

Abstract

The third generation designer amphiphile/surfactant, “Nok” (i.e., SPGS-550-M; β-sitosteryl polyoxoethanylsuccinate), soon to be commercially available from Aldrich, can be prepared in two steps using an abundant plant feedstock, β-sitosterol, together with succinic anhydride and PEG-550-M. Upon dissolution in water it forms nanomicelles that serve as nanoreactors, which can be characterized by both cryo-TEM and dynamic light scattering analyses. Several transition metal-catalyzed reactions have been run under micellar conditions to evaluate this surfactant relative to results obtained in nanoparticles composed of TPGS-750-M (i.e., a second generation surfactant). It is shown that Nok usually affords yields that are, in general, as good or better than those typically obtained with TPGS-750-M, and yet is far less costly.
INTRODUCTION
By far the greatest contributor to organic waste, estimated to be as much as 85% or more, created by the chemical enterprise is in the form of usually hazardous organic solvents.1 Minimizing their use in general,2 at any scale, whether at industrial, governmental, or academic laboratories, is an especially important goal not only insofar as the teachings of green chemistry are concerned, but for sustainability in general. And while several alternative reaction media exist as options,3 the most generally preferred medium is water.4 Of course, substrate solubility issues oftentimes preclude use of water as a true solvent, especially at ambient temperatures, and hence, significant heating (e.g., via microwave)5 may be necessary.6 One attractive alternative, akin to the approach taken by Nature, is to apply the well-known concepts of micellar catalysis,7 where an amphiphile is solubilized in water and (above its critical micelle concentration) spontaneously forms nanoparticles, the cores of which provide the organic medium in which homogeneous catalysis can take place. With the spacial capacity of these nanomicelles being limited (by controlling the amount of amphiphile used), high concentrations of reactants and catalyst result, and the chemistry oftentimes ensues without recourse to heat.8 Reactions run under these very mild conditions are typically very clean.
Since nanomicelles supply, by virtue of their inner lipophilic cores, minimum amounts of the reaction solvent, and given the huge role that organic solvents play in organic reactions,9 the nature of the surfactant can oftentimes be crucial in determining the success of a given reaction. Moreover, the amphiphile should be chosen wisely to align with the “12 Principles of Green Chemistry”,10 that is, it should be “benign by design’,11 and hence, environmentally innocuous. To meet these crucial criteria of solvent assistance and environmental acceptance, our first- and second-generation surfactants were formulated as (racemic) vitamin E derivatives: initially PTS12 and more recently, TPGS-750-M13 (Figure 1). Both, as items of commerce, enable a growing library of “name” (cross-coupling) reactions to be conducted in water at room temperature.8 In an effort to further reduce, in general, the cost of these amphiphiles by avoiding the high variability in the supply of racemic vitamin E, we have designed a third generation surfactant. In this case, we chose the natural phytosterol β-sitosterol (Figure 2), which is a cholesterol mimic14 used widely in the food industry, as the lipophilic portion. Taken together with a succinate (diester) spacer and MPEG-550 (a monomethylated polyethylene glycol), the amphiphile polyoxyethanyl β-sitosteryl succinate (SPGS-550-M) results, more easily referred to as “Nok” (Figure 3). In this report, therefore, we document the design, preparation, and physical features of Nok, and its direct comparison in terms of enabling cross-coupling chemistry in aqueous media with its second-generation precursor TPGS-750-M.
Figure 1.

Structural comparison of first two generations of surfactants: PTS (1), TPGS-750-M (2).
Figure 2.

Structures of several common sterols
Figure 3.

Structure of the 3rd-generation surfactant Nok (average MW 1056).
RESULTS AND DISCUSSION
Nok vs. TPGS-750-M: distinguishing features
Both Nok and TPGS-750-M are composed of a succinic acid spacer group and an MPEG that balance the lipophilicity of the sterol and vitamin E residues, respectively. The hydrophilic-lipophilic balance for Nok is 10, as it is for PTS, while that for TPGS-750-M is 13, indicative of its somewhat greater hydrophilic character. Nok relies on a naturally occurring plant steroid, the hydrocarbon in which is predominantly cyclic in nature, while the latter contains mainly a linear array of nonpolar groups. Secondly, the appearance of neat material is different, as illustrated in Figure 4, although both TPGS-750-M and Nok are waxy solids at room temperature. Dynamic light scattering (DLS)15 experiments indicate that Nok and analog SPGS-600 (prepared with PEG-600 instead of MPEG-550) lead to nanoparticles that are similar in size to those found with TPGS-750-M (Table 1). Elongating the PEG/MPEG chain, on average, leads to smaller particles. Thus, while the commonly used excipient TPGS-1000 exists as ca. 15 nm spheres, TPGS-750-M was engineered to give larger nanomicelles that provide increased internal volume capacity and greater surface area (and hence, greater binding constants) for reactions to occur.13 Likewise, Nok was designed to form nanoparticles in the 45–60 nm range in water, and this was accomplished using MPEG-550. The larger (M)PEGs, such as MPEG-750 and PEG-1000, led to smaller nanomicelles (14 and 25 nm, respectively), the chemistry in which was also, as expected, less effective (vide infra).
Figure 4.

Neat surfactants; (A) TPGS-750-M, (B) Nok.
Table 1.
Average diameter of surfactants in water
| surfactant | average diameter (nm)a |
|---|---|
| TPGS-750-M | 49 |
| TPGS-1000 | 15 |
| Nok (SPGS-550-M) | 46 |
| SPGS-600 | 59 |
| SPGS-750-M | 14 |
| SPGS-1000 | 25 |
| CPGS-750-Mb | 25 |
| PTS | 23 |
| PSSc | 10 |
Micelle size was determined by Dynamic Light Scattering (DLS) at 2 wt % concentration in degassed water
Polyoxyethanyl cholesteryl sebacate.
Polyoxyethanyl β-sitosteryl sebacate.
Although the average size of TPGS-750-M and Nok nanoparticles is roughly comparable (as determined by DLS), the particle shape for each is remarkably dissimilar. Cryo-TEM images show that while TPGS-750-M forms mainly spherical particles,13 Nok forms an intricate array of worm-like micelles (Figure 5). The difference in cost between the two surfactants is also nontrivial: thus, while MPEG-550 and MPEG-750 are comparably priced, β-sitosterol (which is supplied as a mix of sterols; Figure 2), is far less costly than is racemic vitamin E. Since the procedure used to make each surfactant leads to high overall yields, the overriding factor comes down to the choice of the lipophilic component.
Figure 5.

Cryo-TEM image of (A) TPGS-750-M; (B) Nok
Synthesis of Nok
Nok was synthesized in two-steps (Scheme 1) following the published route used for the preparation of TPGS-750-M.13 The isolable solid intermediate, β-sitosterol succinate, was formed initially from treatment of β-sitosterol with succinic anhydride (1.6 equiv) in the presence of Et3N. The resulting carboxylic acid was then esterified with a PEG-diol or monomethylated-PEG (MPEG) to afford several related surfactants varying in their PEG length, including Nok, SPGS-600, SPGS-750-M, and SPGS-1000. This second step was performed using a Dean-stark trap that helped eliminate water efficiently from the reaction. Couplings with PEG-diols PEG-600 and PEG-1000 gave the expected lower yields from double esterification taking place at each terminus of these diols. The structure of each surfactant was elucidated by 1H NMR, 13C NMR, MS, and IR data. Although Nok could be used as obtained from these two steps, it invariably contained ca. 5+% of β-sitosterol, originating from transesterification of the initial succinate ester. If desired, this “impurity” could be removed by simple flushing through a silica gel column with hexanes followed by 50% v/v of EtOAc/hexanes. Both the silica gel and recovered mix of solvents could be recycled for this purpose. Alternatively, its presence in most subsequent coupling reaction mixtures was of no apparent consequence.
Scheme 1.

Two-step synthesis of Nok from β-sitosterol
Representative reactions: Nok vs. TPGS-750-M
As noted earlier (vide supra), Nok was designed to be economically attractive relative to the vitamin E-based surfactants PTS and TPGS-750-M. However, as was true for replacement of PTS by TPGS-750-M, the chemistry in Nok would have to be, in general, as good or better than that enabled by its second-generation precursor. Thus, an extensive study of side-by-side comparison reactions between Nok and TPGS-750-M was undertaken.
Olefin metathesis reactions
An initial screening in a challenging cross-metathesis (CM) reaction involving methyl vinyl ketone (MVK) mediated by the Grubbs-2 catalyst revealed that both surfactants gave the same level of conversion under a given set of conditions (in water at rt; Table 2, entries 2, 3). The background reaction “on water” (i.e., in the absence of a surfactant; entry 1) was minimal and not competitive, as seen in many related situations.16 Nok analogs bearing longer PEG chains (entries 4–6) gave modest results, although in all cases slower reactions and lower levels of conversion were seen. Perhaps most informative is the comparison between Nok and the structurally related cholesterol-derived amphiphiles CPGS-750-M (entry 7) and PSS (entry 9), which form ca. 25 nm particles in water and are not as effective notwithstanding their very similar lipophilic core (Figure 6). Clearly, larger micelles on the order of ca. 50–60 nm in diameter appear to afford the best results.
Table 2.
Surfactant screening: olefin cross-metathesis

| ||
|---|---|---|
| Entry | Surfactant | Conversion (%)a |
| 1 | none | 30 |
| 2 | TPGS-750-M | 77 |
| 3 | Nok | 76 |
| 4 | SPGS-600 | 71 |
| 5 | SPGS-750-M | 63 |
| 6 | SPGS-1000 | 63 |
| 7 | CPGS-750-M | 44 |
| 8 | PTS | 71 |
| 9 | PSS | 62 |
Determined by 1H NMR spectroscopy. Conditions: 0.5 mmol alkene, 1.5 mmol MVK (3 equiv).
Figure 6.
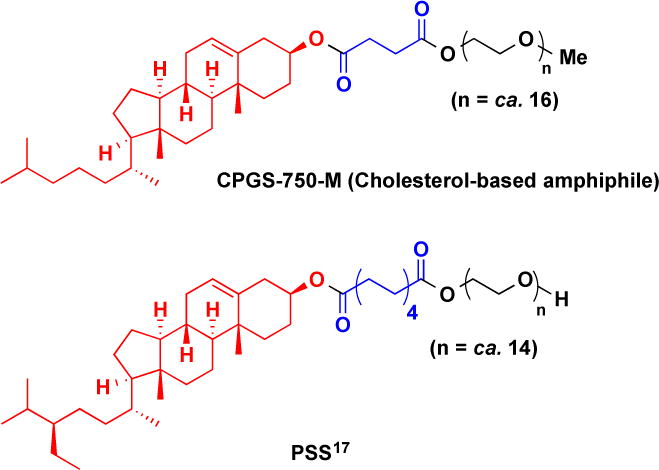
Various amphiphiles studied
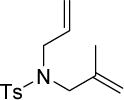
A series of olefin cross- and ring-closing metathesis reactions was conducted to compare and contrast the quality of these couplings in each surfactant, with results shown in Table 3. Under otherwise identical conditions, reactions in two weight percent Nok/H2O provide similar yields without additional optimization. The encouraging results provided an early indication that nanomicelles composed of Nok can readily replace those formed by TPGS-750-M, thereby further reducing the cost of this micellar catalysis. Notably, standard use of chlorinated reaction solvents can be avoided.18
Table 3.
Cross- and Ring-Closing Metathesis: Nok vs. TPGS-750-M
|
| ||||||
|---|---|---|---|---|---|---|
| Entry | Alkene | Partner | Product | TPGS-750-M yield(%)a | Nok yield(%)a | Time (h) |
| 1b |

|

|

|
72 | 74 | 12 |
| 2c |

|

|

|
89 | 86 | 12 |
| 3c |

|

|
|
81 | 84 | 24 |
| 4d |

|

|
81 | 88 | 6 | |
| 5d |
|

|
98 | 96 | 18 | |
Isolated yield of chromatographically pure materials.
Conditions: alkene (0.5 mmol, 1 equiv), MVK (1.5 mmol, 3 equiv).
Reaction: alkene (0.5 mmol, 1 equiv), acrylate (1.0 mmol, 2 equiv).
Reaction: diene (0.25 mmol).
These reactions undergo a change in appearance over time (Figure 7). Initially, the mixture appears quite heterogeneous, becoming brightly colored and pseudo-homogeneous with continued stirring over time. After 4 h, the mixture appears consistently milky.
Figure 7.

Appearance of a cross metathesis reaction (Table 3, entry 1) at different reaction times.
The impact of pH on olefin CM reactions under these aqueous conditions was evaluated, as previously it had been shown that by lowering the pH to ca. 2–3, the desired alkenes can be formed at a far greater rate by protonation of a phosphine ligand, thereby freeing a coordination site on the ruthenium catalyst.13a Hence, by adding a small amount of KHSO4 salt (0.02 M) into each reaction mixture, the coupling rate of the otherwise challenging type II olefin MVK is significantly enhanced (Table 4). Again, both amphiphiles gave comparable yields.
Table 4.
Effect of pH on olefin cross-metathesis reactions in water at rt

| ||
|---|---|---|
| Surfactant | Time (h) | Conversion (%)a |
| 2.0 wt % TPGS-750-M | 12 | 77 |
| 2.0 wt % Nok | 12 | 76 |
| 0.02 M KHSO4 /2.0 wt % TPGS-750-M | 4 | 86 |
| 0.02 M KHSO4 /2.0 wt % Nok | 4 | 84 |
Determined by 1H NMR spectroscopy; conditions: 0.5 mmol alkene and 1.5 mmol MVK (3 equiv).
Pd-catalyzed cross-couplings
An extensive study comparing aqueous solutions of Nok with those containing TPGS-750-M was made for several Pd-catalyzed cross-couplings involving both C-C as well as C-heteroatom bond constructions. Thus, in the former category the following name reactions were examined: Suzuki-Miyaura, Sonogashira, Heck, Stille, and Negishi-like couplings. Studies on carbonheteroatom bond formation included aminations (C-N) and Miyaura borylations (C-B). As with prior studies (vide supra), all reactions were run on the same scale, and in water at ambient temperature (ca. 23 °C).
Suzuki-Miyaura reactions between aryl bromides and arylboronic acids19 in two weight percent surfactant in water are shown in Table 5. Both activated and deactivated substrates provided nearly quantitative yields of desired products. Reactions in Nok give results comparable to those seen using TPGS-750-M in all cases. These reactions are complete in 2–11 hours without by-product formation at room temperature (other than some homocoupling from the excess boronic acid present). These reactions tolerate a variety of functional groups, including cyano- and trifluoromethyl moieties (entries 1, 4). The reaction conditions are also applicable to heteroaromatics in both coupling partners (entry 3).
Table 5.
Suzuki-Miyaura Couplings Reactions: Nok vs. TPGS-750-M

| ||||||
|---|---|---|---|---|---|---|
| Entry | Aryl halide | Boronic acid | Product | TPGS-750-Ma yield(%) | Noka yield(%) | Time (h) |
| 1 |

|

|

|
99 | 99 | 2 |
| 2 |

|

|

|
94 | 99 | 2 |
| 3 |

|

|

|
90 | 95 | 4 |
| 4 |

|
|

|
94 | 99 | 4 |
Isolated yields of chromatographically pure materials; reaction conditions: alkyl bromide (0.5 mmol, 1 equiv), boronic acid (0.75–1.00 mmol), triethylamine (3 equiv), catalyst Pd(dtbpf)Cl2 (2 mol %), and 2 wt % of surfactant/H2O.
A similar trend was observed in Sonogashira couplings. Previously reported comparisons between TPGS-750-M and PTS were made in 3.0 wt % surfactant/water.20 In this study, all the reactions have been done with a lower concentration of amphiphile, at 2.0 wt % surfactant/water, and the yields were maintained (Table 6). Aromatic and heteroaromatic substrates bearing electron-donating or electron-withdrawing groups readily participated. Only with the phenylacetylene/p-bromobenzonitrile pair (entry 3) did a lower isolated yield result, although this is likely due to the highly crystalline nature of the bromide that can give varying results based on the effectiveness of stirring from reaction to reaction.
Table 6.
Sonogashira Couplings: Nok vs. TPGS-750-M

| ||||||
|---|---|---|---|---|---|---|
| Entry | Aryl halide | Alkyne | Product | TPGS-750-Ma yield(%) | Noka yield(%) | Time (h) |
| 1 |

|
|

|
98 | 98 | 24 |
| 2 |

|

|

|
94 | 95 | 28 |
| 3 |

|

|

|
97 | 90 | 2 |
| 4 |

|
|

|
93 | 93 | 15 |
Isolated yields of chromatographically pure materials. Reaction conditions: alkyl bromide (0.5 mmol, 1 equiv), boronic acid (0.65–1.00 mmol), XPhos (2.5 mol %), triethylamine (3 equiv), catalyst Pd(CH3CN)2Cl2 (2 mol %), and 2 wt % of surfactant/H2O.
Heck couplings of both aryl bromides and aryl iodides in 2.0 wt % surfactant/water were also compared. Aryl bromides reacted more slowly than did aryl iodides, as expected. Thus, while the coupling shown in Table 7 (entry 5) required 72 hours to reach completion in 2 wt. % Nok/H2O; with 5 wt % Nok in water and the trivial addition of NaCl the reaction time was reduced to 19 hours (Table 8, entry 4). Both acrylate and styrene-type partners react smoothly in both surfactants, giving comparable yields. As with PTS and TPGS-750-M, the presence of NaCl in Nok/water leads to a “salting out” effect21, which increases reaction rates in Heck couplings.12 This may be due to increased particle size, and the resulting opportunity for educts and catalyst to spend more time within the lipophilic cores of these nanoparticles and less time exchanging between smaller micelles (i.e., have greater binding constants). The specific impact on Nok is illustrated in Figure 8, documenting (via DLS) that with increasing levels of salt concentration, particle size goes up correlating with, in the case of Heck reactions in 5 wt % Nok, an increasing rate of reaction: no NaCl, 1 M, 2 M, and 3 M gave 29, 57, 76, and 90% yield, respectively (Table 8).
Table 7.
Heck Couplings in Nok vs. TPGS-750-M
| Entry | Aryl halide | Partner | Product | TPGS-750-Ma yield(%) | Noka yield(%) | Time (h) |
|---|---|---|---|---|---|---|
| 1 |

|

|

|
99 | 99 | 4 |
| 2 |

|

|
|
92 | 93 | 5 |
| 3 |

|

|
|
99 | 99 | 5 |
| 4 |

|

|

|
99 | 99 | 5 |
| 5 |

|

|

|
84 | 79 | 72 |
Isolated yields of chromatographically pure materials. Reaction conditions: alkyl bromide (0.5 mmol, 1 equiv), acrylate/styrene (1.00 mmol, 2 equiv), triethylamine (3 equiv), catalyst, Pd(t-Bu3P)2 (2 mol %), and 2 wt % of surfactant/H2O.
Table 8.
Effect of NaCl concentration on a Heck reaction of an aryl bromide

| ||
|---|---|---|
| Entry | NaCl concentration (M) | Yield (%)a |
| 1 | 0 | 29 |
| 2 | 1 | 57 |
| 3 | 2 | 76 |
| 4 | 3 | 90 |
Isolated yield of chromatographically pure materials. Reaction conditions: bromobenzene (0.5 mmol, 1 equiv), t-butyl acrylate (1.00 mmol, 2 equiv), triethylamine (3 equiv), catalyst, Pd(t-Bu3P)2 (2 mol %), NaCl, and 5 wt % of surfactant/H2O.
Figure 8.

Particle size of Nok in water as a function of wt. % and NaCl concentration
Stille couplings22 proceeded smoothly with full conversion in relatively short periods of time, using either surfactant. Similar to Heck reactions (vide supra), the presence of NaCl accelerates these couplings. A variety of substrates has been examined (Table 9), with most leading to high yields of the desired products. The enol ether obtained in moderate yield (entry 3) reflects its volatility on isolation and not the extent of conversion or the quality of bond formation.
Table 9.
Comparison of Nok vs. TPGS-750-M in Stille Couplings
|
| ||||||
|---|---|---|---|---|---|---|
| Entry | Aryl halide | Partner | Product | TPGS-750-Ma yield(%) | Noka yield(%) | Time (h) |
| 1 |

|

|

|
98 | 99 | 2 |
| 2 |

|

|

|
100 | 99 | 4 |
| 3 |

|

|

|
58 | 56 | 8 |
| 4 |

|

|

|
83 | 84 | 18 |
| 5 |

|

|

|
84 | 97 | 4 |
Isolated yields of chromatographically pure materials. Conditions: aryl bromide (0.25 mmol, 1 equiv), stannyl reagent (0.275 mmol, 1.1 equiv), catalyst Pd(t-Bu3P)2 (2 mol %), base DABCO (3 equiv), NaCl (1 equiv) and 2 wt % surfactant/H2O.
For carbon-heteroatom bond formations, Miyaura borylations23 and aminations were selected as representative examples. Borylations of aryl bromides with B2pin2 afford good yields using either TPGS-750-M or Nok (Table 10). Amination reactions using either aryl amines24 or ammonia surrogates25 (entries 3, 4) are presented in Table 11. The former proceed as observed previously at room temperature to give unsymmetrical diarylamines,24 while the latter were conducted at 50 °C. By increasing the catalyst loading from 0.5 to 2 mol %, and an increase in ligand from 2 to 4 mol %, aminations with ammonia equivalents, such as a carbamate, can be conducted in water at room temperature.25 Use of an in-situ generated base derived from KOH/TIPSOH helps to further accelerate this reaction, leading to full conversion in 15 hours. Identical results were obtained in either surfactant, Nok or TPGS-750-M (Table 12).
Table 10.
Nok vs. TPGS-750-M in Miyaura borylations

| ||||
|---|---|---|---|---|
| Entry | Product | TPGS-750-Ma Vield(%) | Noka yield(%) | Time (h) |
| 1 |

|
90 | 94 | 13 |
| 2 |

|
78 | 83 | 4 |
| 3 |

|
74 | 82 | 26 |
| 4 |

|
94 | 95 | 24 |
Isolated yields of chromatographically pure materials. Conditions: aryl bromide (0.5 mmol, 1 equiv), B2pin2 (0.55 mmol, 1.1 equiv), catalyst Pd(t-Bu3P)2 (3 mol %), KOAc (3 equiv), and 2 wt % of surfactant/H2O.
Table 11.
Nok vs. TPGS-750-M in amination reactions
|
| ||||||
|---|---|---|---|---|---|---|
| Entry | Aryl halide | Partner | Product | TPGS-750-Ma yield(%) | Noka yield(%) | Time (h) |
| 1b |

|
|

|
95 | 91 | 21 |
| 2b |

|

|

|
75 | 75 | 10 |
| 3c,d |

|

|

|
85 | 85 | 24 |
| 4c,e |

|

|

|
96 | 93 | 24 |
Isolated yields.
Reaction conditions: aryl bromide (1.00 mmol, 1 equiv), toluidine (1.20 mmol, 1.2 equiv), catalyst [(π-allyl)PdCl]2 (0.5 mol %), cBRIDP (2 mol %), K-O-t-Bu (1.5 equiv) and 2 wt % of surfactant/H2O; at rt.
Na-O-t-Bu (1.5 equiv); reaction was run at 50 °C, 24 h using [(π-allyl)PdCl]2 (0.5 mol %), cBRIDP (2 mol %), K-O-t-Bu (1.5 equiv) and 2 wt % of surfactant/H2O.
Reaction conditions: aryl bromide (1.00 mmol, 1 equiv), t-butyl carbamate (1.50 mmol, 1.5 equiv).
Reaction conditions: aryl bromide (0.50 mmol, 1 equiv), ethyl carbamate (0.60 mmol, 1.2 equiv).
Table 12.
Amination in Nok vs. TPGS-750-M

| |
|---|---|
| surfactant | Yield (%)a |
| TPGS-750-M | 98 |
| Nok (SPGS-550-M) | 98 |
Isolated yields.
Another example of a valued Pd-catalyzed cross-coupling is that between an aryl and alkyl halide, mediated by in situ-generated organozinc halides (i.e., Negishi-like couplings), as we previously reported (Table 13).13a In order to avoid prior formation of an organozinc halide, portion-wise addition of an alkyl halide partner to the Pd catalyst, Zn dust, and TMEDA, leads to the desired cross-coupling reaction in both surfactants over 48 hours. Dehalogenated by-products were observed by GCMS, albeit in less than 5%. After chromatographic purification, 85 and 86% of the product was obtained in TPGS-750-M and Nok, respectively.
Table 13.
Negishi-like coupling in Nok vs. TPGS-750-M

| |
|---|---|
| surfactant | Yield (%)a |
| TPGS-750-M | 85 |
| Nok (SPGS-550-M) | 86 |
Isolated yields.
Recycling of Nok
To assess the opportunity to recycle the aqueous phase containing Nok, a Suzuki-Miyaura coupling was examined as a representative reaction. Each cycle was followed by a standard inflask extraction of the product using minimal amounts of an organic solvent (e.g., EtOAc, 3 times). To the aqueous phase remaining in the flask, fresh catalyst, base, and coupling partner were reintroduced. After six cycles, the reaction afforded the same full conversion to the desired product (Table 14), confirming that the surfactant remains available in the water for continuous service in this micellar catalysis.
Table 14.
Recycling of Nok in water

| |||||
|---|---|---|---|---|---|
| Cycle(%conversion)a
| |||||
| 1 | 2 | 3 | 4 | 5 | 6 |
| >99 | >99 | >99 | >99 | >99 | >99 |
Determined by 1H NMR spectroscopy at 400 MHz. Reaction conditions: alkyl bromide (0.5 mmol, 1 equiv), boronic acid (1.00 mmol), triethylamine (3 equiv), Pd(dtbpf)Cl2 (2 mol %), and 2 wt % of surfactant/H2O.
E Factor determination
As an indication of the potential environmental savings these couplings offer insofar as organic waste is concerned, an E Factor4,26 determination was made using a Suzuki-Miyaura coupling as a model case (Scheme 2) based on (1) the total organic solvent used in the reaction and extractive workup, and (2) the combined organic solvent use and waste water associated with the coupling. Although this latter number is historically not part of the E Factor calculation given the oftentimes large volumes of water involved,4 it is useful nonetheless since Suzuki-Miyaura cross-couplings are routinely run in water-miscible solvents, and hence, the waste water produced contains organic waste Moreover, calculation of an associated E Factor can lead to insight as to how low these numbers can be even when water is included, since these reactions typically run at high global concentrations. As illustrated in Scheme 2, the E Factors are very low, reflecting both the high yield of the coupling and the minimum investment of both organic solvent and water in the overall process. Clearly, with options for recycling of the aqueous reaction medium, E Factors that include waste water go down even further. By way of comparison, typical E Factors (organic solvent only) in the pharmaceutical industry range from 25–100.4
Scheme 2.

E Factor model reaction
CONCLUSIONS
The third generation sterol-based surfactant, Nok (SPGS-550-M), has been efficiently prepared in two steps via two successive esterification reactions of inexpensive β-sitosterol with succinic anhydride, followed by esterification of the resulting acid with MPEG-550. The resulting surfactant, which is currently an item of commerce (Aldrich catalog number 776033) forms nanometer-sized micelles in water that accommodate various types of Ru- and Pd-catalyzed coupling reactions, almost all of which take place at room temperature. These studies document that the newly engineered surfactant Nok can substitute in most types of reactions for the second-generation amphiphile TPGS-750-M that is based on the more expensive (racemic) vitamin E.
EXPERIMENTAL SECTION
β-Sitosterol succinate (<10 g scale)
A 100 mL round-bottom flask with magnetic stir bar and septum was charged with β-sitosterol (≥70% purity) (8.33 g, 20.00 mmol) and succinic anhydride (3.20 g, 32.00 mmol). A rubber septum was put on and toluene (40 mL) was added via syringe as solvent. To this well-stirring mixture, triethylamine (0.7 mL, 5.00 mmol) was added via syringe and the reaction flask was placed in a 60°C oil bath and the stirring continued until the reaction reached completion (followed by TLC). The resulting solution was allowed to warm to rt, at which point it was treated with water and extracted with DCM. The combined organic extracts were washed with 2 M HCl (3 × 50 mL), water (3 × 50 mL) and brine (80 mL), and then dried over anhydrous Na2SO4. Concentration of the solvent in vacuo was followed by exposure to high vacuum overnight to afford β-sitosterol succinate (10.16 g, 98%) as a white solid. The structure of this intermediate was confirmed by mp 150–151 °C, lit27 mp 151–153 °C; IR(neat) 2938, 2867, 1731, 1711, 1466, 1442, 1378, 1179, 1033, 802 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.32–5.28 (d, J = 4.6 Hz, 1H), 4.62–4.51 (m, 1H), 2.64–2.57 (t, J = 6.1 Hz, 2H), 2.57–2.50 (t, J = 6.1 Hz, 2H), 2.78–2.21 (d, J = 7.6 Hz, 2H), 1.97–0.51 (m, 45H); 13C NMR (500 MHz, CDCl3) δ; 177.92, 171.51, 139.52, 122.72, 74.54, 56.68, 56.04, 50.00, 45.83, 42.30, 39.71, 38.00, 36.94, 36.57, 36.15, 33.92, 31.85, 28.77, 28.23, 27.68, 26.09, 24.28, 23.06, 21.02, 19.81, 19.30, 19.03, 18.77, 11.89; MS (ESI) m/z 537 [M+Na]+; HRMS (ESI) calcd for C33H54O4Na [M+Na]+ 537.3920, found 537.3909 (∆ = 1.1 mDa, 2.0 ppm).
Nok (SPGS-550-M; <10 g scale)
β-Sitosterol succinate (4.00 g, 7.77 mmol), MPEG-550-M (6.41 g, 11.7 mmol), and p-TsOH (0.21g, 1.11 mmol), were added into a 100 mL round-bottom flask. Toluene (40 mL) was added via syringe, and then the mixture was refluxed using a Dean-Stark trap until complete. After cooling to rt, the mixture was poured into saturated aqueous NaHCO3 and extracted with DCM. The combined organic extracts were washed with saturated aqueous NaHCO3 (3 × 50 mL), brine (2 × 80 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford a pale-yellow, viscous liquid. The oil was poured on top of a silica gel bed, and then first eluted with 50% v/v EtOAc/hexane to remove an impurity, followed by 10% MeOH/DCM to obtain the product. Concentration under vacuum followed by storage under high vacuum overnight afforded Nok as an off-white waxy solid (7.61 g, 92%). IR (neat) 2935, 2868, 1731, 1465, 1345, 1280, 1242, 1107, 1030, 1003, 963, 843, 668, 556 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.37–5.33 (d, J = 3.7 Hz, 1H), 4.66–4.54 (m, 1H), 4.28–4.18 (t, J = 4.6 Hz, 2H), 3.74–3.51 (m, PEG), 3.39 (s, 3H), 2.66–2.56 (m, 4H), 2.32–2.26 (d, J = 7.6 Hz, 2H), 2.02–0.61 (m, 45H); 13C NMR (500 MHz, CDCl3) δ; 171.85, 171.10, 139.15, 122.23, 73.85, 71.54, 70.18, 68.65, 63.37, 58.57, 56.28, 55.64, 49.62, 45.41, 41.91, 39.33, 37.67, 36.58, 36.17, 35.74, 33.54, 31.55, 29.00, 28.66, 27.85, 27.34, 25.70, 23.90, 22.68, 20.05, 19.47, 18.92, 18.71, 18.43, 11.61, 11.49; MS (ESI) m/z 1079 [M+Na]+.
General procedure for cross metathesis (Table 3)
Grubbs second generation catalyst (8.5 mg, 0.010 mmol) was charged under an Ar atmosphere into a 2–5 mL microwave vial containing a magnetic stir bar and Teflon-lined septum. The alkene (0.50 mmol) and acrylate (1.00 mmol) or ketone (1.50 mmol) were added sequentially into the vial, then an aliquot of surfactant solution (1.0 mL, 2 wt. % in degassed water) was added via syringe under a positive flow of Ar. The reaction was allowed to vigorously stir for 12–18 h at rt. The reaction mixture was then diluted with EtOAc and passed through a silica gel bed and further washed with EtOAc to collect the coupling product. All volatile solvents were removed in vacuo to obtain crude product, which was further purified by flash chromatography on silica gel.
(E)-5-(2-((t-Butyldimethylsilyl)oxy)phenyl)pent-3-en-2-one (Table 3, entry 1)
Following the general procedure using (2-allylphenoxy)(t-butyl)dimethylsilane (124 mg, 0.50 mmol), methyl vinyl ketone (106 mg, 1.50 mmol), and Grubbs second generation catalyst (8.5 mg, 0.010 mmol), the reaction was allowed to stir for 12 h. After column chromatography, the product was obtained as a colorless liquid (110 mg, 74%). 1H NMR (400 MHz, CDCl3) δ 7.15–7.09 (td, J = 7.6, 1.7 Hz, 1H), 7.09–7.06 (dd, J = 7.6, 1.7 Hz, 1H), 6.98–6.85 (m, 2H), 6.83–6.78 (dd, J = 9.0, 1.0 Hz, 1H), 6.03–5.95 (dt, J = 1.7, 16.1 Hz, 1H), 3.54–3.48 (dd, J = 8.0, 1.5 Hz, 2H), 2.21 (s, 3H), 0.98 (s, 9H), 0.26 (s, 6H).13a
(E)-t-Butyl 4-(2-((t-butyldimethylsilyl)oxy)phenyl)but-2-enoate (Table 3, entry 2)
Following the general procedure using (2-allylphenoxy)(t-butyl)dimethylsilane (124 mg, 0.50 mmol), t-butyl acrylate (128 mg, 1.00 mmol), and Grubbs second generation catalyst (8.5 mg, 0.010 mmol), the reaction was allowed to stir for 12 h. After column chromatography, the product was obtained as a colorless liquid (149 mg, 86%). 1H NMR (400 MHz, CDCl3) δ 7.16–7.08 (m, 2H), 7.05–6.96 (dt, J = 15.4, 6.6 Hz, 1H), 6.94–6.88 (td, J = 7.5, 1.2 Hz, 1H), 6.84–6.79 (dd, J = 7.9, 1.2 Hz, 1H), 5.72–5.65 (dt, J = 15.6, 1.7 Hz, 1H), 3.50–3.45 (dd, J = 6.5, 1.7 Hz, 2H), 1.47 (s, 9H), 1.01 (s, 9H), 0.25 (s, 6H).16
t-Butyl (E)-4-(4-methoxyphenyl)but-2-enoate (Table 3, entry 3)
Following the general procedure using 1-allyl-4-methoxybenzene (74 mg, 0.50 mmol), t-butyl acrylate (128 mg, 1.00 mmol), and Grubbs second generation catalyst (8.5 mg, 0.010 mmol), the reaction was allowed to stir for 24 h. After column chromatography, the product could not be fully separated from excess acrylate; it was obtained as a colorless liquid (105 mg, 84%). 1H NMR (400 MHz, CDCl3) δ 7.13–7.07 (d, J = 8.5 Hz, 2H), 7.03–6.94 (dt, J = 15.6, 6.6 Hz, 1H), 6.89–6.84 (d, J = 8.5 Hz, 2H), 5.75–5.68 (d, J = 15.6 Hz, 1H), 3.80 (s, 3H), 3.47–3.42 (d, J = 6.6 Hz, 2H), 1.48 (s, 3H)16
General procedure for ring-closing metathesis (Table 3)
Grubbs second generation catalyst (3.4 mg, 0.004 mmol) was charged under an Ar atmosphere in a 2–5 mL microwave vial with magnetic stir bar and Teflon-lined septum. Diene (0.20 mmol) was added to the vial and then an aliquot of surfactant solution (2.0 mL, 2 wt. % in degassed water) was added via syringe under positive flow of Ar. The reaction was allowed to vigorously stir for 6–18 h at rt. The reaction mixture was then diluted with EtOAc and passed through silica gel bed and further washed with EtOAc to collect the coupling product. All volatile solvent was removed in vacuo to obtain crude product that was further purified by flash chromatography on silica gel.
1-Tosyl-1,2,3,6-tetrahydropyridine (Table 3, entry 4)
Following the general procedure using N-allyl-N-(but-3-en-1-yl)-4-methylbenzenesulfonamide (53 mg, 0.20 mmol), the reaction was allowed to stir for 6 h. After column chromatography, the product was obtained as a white solid (47 mg, 88%). 1H NMR (400 MHz, CDCl3) δ 7.70–7.66 (d, J = 8.0 Hz, 2H), 7.35–7.30 (d, J = 8.0 Hz, 2H), 5.79–5.73 (m, 1H), 5.65–5.59 (m, 1H), 3.60–3.56 (m, 2H), 3.20–3.15 (t, J = 5.7 Hz, 2H), 2.44 (s, 3H), 2.25–2.19 (m, 2H).28
3-Methyl-1-tosyl-2,5-dihydro-1H-pyrrole (Table 3, entry 5)
Following the general procedure using N-allyl-4-methyl-N-(2-methylallyl)benzenesulfonamide (53 mg, 0.20 mmol), the reaction was allowed to stir for 18 h. After column chromatography, the product was obtained as a white solid (46 mg, 96%). 1H NMR (400 MHz, CDCl3) δ 7.75–7.71 (d, J = 8.0 Hz, 2H), 7.35–7.30 (d, J = 8.0 Hz, 2H), 5.27–5.23 (m, 1H), 4.11–4.05 (m, 1H), 4.00–3.95 (m, 2H), 2.45–2.42 (s, 3H), 2.44 (s, 3H), 1.66 (s, 3H).29
General procedure for Suzuku-Miyaura Couplings (Table 5)
Arylboronic acid (0.75–1.00 mmol) and Pd(dtbpf)Cl2 (6.5 mg, 0.01 mmol) were added under an Ar atmosphere in a 5.0 mL microwave vial with magnetic stir bar and Teflon-lined septum. Aryl bromide (0.50 mmol), triethylamine (0.21 mL, 1.50 mmol) and surfactant solution (1.0 mL, 2 wt. % in degassed water) were sequentially added into the reaction under a flow of Ar. The reaction was allowed to stir vigorously for 2–11 h. The reaction mixture was then diluted with EtOAc and passed through a silica gel bed and further washed with EtOAc to collect the coupling product. All volatile solvent was removed in vacuo to obtain crude product that was further purified by flash chromatography on silica gel.
[1,1′-Biphenyl]-3-carbonitrile (Table 5, entry 1)
Following the general procedure using 3-bromobenzonitrile (91 mg, 0.50 mmol) and phenylboronic acid (91 mg, 0.75 mmol) the reaction was allowed to stir for 2 h. After column chromatography, the product was obtained as a colorless liquid (61 mg, 99%). 1H NMR (400 MHz, CDCl3) δ 7.89–7.86 (t, J = 1.5 Hz, 1H), 7.85–7.80 (dt, J = 7.8, 1.5 Hz, 1H), 7.66–7.62 (dt, J = 7.8, 1.5 Hz, 1H), 7.60–7.54 (dt, J = 8.0, 1.5 Hz, 3H), 7.52–7.46 (tt, J = 7.3, 1.5 Hz, 2H), 7.46–7.40 (tt, J = 7.3, 1.5 Hz, 1H).30
1-Methyl-4-phenylnaphthalene (Table 5, entry 2)
Following the general procedure using 1-bromo-4-methylnaphthalene (111 mg, 0.50 mmol), and phenylboronic acid (122 mg, 1.00 mmol), the reaction was allowed to stir for 2 h. After column chromatography, the product was obtained as a colorless liquid (109 mg, 99%). 1H NMR (400 MHz, CDCl3) δ 8.14–8.06 (dd, J = 8.5, 0.5 Hz, 1H), 7.98–7.91 (dd, J = 8.3, 0.5 Hz, 1H), 7.63–7.32 (m, 8H), 2.77 (s, 3H).31
3-(Thiophen-3-yl)quinoline (Table 5, entry 3)
Following the general procedure using 3-bromoquinoline (105 mg, 0.50 mmol) and thiophen-3-ylboronic acid (96 mg, 0.75 mmol) the reaction was allowed to stir for 4 h. After column chromatography, the product was obtained as a white solid (102 mg, 95%). 1H NMR (400 MHz, CDCl3) δ 9.25–9.20 (d, J = 2.0 Hz, 1H), 8.34–8.30 (d, J = 2.2 Hz, 1H), 8.18–8.11 (d, J = 8.3 Hz, 1H), 7.90–7.85 (d, J = 7.8 Hz, 1H), 7.75–7.70 (m, 1H), 7.70–7.67 (dd, J =2.9, 1.5 Hz, 1H), 7.62–7.57 (m, 1H), 7.56–7.53 (dd, J = 5.1, 1.5 Hz, 1H), 7.53–7.50 (dd, J = 5.1, 2.9 Hz, 1H).32
2-Methoxy-6-(2-(trifluoromethyl)phenyl)naphthalene (Table 5, entry 4)
Following the general procedure using 1-bromo-2-(trifluoromethyl)benzene (113 mg, 0.50 mmol) and (6-methoxynaphthalen-2-yl)boronic acid (152 mg, 0.75 mmol) the reaction was allowed to stir for 4 h. After column chromatography, the product was obtained as a white solid (152 mg, 99%). mp 68–70 °C; 1H NMR (400 MHz, CDCl3) δ 7.81–7.75 (m, 3H), 7.73 (s, 1H), 7.62–7.56 (t, J = 7.3 Hz, 1H), 7.53–7.49 (t, J = 7.3 Hz, 1H), 7.46–7.40 (t, J = 6.8 Hz, 2H), 7.22–7.18 (m, 2H), 3.96 (s, 3H); 13C NMR (500 MHz, CDCl3) δ ; 157.97, 135.13, 133.79, 132.38, 131.27, 129.65, 128.30, 127.69, 127.26, 126.13, 126.1, 119.24, 105.61, 55.37; HRMS (FI) calcd for C18H13F3O [M]+ 302.0918, found 302.0905 (∆ = 1.3 mDa, 4.3 ppm).
General procedure for Sonogashira couplings (Table 6)
Pd(CH3CN)2Cl2 catalyst (1.3 mg, 0.005 mmol) and XPhos ligand (6.2 mg, 0.013 mmol) were combined under an Ar atmosphere in a 5.0 mL microwave vial with magnetic stir bar and Teflon-lined septum. Aryl bromide (0.50 mmol), alkyne (0.65–1.00 mmol), triethylamine (0.14 mL, 1.00 mmol) and surfactant solution (1.0 mL, 2 wt. % in degassed water) were added, respectively, via syringe into the reaction under flow of Ar. The reaction was allowed to stir vigorously for 4–28 h. The reaction mixture was then diluted with EtOAc and passed through silica gel bed and further washed with EtOAc to collect the coupling product. All volatile solvent was removed in vacuo to obtain crude product that was further purified by flash chromatography on silica gel.
2-(Dodec-1-yn-1-yl)-1-methyl-4-nitrobenzene (Table 6, entry 1)
Following the general procedure using 2-bromo-1-methyl-4-nitrobenzene (109 mg, 0.50 mmol) and 1-dodecyne (109 mg, 0.65 mmol) the reaction was allowed to stir for 24 h. After column chromatography, the product was obtained as a brown liquid (150 mg, 98%). 1H NMR (400 MHz, CDCl3) δ 8.24–8.19 (d, J = 2.0 Hz, 1H), 8.04–7.98 (dd, J = 8.6, 2.2 Hz, 1H), 7.36–7.30 (d, J = 8.6 Hz, 1H), 2.51 (s, 3H), 2.50–2.45 (t, J = 6.8 Hz, 2H), 1.69–1.59 (m, 2H), 1.53–1.42 (m, 2H), 1.40–1.20 (m, 12H), 0.93–0.85 (t, J = 6.7 Hz, 3H).22
1-Methoxy-4-(phenylethynyl)benzene (Table 6, entry 2)
Following the general procedure using 1-bromo-4-methoxybenzene (94 mg, 0.50 mmol) and ethynylbenzene (77 mg, 0.75 mmol) the reaction was allowed to stir for 28 h. After column chromatography, the product was obtained as a brown solid (99 mg, 95%). 1H NMR (400 MHz, CDCl3) δ 7.54–7.45 (m, 4H), 7.36–7.30 (m, 3H), 6.92–6.85 (d, J = 8.8 Hz, 2H), 3.84 (s, 3H).33
4-(Phenylethynyl)benzonitrile (Table 6, entry 3)
Following the general procedure using 4-bromobenzonitrile (91 mg, 0.50 mmol) and ethynylbenzene (77 mg, 0.75 mmol) the reaction was allowed to stir for 2 h. After column chromatography, the product was obtained as a yellow solid (89 mg, 88%). 1H NMR (400 MHz, CDCl3) δ 7.67–7.60 (m, 4H), 7.58–7.52 (m, 2H), 7.41–7.37 (m, 3H).34
3-(6-Chlorohex-1-yn-1-yl)quinoline (Table 6, entry 4)
Following the general procedure using 3-bromoquinoline (104 mg, 0.50 mmol) and 6-chlorohex-1-yne (117 mg, 1.00 mmol) the reaction was allowed to stir for 15 h. After column chromatography, the product was obtained as a yellow liquid (113 mg, 93%). 1H NMR (400 MHz, CDCl3) δ 8.90–8.86 (d, J = 2.0 Hz, 1H), 8.20–8.17 (d, J = 2.0 Hz, 1H), 8.11–8.07 (d, J = 8.6 Hz, 1H), 7.80–7.75 (d, J = 8.1 Hz, 1H), 7.74–7.68 (m, 1H), 7.59–7.53 (m, 1H), 3.67–3.61 (t, J = 6.5 Hz, 2H), 2.58–2.52 (t, J = 7.0 Hz, 2H), 2.06–1.97 (m, 2H), 1.91–1.79 (m, 3H). 13C-NMR (500 MHz, CDCl3) δ 152.59, 146.81, 138.28, 129.96, 129.54, 127.64, 127.54, 127.38, 118.16, 93.16, 78.80, 44.66, 31.89, 26.02, 19.02; HRMS (FI) calcd for C15H14ClN [M]+ 243.0815, found 243.0806 (∆ = 0.9 mDa, 3.7 ppm).
General procedure for Heck couplings (Table 7)
Under an Ar atmosphere, Pd(t-Bu3P)2 (5.1 mg, 0.010 mmol) was weighed into a 5.0 mL microwave vial containing a magnetic stir bar and Teflon-lined septum. An aryl halide (0.50 mmol) and acrylate/styrene (1.00 mmol) were added under a positive flow of Ar followed by surfactant solution (1.0 mL of 2 wt. % in degassed water). Triethylamine (0.21 mL, 1.50 mmol) was then added via syringe as the stoichiometric base. The mixture was stirred vigorously for 4–72 h and then diluted with EtOAc, passed through a silica gel bed and washed with EtOAc to collect the product. All volatile solvent was removed in vacuo to obtain the crude product that was further purified by flash chromatography on silica gel.
(E)-t-Butyl 3-(4-methoxyphenyl)acrylate (Table 7, entry 1)
Following the general procedure using 1-iodo-4-methoxybenzene (117 mg, 0.50 mmol), t-butyl acrylate (128 mg, 1.00 mmol) the reaction was allowed to stir for 4 h. After column chromatography, the product was obtained as a brown liquid (117 mg, 99%). 1H NMR (400 MHz, CDCl3) δ 7.58–7.52 (d, J = 16.1 Hz, 1H), 7.49–7.44 (dt, J = 8.6, 2.0 Hz, 2H), 6.93–6.87 (dt, J = 8.8, 2.0 Hz, 2H), 6.28–6.22 (d, J = 16.1 Hz, 1H), 3.84 (s, 3H), 1.54 (s, 9H).35
(E)-2-Ethylhexyl 3-(4-methoxyphenyl)acrylate (Table 7, entry 2)
Following the general procedure using 1-iodo-4-methoxybenzene (117 mg, 0.50 mmol) and 2-ethylhexyl acrylate (184 mg, 1.00 mmol), the reaction was allowed to stir for 5 h. After column chromatography, the product was obtained as yellow-brown liquid (135 mg, 93%). 1H NMR (400 MHz, CDCl3) δ 7.68–7.60 (d, J = 16.1 Hz, 1H), 7.53–7.45 (dt, J = 8.6, 2.0 Hz, 2H), 6.95–6.87 (dt, J = 8.8, 2.0 Hz, 2H), 6.36–6.28 (d, J = 15.9 Hz, 1H), 4.16–4.07 (m, 2H), 3.85 (s, 3H), 1.71–1.55 (m, 1H), 1.48–1.24 (m, 8H), 0.99–0.86 (t, J = 7.3 Hz, 6H).36
2-Ethylhexyl cinnamate (Table 7, entry 3)
Following the general procedure using iodobenzene (102 mg, 0.50 mmol) and 2-ethylhexyl acrylate (184 mg, 1.00 mmol), the reaction was allowed to stir for 5 h. After column chromatography, the product was obtained as colorless liquid (129 mg, 99%). 1H NMR (400 MHz, CDCl3) δ 7.72–7.65 (d, J =15.9 Hz, 1H), 7.58–7.51 (m, 2H), 7.42–7.36 (m, 2H), 6.50–6.43 (d, J = 15.9 Hz, 1H), 4.15–4.12 (m, 2H), 1.72–1.60 (m, 1H), 1.49–1.26 (m, 9H), 0.98–0.87 (m, 6H).37
(E)-1-Chloro-2-(4-methoxystyryl)benzene (Table 7, entry 4)
Following the general procedure using 1-iodo-4-methoxybenzene (58.5 mg, 0.25 mmol) and 1-chloro-2-vinylbenzene (69 mg, 0.50 mmol), the reaction was allowed to stir for 5 h. After column chromatography, the product was obtained as yellow liquid (61 mg, 99%, 77:23 E/Z). 1H NMR (400 MHz, CDCl3) δ 7.69–7.64 (dd, J = 7.8, 1.3 Hz, 1H), 7.52–7.47 (d, J = 8.8 Hz, 2H), 7.42–7.34 (dd, J = 11.7, 3.4 Hz, 2H), 7.23–7.33 (m, 2H), 7.21–7.14 (m, 1H), 7.07–7.00 (d, J = 16.3 Hz, 1H), 6.94–6.89 (d, J = 8.8 Hz, 2H), 6.85–6.81 (d, J = 8.8 Hz, 2H), 3.84 (s, 3H), 3.80 (s, 3H).38
t-Butyl cinnamate (Table 7, entry 5)
Following the general procedure using bromobenzene (78 mg, 0.50 mmol) and t-butyl acrylate (128 mg, 1.00 mmol), the reaction was allowed to stir for 72 h. After column chromatography, the product was obtained as a colorless liquid (81 mg, 79%). 1H NMR (400 MHz, CDCl3) δ 7.63–7.56 (d, J = 16.1 Hz, 1H), 7.55–7.49 (m, 2H), 7.42–7.34 (m, 3H), 6.41–6.35 (d, J = 16.1 Hz, 1H), 1.55 (s, 9H).39
General procedure for Stille couplings (Table 9)
Pd(t-Bu3P)2 catalyst (2.6 mg, 0.005 mmol) and DABCO base (84 mg, 0.75 mmol) were added under an Ar atmosphere in a 5.0 mL microwave vial containing a magnetic stir bar and Teflon-lined septum. NaCl (15 mg, 0.25 mmol) was added as an additive under a positive flow of Ar, followed by aryl halide (0.25 mmol), stannyl reagent (0.275 mmol) and surfactant solution (0.5 mL of 2 wt. % in degassed water), respectively. The mixture was allowed to stir vigorously for 2–18 h. After confirming full conversion by TLC, the reaction mixture was then diluted with 2.0 mL of EtOAc, and 0.25 mL of triethylamine. The mixture was passed through a silica gel bed and washed with EtOAc to collect the product. All volatile solvent was removed in vacuo to obtain the crude product that was further purified by flash chromatography on silica gel.
1-Methoxy-3-(phenylethynyl)benzene (Table 9, entry 1)
Following the general procedure using 1-bromo-3-methoxybenzene (47 mg, 0.25 mmol) and tributyl(phenylethynyl)stannane (108 mg, 0.275 mmol) the reaction was allowed to stir for 2 h. After column chromatography, the product was obtained as a yellow liquid (52 mg, 99%). 1H NMR (400 MHz, CDCl3) δ 7.58–7.52 (m, 2H), 7.40–7.33 (m, 3H), 7.29–7.25 (t, J = 7.6 Hz, 1H), 7.17–7.12 (d, J = 7.6 Hz, 1H), 7.10–7.06 (s, 1H), 6.94–6.88 (dd, J = 8.3, 2.4 Hz, 1H), 3.84 (s, 3H).40
2-Methoxy-1-(phenylethynyl)naphthalene (Table 9, entry 2)
Following the general procedure using 1-bromo-2-methoxynaphthalene (59 mg, 0.25 mmol) and tributyl(phenylethynyl)stannane (108 mg, 0.275 mmol), the reaction was allowed to stir for 4 h. After column chromatography, the product was obtained as a pale-yellow solid (63 mg, 99%). 1H NMR (400 MHz, CDCl3) δ 8.40–8.35 (d, J = 8.3 Hz, 1H), 7.88–7.83 (d, J = 9.0 Hz, 1H), 7.83–7.79 (d, J = 8.1 Hz, 1H), 7.72–7.67 (dd, J = 8.1, 1.7 Hz, 2H), 7.61–7.55 (td, J = 8.3, 1.2 Hz, 1H), 7.44–7.33 (m, 4H), 7.32–7.28 (d, J = 9.0 Hz,1H), 4.08 (s, 3H).41
(Z)-2-(2-Ethoxyvinyl)-1,3-dimethylbenzene (Table 9, entry 3)
Following the general procedure using 2-bromo-1,3-dimethylbenzene (47 mg, 0.25 mmol) and (Z)-tributyl(2-ethoxyvinyl)stannane (99 mg, 0.275 mmol), the reaction was allowed to stir for 8 h. After column chromatography, the product was obtained as a yellow liquid (24 mg, 56%). 1H NMR (400 MHz, CDCl3) δ 7.10–7.00 (m, 3H), 6.25–6.16 (d, J = 6.8 Hz, 1H), 5.26–5.19 (d, J = 7.0 Hz, 1H), 3.93–3.84 (q, J = 7.0 Hz, 2H), 2.29 (s, 6H), 1.31–1.21 (d, J = 7.0 Hz, 3H); 13C-NMR (500 MHz, CDCl3) δ 145.11, 136.69, 133.96, 127.00, 126.27, 103.26, 68.02, 20.68, 15.39; HRMS (EI) calcd for C12H16O [M]+ 176.1201, found 176.1209 (∆ = 0.8 mDa, 4.5 ppm).
(Z)-1-(2-Ethoxyvinyl)-4-methylnaphthalene (Table 9, entry 4)
Following the general procedure using 1-bromo-4-methylnaphthalene (55 mg, 0.25 mmol) and (Z)-tributyl(2-ethoxyvinyl)stannane (99 mg, 0.275 mmol), the reaction was allowed to stir for 18 h. After column chromatography, the product was obtained a yellow liquid (44 mg, 84%). 1H NMR (400 MHz, CDCl3) δ 8.19–8.11 (t, J = 4.9 Hz, 1H), 8.03–7.98 (t, J = 4.9 Hz, 1H), 7.98–7.94 (d, J = 7.6 Hz, 1H), 7.56–7.47(t, J = 4.9 Hz, 2H), 7.35–7.29 (d, J = 7.6 Hz, 1H), 6.45–6.40 (d, J = 7.1 Hz, 1H), 5.91–5.87 (d, J = 7.1 Hz, 1H), 4.06–3.95 (q, J = 7.1 Hz, 2H), 2.69 (s, 3H), 1.43–1.30 (t, J = 7.1 Hz, 3H); 13C-NMR (500 MHz, CDCl3) δ 146.79, 132.73, 132.35, 131.19, 130.04, 126.47, 126.44, 125.15, 125.10, 124.55, 124.46, 101.47, 68.93, 19.57, 15.43; HRMS (EI) calcd for C15H16O [M]+ 212.1201, found 212.1203 (∆ = 0.2 mDa, 0.9 ppm).
2-(2-Methoxynaphthalen-1-yl)furan (Table 9, entry 5)
Following the general procedure using 1-bromo-2-methoxynaphthalene (59 mg, 0.25 mmol) and tributyl(furan-2-yl)stannane (98 mg, 0.275 mmol), the reaction was allowed to stir for 4 h. After column chromatography, the product was obtained as a brown liquid (55 mg, 97%). 1H NMR (400 MHz, CDCl3) δ 7.94–7.89 (d, J = 9.0 Hz, 1H), 7.89–7.84 (d, J = 8.3 Hz, 1H), 7.84–7.80 (d, J = 8.1 Hz, 1H), 7.69–7.64 (t, J = 1.5 Hz, 1H), 7.48–7.42 (td, J = 6.8, 1.5 Hz, 1H), 7.40–7.33 (td, J = 6.8, 1.2 Hz, 2H), 6.65–6.62 (d, J = 1.5 Hz, 2H), 3.94 (s, 3H).24
General procedure for Miyaura borylations (Table 10)
The catalyst (Pd(t-Bu3P)2, 7.7 mg, 0.015 mmol) was added under an Ar atmosphere in a 5.0 mL microwave vial with magnetic stir bar and Teflon-lined septum. B2pin2 (140 mg, 0.55 mmol) and KOAc (147 mg, 1.50 mmol) were added under a positive flow of Ar followed by 1.0 mL of surfactant solution (2 wt. % in degassed water). The mixture was stirred vigorously for about 10 min, and then the aryl bromide (0.50 mmol) was added followed by an additional 1.0 mL of surfactant solution. The reaction was allowed to stir vigorously for 4–26 h. The reaction mixture was then diluted with EtOAc and passed through silica gel bed and washed with EtOAc to collect the product. All volatile solvent was removed in vacuo to obtain the crude product that was further purified by flash chromatography on silica gel.
Methyl 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate (Table 10, entry 1)
Following the general procedure using methyl 3-bromobenzoate (109 mg, 0.50 mmol), 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(1,3,2-dioxaborolane) (140 mg, 0.55 mmol), the reaction was allowed to stir for 4 h. After column chromatography, the product was obtained as a yellow solid (125 mg, 94%). 1H NMR (400 MHz, CDCl3) δ 8.48 (s, 1H), 8.17–8.09 (dt, J = 7.8, 1.6 Hz, 1H), 8.03–7.95 (dt, J = 7.3, 1.2 Hz, 1H), 7.50–7.40 (t, J = 7.6 Hz, 1H), 3.92 (s, 3H), 1.36 (s, 12H).42
2-(4-Methoxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (Table 10, entry 2)
Following the general procedure using 1-bromo-4-methoxybenzene (93 mg, 0.50 mmol) and 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(1,3,2-dioxaborolane) (140 mg, 0.55 mmol), the reaction was allowed to stir for 4 h. After column chromatography, the product was obtained as a yellow liquid (97 mg, 83%). 1H NMR (400 MHz, CDCl3) δ 7.79–7.73 (dt, J = 8.6, 2.0 Hz, 2H), 6.93–6.88 (dt, J = 8.8 Hz, 2.0 Hz, 2H), 3.84 (s, 3H), 1.34 (s, 12H).43
2-(2,6-Dimethylphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (Table 10, entry 3)
Following the general procedure using 2-bromo-1,3-dimethylbenzene (93 mg, 0.50 mmol) and 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(1,3,2-dioxaborolane) (140 mg, 0.55 mmol), the reaction was allowed to stir for 26 h. After column chromatography, the product was obtained as a yellow liquid (92 mg, 82%). 1H NMR (400 MHz, CDCl3) δ 7.16–7.10 (t, J = 7.6 Hz, 1H), 6.98–6.92 (d, J = 7.8 Hz, 2H), 2.40 (s, 6H), 1.40 (s, 12H).44
2-([1,1′-Biphenyl]-4-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (Table 10, entry 4)
Following the general procedure using 4-bromo-1,1′-biphenyl (117 mg, 0.50 mmol) and 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(1,3,2-dioxaborolane) (140 mg, 0.55 mmol), the reaction was allowed to stir for 24 h. After column chromatography, the product was obtained as a pale-yellow solid (133 mg, 95%). 1H NMR (400 MHz, CDCl3) δ 7.92–7.87 (d, J = 8.3 Hz, 2H), 7.65–7.60 (m, 4H), 7.49–7.42 (t, J = 7.8 Hz, 2H), 7.40–7.34 (m, 1H), 1.37 (s, 12H).45
General procedure for aminations (Table 11)
[(π-allyl)PdCl]2 catalyst (1.8 mg, 0.005 mmol), cBRIDP ligand (7.0 mg, 0.020 mmol), and KO-t-Bu as base (168 mg, 1.50 mmol) were added under an Ar atmosphere into a 5.0 mL microwave vial containing a magnetic stir bar and Teflon-lined septum. p-Toluidine (1.20 mmol) was added under a positive flow of Ar, followed by surfactant solution (1.0 mL of 2 wt. % in degassed water), and aryl bromide (1.00 mmol), respectively. The mixture was stirred vigorously for 3–24 h. The reaction mixture was then diluted with EtOAc and passed through a silica gel bed and further washed with EtOAc to collect the coupling product. All volatile solvent was removed in vacuo to obtain the crude product that was further purified by flash chromatography on silica gel.
4-Methoxy-N-(p-tolyl)aniline (Table 11, entry 1)
Following the general procedure using 1-bromo-4-methoxybenzene (187 mg, 1.00 mmol) and p-toluidine (129 mg, 1.20 mmol), the reaction was allowed to stir for 21 h. After column chromatography, the product was obtained as an off-white solid (195 mg, 91%). 1H NMR (400 MHz, CDCl3) δ 7.08–7.00 (m, 4H), 6.90–6.82 (m, 4H), 5.41 (s, 1H), 3.80 (s, 3H), 2.29 (s, 3H).46
N-(p-Tolyl)naphthalen-2-amine (Table 11, entry 2)
Following the general procedure using 2-bromonaphthalene (207 mg, 1.00 mmol) and p-toluidine (129 mg, 1.20 mmol), the reaction was allowed to stir for 10 h. After column chromatography, the product was obtained as an off-white solid (176 mg, 75%). 1H NMR (400 MHz, CDCl3) δ 7.75–7.71 (d, J = 8.8 Hz, 2H), 7.65–7.60 (d, J = 8.3 Hz, 1H), 7.42–7.36 (td, J = 6.8, 1.2 Hz, 2H), 7.31–7.27(td, J = 6.8, 1.2 Hz, 1H), 7.21–7.17 (dd, J = 8.8, 2.4 Hz, 1H), 7.17–7.08 (q, J = 8.3, 4H), 5.96 (s, 1H), 2.35 (s, 3H).24
Ethyl 4-((t-butoxycarbonyl)amino)benzoate (Table 11, entry 3)
The general procedure was followed using ethyl 4-bromobenzoate (231 mg, 1.00 mmol), t-butyl carbamate (176 mg, 1.50 mmol), and Na-O-t-Bu (144 mg, 1.50 mmol) as base. The reaction was allowed to stir in oil bath at 50 °C for 24 h. After column chromatography, the product was obtained as a white solid (228 mg, 85%). H NMR (400 MHz, CDCl3) δ 8.01–7.96 (d, J = 8.8 Hz, 2H), 7.46–7.40 (d, J = 8.8 Hz, 2H), 6.66 (s, 1H), 4.39–4.32 (q, J = 7.2 Hz, 2H), 1.54 (s, 9H), 1.42–1.36 (t, J = 7.2 Hz, 3H).47
Ethyl (4-benzoylphenyl)carbamate (Table 11, entry 4)
The general procedure was followed using (4-bromophenyl)(phenyl)methanone (131 mg, 0.50 mmol), ethyl carbamate (53 mg, 0.60 mmol), and Na-O-t-Bu 72 mg, 0.75 mmol) as base. The reaction was allowed to stir in oil bath at 50 °C for 24 h. After column chromatography, the product was obtained as a yellow solid (126 mg, 93%), mp 187–189 °C, lit48 mp 189 °C; H NMR (400 MHz, CDCl3) δ 7.85–7.81 (d, J = 8.8 Hz, 2H), 7.80–7.76 (d, J = 8.6 Hz, 2H), 7.62–7.56 (tt, J = 7.3, 1.2 Hz, 1H), 7.53–7.46 (m, 4H), 6.81 (s, 1H), 4.31–4.24 (q, J = 7.1 Hz, 2H), 1.38–1.32 (t, J = 7.1 Hz, 3H); 13C-NMR (500 MHz, CDCl3) δ 195.62, 153.17, 142.14, 137.95, 132.11, 131.78, 129.81, 128.23, 117.47, 61.63, 14.49; HRMS (ESI) calcd for C16H15NO3 [M+Na]+ 292.0950, found 292.0946 (∆ = 0.4 mDa, 1.4 ppm).
t-Butyl (2-methyl-5-nitrophenyl)carbamate (Table 12)
Following the general procedure for amination using [(π-allyl)PdCl]2 catalyst (1.8 mg, 0.020 mmol), cBRIDP ligand (3.5 mg, 0.040 mmol), KOH (21 mg, 0.375 mmol)/TIPSOH (65 mg, 0.375 mmol), and using 2-bromo-1-methyl-4-nitrobenzene (54 mg, 0.25 mmol) and t-butyl carbamate (35 mg, 0.30 mmol), the reaction was allowed to stir at rt for 15 h. After column chromatography, the product was obtained as a pale yellow solid (63 mg, 98%). 1H NMR (400 MHz, CDCl3) δ 8.86 (s, 1H), 7.89–7.80 (dd, J = 8.4, 2.3 Hz, 1H), 7.31–7.27 (d, J = 8.3 Hz, 1H), 6.41 (s, 1H), 2.35 (s, 3H), 1.56 (s, 9H).49
in situ-Derived organozinc halide-mediated cross-couplings. Ethyl 4-cyclohexylbenzoate (Table 13)
In a 5.0 mL microwave vial under Ar containing zinc dust (65 mg, 1 mmol) and PdCl2(Amphos)2 (1.2 mg, 0.0017 mmol), a surfactant solution (1.0 mL of 2 wt. % in degassed water) and N,N,N′,N′-tetramethylethylenediamine (TMEDA) (39 mg, 0.33 mmol) were added at rt followed by the addition of bromocyclohexane (136 mg, 0.83 mmol) and ethyl 4-bromobenzoate (77 mg, 0.33 mmol). The flask was stirred vigorously at rt for 24 h. Another portion of Zn dust (22 mg, 0.33 mmol), PdCl2(Amphos)2 (0.6 mg, 0.00085 mmol), TMEDA (39 mg, 0.33 mmol) and bromocyclohexane (26 mg, 0.16 mmol) were added to the reaction mixture and the reaction was continuously stirred for another 24 h. The reaction mixture was then filtered through a plug of silica gel and washed with diethyl ether, after which the solvents were removed under vacuum. After column chromatography on silica gel, the product was obtained as a white solid (66 mg, 86%). 1H NMR (400 MHz, CDCl3) δ 8.00–7.91 (d, J = 8.3 Hz, 2H), 7.30–7.20 (d, J = 8.1 Hz, 2H), 4.43–4.27 (q, J = 7.2 Hz, 2H), 2.62–2.46 (m, 1H), 1.92–1.78 (m, 4H), 1.48–1.31 (m, 9H).50
Supplementary Material
Acknowledgments
We would like to warmly thank the NIH for support of our program in green chemistry (GM 86485), and Drs. Thomas J. Colacot (Johnson Matthey) and Richard Pederson (Materia) for supplying the palladium catalysts and Grubbs catalyst, respectively, used in this study. The cryo-TEM experiments were performed by Dr. Wei Zhang (University of Minnesota), for which we are most appreciative.
Footnotes
Supporting information. Copies of MS and IR spectra of all new compounds, copies of 1H and 13C NMR spectra of all new compounds and copies of 1H NMR spectra of all known compounds. These materials are available free of charge at http://pubs.acs.org.
References
- 1.(a) Dunn P, Henderson R, Mergelsberg I, Wells A. “Moving towards Greener Solvents for Pharmaceutical Manufacturing – An Industry Perspective”, College Park, Maryland. ACS GCI®Pharmaceutical Roundtable. 2009 Jun 23–25; http://acs.confex.com/acs/green09/recordingredirect.cgi/id/510.; (b) Shaughnessy KH, DeVasher RB. Curr Org Chem. 2005;9:585–604. [Google Scholar]; (c) Constable DJC, Curzons AD, Cunningham VL. Green Chem. 2002;4:521–527. [Google Scholar]
- 2.(a) Li C-J, Chan T-H. Comprehensive Organic Reactions in Aqueous Media. 2. Wiley-VCH; Hoboken, NJ: 2007. [Google Scholar]; (b) Lindström UM, editor. Organic Reactions in Water: Principles, Strategies, and Applications. Blackwell Publishing; Oxford, U.K.: 2007. [Google Scholar]
- 3.(a) Sheldon RA. Green Chem. 2005;7:267–278. [Google Scholar]; (b) Leitner W, Seddon KR, Wasserscheid P, editors. Green Chem. 2003;5:99–284. (Special Issue on Green Solvents for Catalysis) [Google Scholar]
- 4.Sheldon RA, Arens IWCE, Hanefeld U. Green Chemistry and Catalysis. Wiley-VCH; Weinheim, Germany: 2007. [Google Scholar]
- 5.Bai L, Wang J-X, Zhang Y. Green Chem. 2003;5:615–617. [Google Scholar]
- 6.(a) Li CJ. Acc Chem Res. 2010;43:581–590. doi: 10.1021/ar9002587. [DOI] [PubMed] [Google Scholar]; (b) Li C-J. Metal-Mediated C–C Bond Formation in Aqueous Media. In: Lindström UM, editor. Organic Reactions in Water: Principles, Strategies, and Applications. Blackwell Publishing; Oxford, U.K.: 2007. [Google Scholar]; (c) Li CJ, Chen L. Chem Soc Rev. 2006;35:68–82. doi: 10.1039/b507207g. [DOI] [PubMed] [Google Scholar]
- 7.Dwars T, Paetzold E, Oehme G. Angew Chem, Int Ed. 2005;44:7174–7199. doi: 10.1002/anie.200501365. [DOI] [PubMed] [Google Scholar]
- 8.Lipshutz BH, Ghorai S. Aldrichimica Acta. 2012;45:3–16. [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Usachev BI, Obydennov DL, Kodess MI, Sosnovskikh VY. Tetrahedron Lett. 2009;50:4446–4448. [Google Scholar]; (b) Ackermann L, Fenner S. Org Lett. 2011;13:6548–6551. doi: 10.1021/ol202861k. [DOI] [PubMed] [Google Scholar]
- 10.Anastas PT, Farris CA, editors. Benign by Design: Alternative Synthetic Design for Pollution Prevention. American Chemical Society; Washington, DC: 1994. (ACS Symposium Series 557). [Google Scholar]
- 11.Anastas P, Eghbali N. Chem Soc Rev. 2010;39:301–312. doi: 10.1039/b918763b. [DOI] [PubMed] [Google Scholar]
- 12.Lipshutz BH, Ghorai S, Leong WWY, Taft BR, Krogstad DV. J Org Chem. 2011;76:5061–5073. doi: 10.1021/jo200746y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Aldrich catalog number 698717.
- 13.Lipshutz BH, Ghorai S, Abela AR, Moser R, Nishikata T, Duplais C, Krasovskiy A, Gaston RD, Gadwood RC. J Org Chem. 2011;76:4379–4391. doi: 10.1021/jo101974u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Aldrich catalog number 763918.
- 14.(a) Moreau RA, Whitaker BD, Hicks KB. Prog Lipid Res. 2002;41:457–500. doi: 10.1016/s0163-7827(02)00006-1. [DOI] [PubMed] [Google Scholar]; (b) Quílez J, García-Lorda P, Salas-Salvado J. Clin Nutr. 2003;22:343–351. doi: 10.1016/s0261-5614(03)00060-8. [DOI] [PubMed] [Google Scholar]; (c) Kritchevsky D, Chen SC. Nutr Res. 2005;25:413–428. [Google Scholar]; (d) Brufau G, Canela MA, Rafecas M. Nutr Res. 2008;28:217–225. doi: 10.1016/j.nutres.2008.02.003. [DOI] [PubMed] [Google Scholar]; (e) García-Llatas G, Rodríguez-Estrada MT. Chem Phys Lipids. 2011;164:607–624. doi: 10.1016/j.chemphyslip.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 15.(a) Schmitz KS. Dynamic Light Scattering by Macromolecules. Academic Press; Boston: 1990. [Google Scholar]; (b) Evans DF, Wennerström H. The Colloidal Domain. VCH; New York: 1994. [Google Scholar]; (c) Borkovec M. Measuring Particle Size by Light Scattering. In: Holmberg K, editor. Handbook of Applied Surface and Colloid Chemistry. Wiley; Chichester, U.K.: 2002. [Google Scholar]
- 16.Lipshutz BH, Aguinaldo GT, Ghorai S, Voigtritter K. Org Lett. 2008;10:1325–1328. doi: 10.1021/ol800028x. [DOI] [PubMed] [Google Scholar]
- 17.(a) Borowy-Borowski H, Sikorska-Walker M, Walker PR. Water-Soluble Compositions of Bioactive Lipophilic Compounds. 6,045,826. U.S. Patent. 2000 Apr 4;; (b) Borowy-Borowski H, Sikorska-Walker M, Walker PR. Water-Soluble Compositions of Bioactive Lipophilic Compounds. 6,191,172. U.S. Patent. 2001 Feb 20;; (c) Borowy-Borowski H, Sikorska-Walker M, Walker PR. Water-Soluble Compositions of Bioactive Lipophilic Compounds. 6,632,443. U.S. Patent. 2003 Oct 14;
- 18.Connon SJ, Blechert S. Angew Chem, Int Ed. 2003;42:1900–1923. doi: 10.1002/anie.200200556. [DOI] [PubMed] [Google Scholar]
- 19.Lipshutz BH, Petersen TB, Abela AR. Org Lett. 2008;10:1333–1336. doi: 10.1021/ol702714y. [DOI] [PubMed] [Google Scholar]
- 20.Lipshutz BH, Chung DW, Rich B. Org Lett. 2008;10:3793–3796. doi: 10.1021/ol801471f. [DOI] [PubMed] [Google Scholar]
- 21.(a) Nishikido N, Matuura R. Bull Chem Soc Jap. 1977;50:1690–1694. [Google Scholar]; (b) Schott H. J Colloid Interface Sci. 1995;173:265–277. [Google Scholar]
- 22.Lu G-P, Cai C, Lipshutz BH. Green Chem. 2013;15:105–109. [Google Scholar]
- 23.Lipshutz BH, Moser R, Voigtritter KR. Isr J Chem. 2010;50:691–695. doi: 10.1002/ijch.201000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lipshutz BH, Chung DW, Rich B. Adv Synth Catal. 2009;351:1717–1721. doi: 10.1002/adsc.200900323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Isley, N. A.; Dobarco, S.; Lipshutz, B. H. unpublished data.
- 26.Sheldon RA. Green Chem. 2007;9:1273–1283. [Google Scholar]
- 27.Song YH, Hong S, Lim H, Seo J, Chung S, No I, Lee K, Yoon M. Chem Pharm Bull (Tokyo) 2004;52:597–601. doi: 10.1248/cpb.52.597. [DOI] [PubMed] [Google Scholar]
- 28.Akiyama R, Kobayashi S. Angew Chem, Int Ed. 2002;41:2602–2604. doi: 10.1002/1521-3773(20020715)41:14<2602::AID-ANIE2602>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 29.Castarlenas R, Vovard C, Fischmeister C, Dixneuf PH. J Am Chem Soc. 2006;128:4079–4089. doi: 10.1021/ja0579762. [DOI] [PubMed] [Google Scholar]
- 30.Moreno-Manas M, Pleixats R, Serra-Muns A. Synlett. 2006:3001–3004. [Google Scholar]
- 31.Glass AC, Morris BB, Zakharov LN, Liu SY. Org Lett. 2008;10:4855–4857. doi: 10.1021/ol8019617. [DOI] [PubMed] [Google Scholar]
- 32.Noël T, Musacchio AJ. Org Lett. 2011;13:5180–5183. doi: 10.1021/ol202052q. [DOI] [PubMed] [Google Scholar]
- 33.Ma Y, Song C, Jian W, Wu Q, Wang Y, Liu X, Andrus MB. Org Lett. 2003;5:3317–3319. doi: 10.1021/ol035147k. [DOI] [PubMed] [Google Scholar]
- 34.Hundertmark T, Littke AF, Buchwald SL, Fu GC. Org Lett. 2000;2:1729–1731. doi: 10.1021/ol0058947. [DOI] [PubMed] [Google Scholar]
- 35.Tang Y, Yu Y, Xia W, Song Y, Huang Z. J Org Chem. 2002;67:3096–3103. doi: 10.1021/jo025586h. [DOI] [PubMed] [Google Scholar]
- 36.Wang P, Liu C-R, Sun X-L, Chen SS, Li J-F, Xie Z, Tang Y. Chem Commun. 2012;48:290–292. doi: 10.1039/c1cc16747b. [DOI] [PubMed] [Google Scholar]
- 37.Leng Y, Yang F, Wei K, Wu Y. Tetrahedron. 2010;66:1244–1248. [Google Scholar]
- 38.Brown C, Sikkel BJ, Carvalho CF, Sargent MV. J Chem Soc Perkin Trans. 1982;1:3007–3010. [Google Scholar]
- 39.Gottumukkala AL, Teichert JF, Heijnen D, Eisink N, Dijk Sv, Ferrer C, Hoogenband AVD, Minnaard AJ. J Org Chem. 2011;76:3498–3501. doi: 10.1021/jo101942f. [DOI] [PubMed] [Google Scholar]
- 40.Feng C, Loh TP. Chem Commun. 2010;46:4779–4781. doi: 10.1039/c0cc00403k. [DOI] [PubMed] [Google Scholar]
- 41.Feuerstein M, Berthiol F, Doucet H, Santelli M. Synthesis. 2004;8:1281–1289. [Google Scholar]
- 42.Chow WK, Yuen OY, So CM, Wong WT, Kwong FY. J Org Chem. 2012;77:3543–3548. doi: 10.1021/jo202472k. [DOI] [PubMed] [Google Scholar]
- 43.Kleeberg C, Dang L, Lin Z, Marder TB. Angew Chem. 2009;121:5454–5458. doi: 10.1002/anie.200901879. [DOI] [PubMed] [Google Scholar]
- 44.Billingsley KL, Barder TE, Buchwald SL. Angew Chem, Int Ed. 2007;46:5359–5363. doi: 10.1002/anie.200701551. [DOI] [PubMed] [Google Scholar]
- 45.Qiu D, Jin L, Zheng Z, Meng H, Mo F, Wang X, Zhang Y, Wang J. J Org Chem. 2013;78:1923–1933. doi: 10.1021/jo3018878. [DOI] [PubMed] [Google Scholar]
- 46.Zhang L, Wang W, Fan R. Org Lett. 2013;15:2018–2021. doi: 10.1021/ol4007162. [DOI] [PubMed] [Google Scholar]
- 47.Ma F, Xie X, Zhang L, Peng Z, Ding L, Fu L, Zhang Z. J Org Chem. 2012;77:5279–5285. doi: 10.1021/jo3005827. [DOI] [PubMed] [Google Scholar]
- 48.Doebner O. Justus Liebigs Ann Chem. 1881;210:246–284. [Google Scholar]
- 49.Yu C, Liu B, Hu L. J Org Chem. 2001;66:919–924. doi: 10.1021/jo005666q. [DOI] [PubMed] [Google Scholar]
- 50.Luo X, Zhang H, Duan H, Liu Q, Zhu L, Zhang T, Lei A. Org Lett. 2007;9:4571–4574. doi: 10.1021/ol701995t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.