris into the necrotic core. The immunohistochemical analysis for activated microglia using anti-Iba-1 showed weaker signal in necrotic tissue in both species at both time points.
Input Function Determination
Real-time sampling of whole blood yielded TACs similar to the corresponding image-derived curves in the two animals (Figure 3A). A visible time shift is attributable to the delay in the sampling setup (Figure 3B) caused by transit of blood from the heart to the scintillation counter. Manual sampling also yielded results similar to image-derived curves but with more variability at the initial time points (Figure 3C). The activity in plasma for 12 animals at a range of time points from 0 to 30 minutes was 48.0%±6.1% (s.d.) of whole blood. The plasma protein fraction of plasma activity in two animals was 2.2%±0.8% and 1.4%±0.3% (mean±s.e.m., two replicates).
Figure 3.

Results from different whole blood measurement techniques. (A) Example comparison of [18F]fluoroacetate ([18F]FACE) whole blood curves derived from real-time sampling (B) and concurrent imaging. (C) Manually sampled whole blood with plasma and red/white blood cell fractions denoted as ‘supernatant' and ‘pellet', respectively. (D) Chromatograms of: plasma samples from animals (n=2, 1 replicate each) 30 minutes post [18F]FACE injection; 0.1 mg/mL fluorocitrate standard; 1 mg/mL FACE standard. Signal from plasma samples are represented as percent of the median-adjusted maximum analog-to-digital converter signal from the 0–5 minute retention times. Chromatograms of fluorocitrate and FACE standards are represented as percent of median-adjusted maximum extracted ion count between the 0 to 5 minutes retention times.
Metabolite analysis by high performance liquid chromatography-mass spectrometry (Figure 3D) suggests no significant presence of [18F]fluoride or [18F]fluorocitrate at 30 minutes post injection of [18F]FACE, and that the activity in plasma is due to parent alone. Chromatograms of plasma samples showed peaks around the expected 3.5-minute retention time for [18F]FACE.
Based on these data, the plasma input function was estimated as 50% of the image-based whole blood time-activity curve.
Kinetic Modeling of [18F]fluoroacetate Lesion Uptake
Results of kinetic modeling for all data are shown in Figure 4.
Figure 4.

[18F]fluoroacetate kinetic modeling parameters for both mouse and rat cerebral H-I for both 3 and 24 hours post insult imaging groups. 1T2k, 2T3k, and 2T4k-k5 models (shown in rows) were fit to the data. An asterisk (*) over a pair of bars indicates paired t-test of difference between contralateral versus lesion mean yielded P<0.05. A dashed, capped line over a pair of time points indicates an unpaired t-test difference between 3- and 24- hour means yielded P<0.05.
C57BL/6 mouse
In the 1T2k model, K1 was significantly lower in lesion at the 24-hour time point. k2 was significantly lower in lesion at both 3- and 24-hour time points. The resulting trend was an elevated K1/k2 in lesion at both time points with significance at 24 hours. There was a significant difference in k2 between the 3- and 24-hour time points in the contralateral ROI (P=0.033). The same trends in K1 and k2 were seen in the 2T3k model, with the addition of significantly lower lesion k2 at the 3 hour time point (P=0.006). k3 was higher in lesion than contralateral at both time points, approaching significance at the 24-hour time point (P=0.066). However, there was a large (>>25%) percent coefficient of variation (%COV) for the estimation of this parameter (Supplementary Table S1). In the 2T3k model, k3 is an order of magnitude lower than K1 and k2, suggesting that uptake is not flow limited and that metabolism of [18F]FACE is slow compared with transport, as has been observed in literature for acetate.28 In the 2T4k-k5 model, as with the other models, K1/k2 was significantly higher in lesion. k3 was not significantly different in either ROI, although k5 was significantly lower in lesion at the 24 hour time point. However, the %COV of estimation for k3 and k5 was quite high (Supplementary Table S1), suggesting poor identifiability of these parameters for these data.
Sprague–Dawley rat
In the 1T2k model, significantly lower lesion k2 at both 3 and 24 hours (P=0.011 and P=0.008, respectively) appeared to drive the significantly higher lesion K1/k2 (P=0.011 at 3 hours, P=0.004 at 24 hours). In addition, K1/k2 was significantly lower at 24 hours compared with 3 hours in both lesion and contralateral ROIs (P=0.033 and P=0.018, respectively). Similar trends were seen in the 2T3k model. k3 was significantly higher in the contralateral tissue compared with lesion at both the 3- and 24-hour time points (P=0.007 and P=0.004, respectively), with %COV values in lesion and contralateral of 30.1±35.2 and 18.2±23.3, respectively. While the %COV values are large, they are low relative to that seen in the mouse data, which suggests that a longer scan time improved parameter identifiability (Supplementary Table S1). In the rat data, we briefly examined the effect of a lengthened acquisition on our ability to estimate k3. Kinetic modeling of the first 30 minutes of rat data resulted in increased variability in estimates but no change in trends between lesion and contralateral. In the 2T3k model, k3 is several orders of magnitude lower than K1 and k2. There was no significant difference between lesion and contralateral ROIs for k3 or k5 in the 2T4k-k5 model. In addition, the %COV values for these parameters were quite large (>>25%, Supplementary Table S1).
In all models, for all data, K1/k2 was higher in lesion, with statistical significance in mouse data for 24 hours and in all rat data. k2 was likewise significantly lower in the majority of cases. This suggests that the uptake of [18F]FACE is primarily transport-driven and is the result of decreased efflux of tracer from the tissue. The low order of magnitude of k3 and k5 in the data suggests that kinetics of the ‘metabolically trapped' compartment are not the main contributor to [18F]FACE uptake in these models. Net influx, Ki=K1k3/(k2+k3), was significantly higher in lesion at 24 hour in mouse (P=0.0146), but was not significantly higher in lesion in rat at either time point (Figure 5).
Figure 5.

Macroparameters K1/K2 and Ki for mouse and rat data modeled with 2T3k. Middle bars represent the means, while the outer bars represent±one s.d. An asterisk (*) over a pair of bars indicates a paired t-test of difference between contralateral versus lesion mean yielded P<0.05. A dashed, capped line over a pair of time points indicates an unpaired t-test difference between 3- and 24- hour means yielded P<0.05.
Comparison of Kinetic Models
(See Supplementary Table S2) The Akaike Information Criterion and Bayesian Criterion, on average, favored the 2T3k and 2T4k-k5 models over the 1T2k model in both species at both 3- and 24-hour times for lesion and contralateral regions of interest. In the F-Test for nested models comparing the 2T3k and 1T2k models, the majority of cases favored 2T3k (with the exception of the contralateral ROI in mouse at 3 hours and the lesion ROI in mouse at 24 hours). In comparison between the 2T4k-k5 and 2T3k models, the results were less conclusive with lesion regions of interest never having more than 50% of cases where 2T4k-k5 is significantly favored over 2T3k (with the exception of the lesion ROI in rat at 24 hours). In the contralateral region, the addition of the k5 parameter was significant in more cases, including four of five rats at 24 hours and four of four mice at 24 hours.
Parametric Maps
Areas of elevated K1/k2 co-localized with the T2 lesion ROI in mouse and rat (Figure 6), suggesting a relationship between edema and transport-driven uptake of [18F]FACE. Literature suggests that for this animal model, there is likely some mixture of cytotoxic edema and vasogenic edema, especially at the 24-hour time point.29
Figure 6.

Mouse and rat T2 maps with [18F]fluoroacetate ([18F]FACE) Vt (2T3k model) overlay. One axial slice, at approximately bregma zero, is shown for each animal.
Discussion
In this study, we have evaluated kinetics of [18F]FACE in rodent models of cerebral H–I. As others have reported, clearance of [18F]FACE from the blood is slow in rats, cynomolgus monkeys, and pigs.17, 18 In our data, blood activity levels remained at or above that of contralateral brain tissue, and were relatively stable from 15 minutes to the end of the scan. Because of the bolus plus infusion–like behavior of the plasma input function, a long scan time is required to properly estimate k3. The similarity between [18F]FACE time-activity curves in blood obtained by ex vivo or in vivo blood measurements and in the image-based cardiac blood ROI allowed direct estimation of whole blood, from which we estimated the plasma input function.
A concern of using [18F]FACE is confounding signal from [18F]fluoride. In rodents, liver enzymes in the presence of glutathione are responsible for most defluorination.30 Extensive defluorination of [18F]FACE has been reported in Sprague–Dawley rats at 4 hours and in nude mice at 2 hours (bone uptake of 1.87±0.15%ID/g and 5.23±1.64 %ID/g, respectively),17 but is significantly lower at earlier times (0.76±0.03 %ID/g at 1 hour for rat, 2.59±0.36 %ID/g at 30 minutes for mice).17 Our data also showed some bone uptake in rat data toward the latter part of acquisition. In addition, hyperintense regions in k3 maps were observed to coincide with skeletal structures, suggesting [18F]fluoride trapping in bone. However, under the assumptions that (1) defluorination of [18F]fluorocitrate is slow and that (2) [18F]fluoride rapidly effluxes from tissue and is taken up by bone, the contribution from this metabolite of [18F]FACE can be considered negligible in brain tissue. We did not detect [18F]fluoride in metabolite analysis of mouse plasma samples taken 30 minutes post injection. In addition, others have reported low levels of bone uptake within 90 minutes post injection in mouse and rat.17, 19 Furthermore, defluorination is not as prevalent in non-human primates, with bone uptake at 3% of injected dose in cynomolgus monkeys but 39% in pigs.18 In baboons, the specific uptake value of iliac bone was reported to be low even 2 hours post [18F]FACE injection.17
Increased blood–brain barrier permeability, as measured by extravasation of Evans blue dye, occurs in cerebral H–I by 24 hours in C57BL/6 mice.29 However, we did not observe significantly elevated K1 or k2 in lesions for either species at 3 or 24 hours post insult, suggesting that uptake changes are not a result of increased transport across the blood–brain barrier.
Elevated Lesion Uptake of [18F]fluoroacetate in Cerebral Ischemia–Hypoxia is Transport-Driven Rather than Metabolism-Driven
Uptake of acetate is largely dictated by diffusional uptake rather than metabolism, as the rate of the former is often much larger than that of the latter.28 This effect is seen in our data, as transport-related changes appeared to be the main driver of elevated [18F]FACE uptake in cerebral H–I lesions. Specifically, K1/k2 was significantly elevated in lesion in nearly all time and species, driven by decreased k2 (Figure 4).
The basis for this change is not clear, but may be due to equalization of H+ gradients after depolarization of the cell membrane following ischemic insult since monocarboxylates are cotransported with H+ by MCTs. The co-localization of elevated K1/k2, decreased ADC, and elevated T2 in 3 and 24-hour mouse groups and in one 3-hour rat suggest a combination of cytotoxic and vasogenic edema, which has been shown to evolve in rodents within the first 24 hours in cerebral H–I and MCAo models.29, 31 The depletion of ATP production following ischemia leads to the failure of ATP-dependent pumps upon which a normal concentration gradient depends, and the resulting influx of ions is accompanied by influx of extracellular water.32 As a small anion, it is possible that [18F]FACE is primarily taken up by this process.
In response to hypoxia, MCT4 is upregulated in glia for the purpose of effluxing excess lactate.33, 34, 35 Unilateral MCAo in Wistar rats demonstrated elevated ipsilateral lactate concentrations.34 It is possible that the observed decrease in k2 is due to competition with elevated lactate efflux, on the basis that MCT1 and MCT4 transporters have an order of magnitude greater affinity for lactate compared with acetate.33
Metabolism-Driven Uptake is Low but Possibly Detectable with [18F]fluoroacetate
Our in vivo estimation of K1 and k2 in the 2T3k model yielded values on the same order of magnitude to those reported in literature by autoradiographic techniques—Lear and Ackermann10 reported values of K1=0.16±0.03 and k2=0.19±0.03. In our rat data (see Figure 4), K1=0.432±0.040 and k2=0.343±0.035 at 3 hours post insult, and K1=0.419±0.064 and k2=0.355±0.058 at 24 hours post insult for contralateral brain tissue. However, we found k3 to be around a factor of 2 lower (see Figure 4: k3=0.006±0.001 at 3 hours and k3=0.004±0.002 at 24 hours, compared with k3=0.01±0.03 as found by Lear and Ackermann10). The significant difference between contralateral and lesion tissue k3 in rat suggests that while metabolism is low and [18F]FACE uptake is driven primarily by K1 and k2, changes in k3 induced by H–I are still detectable by analysis of [18F]FACE kinetics.
Astrocyte metabolism of [11C]acetate was reported to be decreased in rat MCAo models at early time points (0 to 3 hours) during which cell death is expected.12 In our study, GFAP staining of brain tissues showed lower reactivity in the lesion at 3 hours post insult and diffuse staining at 24 hours post insult suggestive of astrocytic death. Depressed incorporation of 14C in glutamate and glutamine has been previously observed in rat striatum following MCAo, slowly recovering within 48 hours post reperfusion.13 In our data, this trend is reflected by k3 in rats, but the opposite trend was seen in mice. However, scan duration was threefold shorter in the mice, and the uncertainty of k3 estimation due to a shorter scan length is reflected in higher %COV values (Figure 4) and is highlighted by known astrocyte behavior in the acute period following insults generated by models such as MCAo and cerebral H–I. For instance, astrocyte viability is decreased, with loss expected even at 48 hours post injury.13 Astrocyte loss does not necessarily imply lack of elevated metabolism in remaining astrocytes—in newborn piglets for example, astrocytes have been found to proliferate 48 hours after cerebral H–I despite decreased numbers as measured by GFAP reactivity36—but more detailed methods would need to be developed to detect changes in cellular acetate metabolism in these remaining cells within a given volume. To assess the increase of glial metabolism in tissues surrounding the ischemic lesion associated with the neuroinflammatory response, the evaluation should be performed at later time points, where a neuroinflammatory response has been observed post-stroke in rodents.37
Upon further examination of the 2T3k results, we found no significant trends in Ki with the exception of elevated lesion Ki in the 24 hour mouse group. However, we do not believe there are implications of this finding with respect to metabolism, due to the uncertainty in k3 estimation for the mouse data. It is more likely that transport effects are responsible for elevated Ki in this case.
Kinetic model comparisons between the 1T2k, 2T3k, and 2T4k-k5 models suggested that the 1T2k model is insufficient to describe [18F]FACE uptake in either lesion or contralateral tissue, despite uptake being driven primarily by K1 and k2. The addition of a metabolite efflux rate, k5, yielded less certain results in model comparison—while residuals were smaller than in the 2T3k model, these differences were small and the significance of the added parameter was not seen in the majority of cases in the lesion ROI. The increased number of significant cases in the contralateral ROI may represent a greater contribution from metabolite production and thus efflux from areas with better glial viability. However, the %COV values for k5 were in most cases larger than the generally accepted 25% threshold (Price, JC et al, personal communication), and the order of magnitude changes in k3 upon introduction of k5 belies the difficulty of parameter identifiability for k3 and especially k5. Relative levels of defluorination in the brain relative to peripheral sources must be taken into consideration when using the additional parameter—in the animal models used in this study, it appears that a 2T3k model is necessary and sufficient for fitting to [18F]FACE uptake.
Conclusions
In conclusion, we have used multi-modal in vivo imaging methods and (1) observed significantly elevated lesion uptake of [18F]FACE in rodent cerebral H–I at 3 and 24 hours post injury, (2) developed and validated an image-based method to extract the [18F]FACE plasma input function for the mouse and rat, (3) developed, used, and assessed kinetic models of [18F]FACE uptake, and (4) found that in rodent cerebral H–I, [18F]FACE uptake is driven primarily by changes in transport rather than in glial metabolism, although the latter may potentially be detectable with [18F]FACE kinetic modeling using the 2T3k model as corroborated by immunohistochemical evidence and previous literature.
The use of [18F]FACE for detecting changes in metabolism must be validated on a case-by-case basis, taking into consideration the tissue(s) of interest and their known behaviors under the conditions of interest. In the case of cerebral H–I, it appears that [18F]FACE uptake alone not an indicator for glial metabolism, but that kinetic parameters estimated by modeling reveal useful information. Additional work must be done for the use of [18F]FACE in PET studies to be tractable and translatable. For instance, [18F]FACE kinetics may be evaluated in animal models with less severe impact on glial viability but with significantly elevated glial acetate metabolism. Studies should also evaluate the effect of edema on [18F]FACE uptake by glia in stroke models in order to ascertain the relative contribution of transport and metabolism effects. As acetate entry into the tricarboxylic acid cycle is an alternative pathway to pyruvate metabolism, animal models with well-understood changes in the relative levels of pyruvate and acetate metabolism should be used for further validation of [18F]FACE. If issues such as these (and those suggested by Lopresti and Mason20) can be addressed, [18F]FACE kinetics may then be a useful tool for investigating glial metabolic changes under a variety of neurological conditions.
Acknowledgments
The authors would like to thank the following current and former members of the Biomedical Imaging group at Genentech, Inc. for radiotracer synthesis and for technical assistance related to the animal model, PET, MRI, and IHC: Herman S Gill, Baby Martin-McNulty, Annie K Ogasawara, Alexander N Vanderbilt, Joan M Greve, Maj Hedehus, Oded Foreman, Sandra Sanabria-Bohórquez, Melissa G Edick, Simon P Williams, and Nicholas van Bruggen.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on the Journal of Cerebral Blood Flow & Metabolism website (http://www.nature.com/jcbfm)
This work was supported jointly by the UC Davis—Genentech Graduate Scholar Program and National Institute of Biomedical Imaging and Bioengineering award number R01 EB000993.
Supplementary Material
References
- Allen NJ, Barres BA. Glia—more than just brain glue. Nature. 2009;457:675–677. doi: 10.1038/457675a. [DOI] [PubMed] [Google Scholar]
- Phelps M, Huang S, Hoffman E, Selin C, Sokoloff L, Kuhl D. Tomographic measurement of local cerebral glucose metabolic rate in humans with (F-18) 2-fluoro-2-deoxy-D-glucose: validation of method. Ann Neurol. 1979;6:371–388. doi: 10.1002/ana.410060502. [DOI] [PubMed] [Google Scholar]
- Reivich M, Kuhl D, Wolf A, Greenberg J, Phelps M, Ido T, et al. The [18F]fluorodeoxyglucose method for the measurement of local cerebral glucose utilization in man. Circ Res. 1979;44:127–137. doi: 10.1161/01.res.44.1.127. [DOI] [PubMed] [Google Scholar]
- Muir D, Berl S, Clarke DD. Acetate and fluoroacetate as possible markers for glial metabolism in vivo. Brain Res. 1986;380:336–340. doi: 10.1016/0006-8993(86)90231-3. [DOI] [PubMed] [Google Scholar]
- Cagnin A, Gerhard A, Banati RB. In vivo imaging of neuroinflammation. Eur Neuropsychopharmacol. 2002;12:581–586. doi: 10.1016/s0924-977x(02)00107-4. [DOI] [PubMed] [Google Scholar]
- Banati RB. Visualising microglial activation in vivo. Glia. 2002;40:206–217. doi: 10.1002/glia.10144. [DOI] [PubMed] [Google Scholar]
- Waniewski RA, Martin DL. Preferential utilization of acetate by astrocytes is attributable to transport. J Neurosci. 1998;18:5225–5233. doi: 10.1523/JNEUROSCI.18-14-05225.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Halestrap AP, Pierre K. Cellular and subcellular distribution of monocarboxylate transporters in cultured brain cells and in the adult brain. J Neurosci Res. 2005;79:55–64. doi: 10.1002/jnr.20307. [DOI] [PubMed] [Google Scholar]
- Moreira TJ, Pierre K, Maekawa F, Repond C, Cebere A, Liljequist S, et al. Enhanced cerebral expression of MCT1 and MCT2 in a rat ischemia model occurs in activated microglial cells. J Cereb Blood Flow Metab. 2009;29:1273–1283. doi: 10.1038/jcbfm.2009.50. [DOI] [PubMed] [Google Scholar]
- Lear JL, Ackermann RF. Evaluation of radiolabeled acetate and fluoroacetate as potential tracers of cerebral oxidative metabolism. Metab Brain Dis. 1990;5:45–56. doi: 10.1007/BF00996977. [DOI] [PubMed] [Google Scholar]
- Hosoi R, Matsuyama Y, Hirose S, Koyama Y, Matsuda T, Gee A, et al. Characterization of 14C-acetate uptake in cultured rat astrocytes. Brain Res. 2009;1253:69–73. doi: 10.1016/j.brainres.2008.11.068. [DOI] [PubMed] [Google Scholar]
- Hosoi R, Kashiwagi Y, Tokumura M, Abe K, Hatazawa J, Inoue O. Sensitive reduction in 14C-acetate uptake in a short-term ischemic rat brain. J Stroke Cerebrovasc Dis. 2007;16:77–81. doi: 10.1016/j.jstrokecerebrovasdis.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Thoren AE, Helps SC, Nilsson M, Sims NR. Astrocytic function assessed from 1-14C-acetate metabolism after temporary focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2005;25:440–450. doi: 10.1038/sj.jcbfm.9600035. [DOI] [PubMed] [Google Scholar]
- Morrison J, Peters R. Biochemistry of fluoroacetate poisoning: the effect of fluorocitrate on purified aconitase. Biochem J. 1954;58:473. doi: 10.1042/bj0580473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett S, Spencer MM. Sodium fluoroacetate (1080) as a rat poison. J Hyg. 1949;47:426–430. doi: 10.1017/s0022172400014765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonnum F, Johnsen A, Hassel B. Use of fluorocitrate and fluoroacetate in the study of brain metabolism. Glia. 1997;21:106–113. [PubMed] [Google Scholar]
- Ponde DE, Dence CS, Oyama N, Kim J, Tai YC, Laforest R, et al. 18F-fluoroacetate: a potential acetate analog for prostate tumor imaging—in vivo evaluation of 18F-fluoroacetate versus 11C-acetate. J Nucl Med. 2007;48:420–428. [PubMed] [Google Scholar]
- Lindhe Ö, Sun A, Ulin J, Rahman O, Långström B, Sörensen J. [18F]fluoroacetate is not a functional analogue of [11C]acetate in normal physiology. Eur J Nucl Med Mol Imaging. 2009;36:1453–1459. doi: 10.1007/s00259-009-1128-7. [DOI] [PubMed] [Google Scholar]
- Marik J, Ogasawara A, Martin-McNulty B, Ross J, Flores JE, Gill HS, et al. PET of glial metabolism using 2-18F-fluoroacetate. J Nucl Med. 2009;50:982. doi: 10.2967/jnumed.108.057356. [DOI] [PubMed] [Google Scholar]
- Lopresti BJ, Mason NS. 2-18F-fluoroacetate: a useful tool for assessing gliosis in the central nervous system. J Nucl Med. 2009;50:841–843. doi: 10.2967/jnumed.108.058875. [DOI] [PubMed] [Google Scholar]
- Vannucci SJ, Willing LB, Goto S, Alkayed NJ, Brucklacher RM, Wood TL, et al. Experimental stroke in the female diabetic, db/db, mouse. J Cereb Blood Flow Metab. 2001;21:52–60. doi: 10.1097/00004647-200101000-00007. [DOI] [PubMed] [Google Scholar]
- Levine S. Anoxic-ischemic encephalopathy in rats. Am J Pathol. 1960;36:1. [PMC free article] [PubMed] [Google Scholar]
- Landaw E, DiStefano JJ. Multiexponential, multicompartmental, and noncompartmental modeling. II. Data analysis and statistical considerations. Am J Physiol. 1984;246:R665–R677. doi: 10.1152/ajpregu.1984.246.5.R665. [DOI] [PubMed] [Google Scholar]
- Akaike H. A new look at the statistical model identification. IEEE Trans Automat Contr. 1974;19:716–723. [Google Scholar]
- Schwarz G. Estimating the dimension of a model. Ann Statist. 1978;6:461–464. [Google Scholar]
- Nishii R, Tong W, Wendt R, Soghomonyan S, Mukhopadhyay U, Balatoni J, et al. Pharmacokinetics, metabolism, biodistribution, radiation dosimetry, and toxicology of 18F-fluoroacetate (18F-FACE) in non-human primates. Mol Imaging Biol. 2012;14:213–224. doi: 10.1007/s11307-011-0485-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassen NA. Neuroreceptor quantitation in vivo by the steady-state principle using constant infusion or bolus injection of radioactive tracers. J Cereb Blood Flow Metab. 1992;12:709–716. doi: 10.1038/jcbfm.1992.101. [DOI] [PubMed] [Google Scholar]
- Hertz L, Dienel GA. Lactate transport and transporters: general principles and functional roles in brain cells. J Neurosci Res. 2004;79:11–18. doi: 10.1002/jnr.20294. [DOI] [PubMed] [Google Scholar]
- Adhami F, Liao G, Morozov YM, Schloemer A, Schmithorst VJ, Lorenz JN, et al. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am J Pathol. 2006;169:566. doi: 10.2353/ajpath.2006.051066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecle B, Casida JE. Enzymatic defluorination and metabolism of fluoroacetate, fluoroacetamide, fluoroethanol, and (-)-erythro-fluorocitrate in rats and mice examined by fluorine-19 and carbon-13 NMR. Chem Res Toxicol. 1989;2:429–435. doi: 10.1021/tx00012a012. [DOI] [PubMed] [Google Scholar]
- Loubinoux I, Volk A, Borredon J, Guirimand S, Tiffon B, Seylaz J, et al. Spreading of vasogenic edema and cytotoxic edema assessed by quantitative diffusion and T2 magnetic resonance imaging. Stroke. 1997;28:419–427. doi: 10.1161/01.str.28.2.419. [DOI] [PubMed] [Google Scholar]
- Liang D, Bhatta S, Gerzanich V, Simard JM. Cytotoxic edema: mechanisms of pathological cell swelling. Neurosurg Focus. 2007;22:E2. doi: 10.3171/foc.2007.22.5.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmer K, Friedrich B, Lang F, Deitmer J, Bröer S. The low-affinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. Biochem J. 2000;350 (Pt 1:219. [PMC free article] [PubMed] [Google Scholar]
- Yager JY, Brucklacher RM, Vannucci RC. Cerebral oxidative metabolism and redox state during hypoxia-ischemia and early recovery in immature rats. Am J Physiol. 1991;261:H1102–H1108. doi: 10.1152/ajpheart.1991.261.4.H1102. [DOI] [PubMed] [Google Scholar]
- Ullah MS, Davies AJ, Halestrap AP. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1α-dependent mechanism. J Biol Chem. 2006;281:9030–9037. doi: 10.1074/jbc.M511397200. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Brambrink AM, Lehmann C, Portera-Cailliau C, Koehler R, Rothstein J, et al. Hypoxia—ischemia causes abnormalities in glutamate transporters and death of astroglia and neurons in newborn striatum. Ann Neurol. 1997;42:335–348. doi: 10.1002/ana.410420310. [DOI] [PubMed] [Google Scholar]
- Harhausen D, Sudmann V, Khojasteh U, Müller J, Zille M, Graham K, et al. Specific imaging of inflammation with the 18kDa translocator protein ligand DPA-714 in animal models of epilepsy and stroke. PloS ONE. 2013;8:e69529. doi: 10.1371/journal.pone.0069529. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.