

Abstract
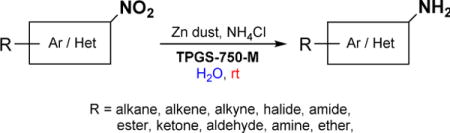
A robust and green protocol for the reduction of functionalized nitroarenes to the corresponding 1° amines has been developed. It relies on inexpensive zinc dust in water containing nanomicelles derived from the commercially available designer surfactant TPGS-750-M. This mild process takes place at room temperature and tolerates a wide range of functionalities. Highly selective reductions can also be achieved in the presence of common protecting groups.
Aromatic and heteroaromatic primary amines have proven to be important building blocks in the synthesis of a wide range of pharmaceuticals, polymers, and fine chemicals, with particular utility in cross-coupling reactions.1 However, the inherent reactivity of this functional group often requires protection from premature participation in multi-step syntheses. While a wide range of protecting groups are available to limit amine reactivity, one popular strategy involves their masking as nitroarenes. This approach provides multiple benefits, including facile nitrogen introduction via early stage nitration, excellent atom economy, good stability of the nitro group under numerous reaction conditions, and directed arene functionalization resulting from the associated electronics of the nitro moiety. Unfortunately, this protection strategy introduces a new challenge; the required chemoselective reduction of nitroarenes in the presence of other functional groups, including those that may be competitively reduced.
Classically, such reductions have been achieved under harsh conditions, including strong acids and various metals, such as iron or tin.2 Although effective in scaled industrial processes,3 these reductions tend to suffer from a limited substrate scope due to their severity, and present significant safety and environmental issues associated with their use. Several protocols have been reported in recent years to address these concerns. Catalytic hydrogenation using transition metal catalysis has received high interest, but this approach exhibits limited selectivity in the presence of other reducible functional groups.4 Even when chemoselectivity is not an issue, this route still requires specialized equipment, pressurized hydrogen gas, and flammable organic solvents, decreasing its safety and ultimate appeal. Additionally, the known energetic nature5 of nitro reduction intermediates often makes working in organic solvent under a pressurized atmosphere of hydrogen particularly undesirable.
Transfer hydrogenation processes of nitroarenes eliminate the use of pressurized hydrogen gas, but often show a decrease in yields in the presence of other reducible groups.6 Most recently, MO3S4 cluster catalysts have been developed that offer impressive chemoselectivity and functional group tolerance, but have limitations due to their cost and complexity.7 Additional exotic solutions, such as the use of copper,8a iron,8b or precious metal8c nanoparticles, or other engineered nanomaterials,9 have demonstrated good activity, but may suffer from a similar lack of selectivity.
One classical solution involves the use of elemental zinc as a reducing agent in various reaction environments, including acetic acid or methanol mixtures, or more recently biphasic solutions.10 While chemoselectivity is impressively high, especially when acidic pH can be avoided, literature methods all require the use of organic solvents or ionic liquids11 in order to solvate nitroarene substrates, and often require heating.2 Standard protocols generally call for 10–20 equivalents of zinc dust, increasing material costs, waste disposal, investment in energy, and limitations in reaction throughput. It follows that if the virtues of zinc could be leveraged by its utilization at ambient temperatures while eliminating organic solvents as the reaction media, a greener and more robust protocol would result, providing a solution to this important and timely synthetic problem.
Our work continues to focus, in part, on the design of nonionic surfactants that enable transition metal-catalyzed reactions to be performed in water at room temperature (rt), rather than in traditional organic solvents.12 Several applications of micellar technology to an array of valued organic transformations have already been developed. To further expand the scope of these micellar surfactant conditions, zinc-mediated reductions of nitro-arenes and nitro-heteroarenes have been studied; we now report the results of this investigation.
A representative hydrophobic nitroarene was examined in a 2 wt. % aqueous solution of the designer surfactant TPGS-750-M, which is an item of commerce (Sigma-Aldrich #733857).13 Addition of zinc dust and ammonium chloride to a solution of 4-nitrobenzophenone 1 resulted in clean conversion to 4-aminobenzophenone 1b upon stirring for six hours at room temperature (Scheme 1).
Scheme 1.
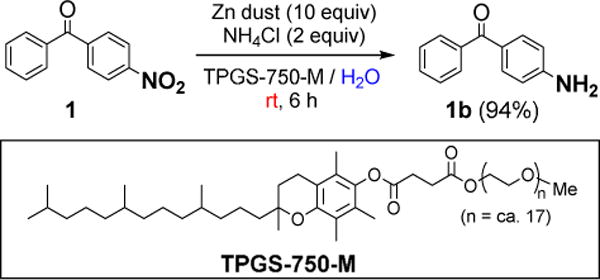
Initial study: reduction of 4-nitrobenzophenone
Monitoring the reaction products by GCMS revealed no residual nitroso or hydroxylamine intermediates, and no unwanted reduction of the carbonyl group at full conversion. A simple workup including filtration through a silica gel plug followed by rinsing with a minimal volume of ethyl acetate afforded the amino product 1b in 94% yield. Alternately, extraction with a minimal volume of organic solvent led to isolated product of similar purity and yield.
Following this initial success, experiments were conducted to optimize the reaction protocol (Table 1) Zinc dust loading was varied from the required 3 equivalents up to 10 equivalents suggested by prior literature.11 While limiting zinc loading to 3 equivalents slowed reaction rates and failed to achieve 100% conversion, with significant amounts of partially reduced hydroxylamine as an impurity, 5 equivalents resulted in clean, complete conversion. The global reaction concentration was established at 0.5 M, affording a reasonable reaction rate. Increasing the concentration to 1 M resulted in a similar rate, while higher concentrations showed poor substrate solubility and failed to reach complete conversion. Attempting the reaction “on-water” without the aid of TPGS-750-M resulted in low and inconsistent yields.
Table 1.
Optimization of zinc-mediated reduction

| ||||
|---|---|---|---|---|
| zinc (equiv) |
additive (2 equiv) |
concentration (M) |
time (h) |
conversion (%) |
| 3 | NH4Cl | 0.5 | 2 | 60 |
| 5 | NH4Cl | 0.5 | 2 | 84 |
| 10 | NH4Cl | 0.5 | 2 | 87 |
| 5 | NH4Cl | 0.5 | 4 | 100 |
| 5 | NH4Cl onlya | 0.5 | 4 | 62 |
| 5b | acetic acid | 0.5 | 2 | 100 |
| 5b | formic acid | 0.5 | 2 | 100 |
| 5 | TMEDA | 0.5 | 24 | 0 |
| 5 | pH 6 phosphate | 0.5 | 4 | 93 |
| 5 | pH 8 phosphate | 0.5 | 24 | 0 |
| 5 | NH4Clc | 1.0 | 4 | 100 |
| 5 | NH4Clc | 1.5 | 4 | 72 |
“on water”, no surfactact
2 batchwise zinc additions
1.2 equiv.
Several alternative additives were screened, with none showing better overall performance than that seen with NH4Cl. Acids (HOAc, HCOOH) resulted in rapid conversion, but also created inherently limited functional group compatibility and poor surfactant emulsion stability. Additionally, these highly acidic conditions resulted in a significant exotherm upon introduction of zinc dust, necessitating slow batch-wise addition and potentially limiting scalability. Basic additives (TMEDA, NH4OH, Et3N) completely shut down the reduction and returned only unreduced starting material.
The test reaction was also run in a 1 M potassium phosphate buffered solution of 2% TPGS-750-M adjusted to various levels of pH. Adequate conversion was observed with no additional additive when the starting pH was set to 6.0 or lower, albeit at significantly slower rates. No activity was seen above a starting pH of 8.0. Final optimized conditions were set at 0.5–1.0 M nitroarene, 5 equivalents fresh zinc dust, and 1.2 equivalents of ammonium chloride.
Using these optimized conditions, a series of functionalized nitroarenes was subjected to this reduction protocol to evaluate its scope and chemoselectivity (Figure 1). Reductions proceeded smoothly in the presence of both electron-withdrawing as well as electron-donating groups. Functional group tolerance was shown to include aldehydes, ketones, esters, amides, allenes, sulfides, and various halogens, suggestive of a strong preference for selective nitro group reduction.
Figure 1.
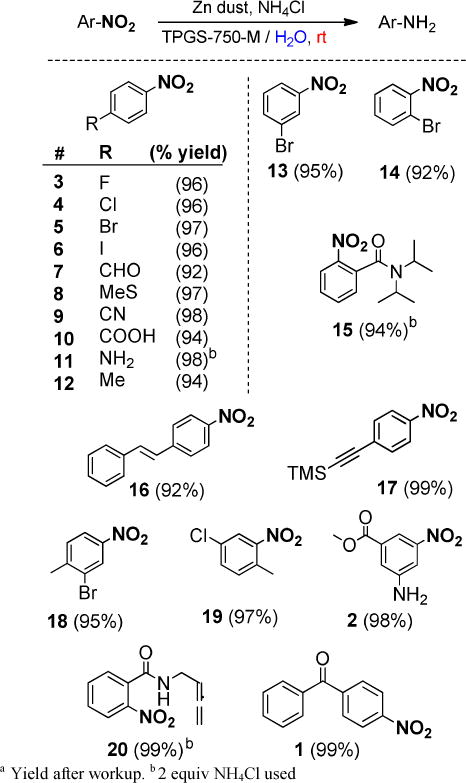
Substrate Scope: Zn-mediated reductions of aromatic nitro groups in TPGS-750-M/water at room temp a
In contrast to classical hydrogenation,5 no complications were encountered due to the presence of other reducible functionalities, including alkenes, alkynes, and carbonyls groups. Nitroarenes bearing halogen reacted particularly cleanly, yielding a variety of potentially synthetically useful building blocks. Most substrates showed complete reduction in 1–4 hours.
Orthogonal blocking group strategies were evaluated using protected nitroanilines and nitrophenols, subjecting each to our standard nitro group reduction conditions (Figure 2). Carbamate 21, benzylic amine 22, and benzyl ether 23 were all found to be stable over a period of 48 hours.
Figure 2.
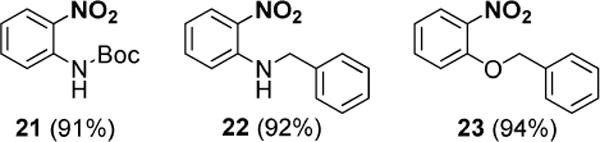
Screening of protecting group stability
Selected heteroaromatic natural products were nitrated and then subjected to reduction (Figure 3). The protocol successfully yielded clean amino derivatives 24b–26b, demonstrating its potential utility for unmasking an NH2 moiety in advanced intermediates in pharmaceutical or natural product synthesis, or other heteroaromatic targets of interest.
Figure 3.
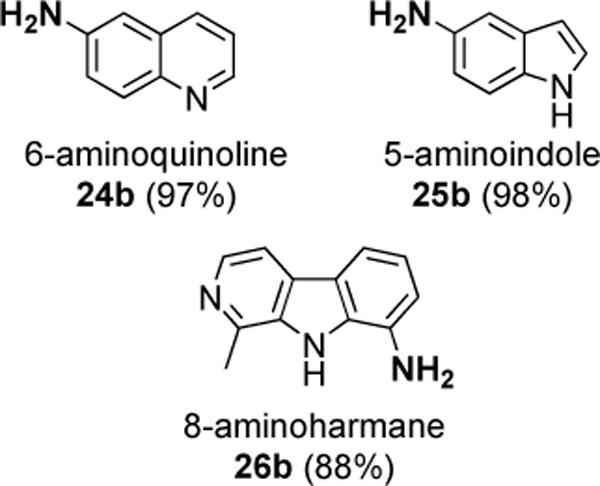
Reductions of natural products / heteroaromatics
To demonstrate the ease of use and flexibility of this protocol, a few modifications were attempted with a focus on robustness and potential scalability. To eliminate the use of silica gel filtration on scale, the reaction was instead extracted with a minimum amount of ethyl acetate. As demonstrated in previous publications from our group,7 a standard in-flask extraction leads to clean product, while leaving the surfactant behind in the aqueous phase to be recycled. As alluded to previously (vide supra), substrate concentration could be increased from 0.5 to 1.5 M to minimize the investment in water, with a slight reduction in rate. Mild heating (60 °C) could be applied to reduce reaction times to <4 hours, if desired. However, sensitive substrates, e.g., a substituted aryl iodide, showed increased lability upon heating, so a balance between throughput, energy input, and product stability should be taken into consideration.
Lastly, this methodology was applied to a two-step synthesis of the antiarrhythmic agent procainamide (Scheme 2). The amide bond was constructed in aqueous 2 wt. % TPGS-750-M with 4-nitrobenzoic acid 28 and N,N-diethyl ethylenediamine 29, utilizing EDCI/HOBt at room temperature. After isolation, the resulting nitrobenzamide 30 was reduced under standard conditions, producing the final API 1-chloro-4-methyl-2-nitrobenz-ene 31 in 83% yield over two steps. E Factors,14 comparing the mass ratio of organic solvent used to product produced, could be dramatically reduced relative to those associated with the patented route that relies on organic solvents.15 This approach also obviated the need for specialized hydrogenation equipment, thereby eliminating this particular process safety risk and its costs associated with such nitro group reductions.
Scheme 2.
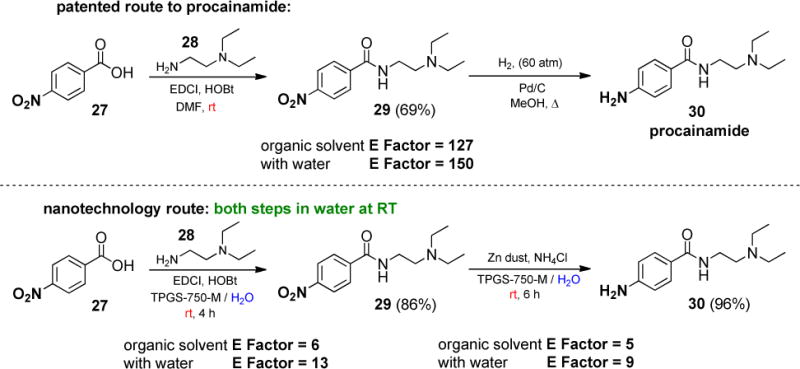
Procainamide synthesis: comparison of route in organic solvents vs. in water at room temperature
Supplementary Material
Acknowledgments
We warmly thank the NIH for supporting our research in green chemistry (GM 86485). Support provided by a New Leaf Grant from UCSB to BHL is also appreciated.
Footnotes
Supporting Information Available: Experimental procedures and characterization information for all new compounds. This information is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Downing RS, Kunkeler PJ, van Bekkum H. Catal Today. 1997;37:121. [Google Scholar]
- 2.Vogel AI, et al. Textbook of Practical Organic Chemistry. 5. Addison Wesley Longman Limited; Edinburgh Gate, England: 1989. p. 953. Chapter 6. [Google Scholar]
- 3.Weissermel K, Arpe HJ. Industrial Organic Chemistry. 4. John Wiley & Sons; Darmstadt, Germany: 2003. p. 376. Chapter 13. [Google Scholar]
- 4.Pandarus V, Ciriminna R, Beland F, Pagliaro M. Catal Sci Technol. 2011;1:1616. [Google Scholar]
- 5.Figueras F, Coq B. J Mol Catal A: Chem. 2001;173:223. [Google Scholar]
- 6.Taleb AB, Jenner G. J Mol Catal. 1994;91:149. [Google Scholar]
- 7.a) Sorribes I, Wienhöfer G, Vicent C, Junge K, Llusar R, Beller M. J Am Chem Soc. 2011;133:12875. doi: 10.1021/ja2061038. [DOI] [PubMed] [Google Scholar]; b) Sorribes I, Wienhöfer G, Vicent C, Junge K, Llusar R, Beller M. Angew Chem, Int Ed. 2012;21:7794. doi: 10.1002/anie.201202584. [DOI] [PubMed] [Google Scholar]
- 8.a) Dey R, Mukherjee N, Ahammed S, Ranu BC. Chem Commun. 2012;48:7982. doi: 10.1039/c2cc30999h. [DOI] [PubMed] [Google Scholar]; b) Kim S, Kim E, Kim BM. Chem Asian J. 2011;6:1921. doi: 10.1002/asia.201100311. [DOI] [PubMed] [Google Scholar]; c) Zhang L, Zhang S, Kang DE, Shin JY, Suh H, Kim I. RSC Advances. 2013;3:4692. [Google Scholar]
- 9.a) Rahman A, Jonnalagadda SB. Catal Lett. 2008;123:264. [Google Scholar]; b) Wang HC, Li BL, Zheng YJ, Wang WY. Bull Korean Chem Soc. 2012;33:2961. [Google Scholar]; c) Kaur B, Tumma M, Srivastava R. Ind Eng Chem Res. 2013;52:11479. [Google Scholar]; d) Tumma M, Srivastava R. Catal Comm. 2013;37:64. [Google Scholar]
- 10.Kabalka GW, Varma RS. In: Comprehensive Organic Synthesis. 1. Trost BM, Fleming I, editors. Vol. 8. Elsevier Ltd; Kidlington, Oxford: 1991. p. 363. Chapter 2.1. [Google Scholar]
- 11.Khan FA, Dash J, Sudheer C, Gupta RK. Tetrahedron Lett. 2003;42:7783. [Google Scholar]
- 12.Lipshutz BH, Ghorai S. Aldrichimica Acta. 2012;45:3. [PMC free article] [PubMed] [Google Scholar]
- 13.Lipshutz BH, Ghorai S, Abela AR, Moser R, NIshikata T, Duplais C, Krasovskiv A, Gaston RD, Gadwood RC. J Org Chem. 2011;76:4379. doi: 10.1021/jo101974u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Sheldon RA. Green Chem. 2007;9:1273. [Google Scholar]; b) Lipshutz BH, Isley NA, Fennewald JC, Slack ED. Angew Chem, Int Ed. 2013;52:10952. doi: 10.1002/anie.201302020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wurster JA, Wang EH, Yee RC, Allergan Inc Kinase Inhibitors. 20070032478. US Patent. 2007 Feb 8;
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.