

Abstract
We have used a “2-color” SERCA (sarco/endoplasmic reticulum calcium ATPase) biosensor and a unique high-throughput fluorescence lifetime plate-reader (FLT-PR) to develop a high-precision live-cell assay designed to screen for small molecules that perturb SERCA structure. A SERCA construct, in which red fluorescent protein (RFP) was fused to the N terminus and green fluorescent protein (GFP) to an interior loop, was stably expressed in an HEK cell line that grows in monolayer or suspension. Fluorescence resonance energy transfer (FRET) from GFP to RFP was measured in the FLT-PR, which increases precision 30-fold over intensity-based plate-readers without sacrificing throughput. FRET was highly sensitive to known SERCA modulators. We screened a small chemical library and identified ten compounds that significantly affected 2-color SERCA FLT. Three of these compounds reproducibly lowered FRET and inhibited SERCA in a dose-dependent manner. This assay is ready for large-scale HTS campaigns, and is adaptable to many other targets.
Keywords: SERCA2a, HEK, FRET, LOPAC, Screening, Enzyme Activation
Muscle contraction and relaxation are controlled primarily by calcium transport proteins.1 The ryanodine receptor initiates contractions by releasing micromolar levels of Ca2+ from the sarcoplasmic reticulum (SR), and relaxation is achieved when the sarcoplasmic reticulum Ca-ATPase (SERCA) pumps Ca2+ back into the SR against a 3-log [Ca2+] gradient. In cardiac muscle, the SERCA2a isoform is regulated by the inhibitory transmembrane protein phospholamban (PLB), which inhibits Ca2+ transport only at low diastolic [Ca2+]. SERCA is maximally active at high, systolic [Ca2+] or when PLB is phosphorylated at S16 or T17 by PKA or CaMKII, respectively. PKA phosphorylation of PLB is a key part of the β-adrenergic response pathway activated when cardiac reserves must be accessed.2
Decreased Ca2+-transport activity associated with many forms of heart failure (HF) is often due to decreased SERCA2a expression or activity,2 so several recent efforts to combat HF aim to correct this dysfunction either by SERCA overexpression3 or by reducing PLB inhibition.2,4 Overexpression of SERCA2a via rAAV gene therapy has been effective in both animal models of HF and human clinical trials3. However, the complexities of gene therapy call for a parallel effort to develop small-molecule activators of SERCA.5
Screens for small-molecule SERCA activators have previously focused on directly measuring ATPase activity, which is a low-throughput, low-precision approach.6,7 We recently reported a FRET-based assay for high-throughput screening of the SERCA-PLB complex in reconstituted membranes.5 This HTS assay was designed to detect changes in FRET as an indicator of structural changes caused by compounds that bind to, and structurally alter, the SERCA-PLB complex. A pilot screen was carried out using this method with steady-state (intensity) detection of fluorescence, and hits were resolved, some of which turned out to be SERCA activators.8 This outcome validated our approach, but that paper also suggested that fluorescence lifetime (FLT) detection should provide a much higher precision than steady-state detection, thus having the potential to substantially lower the rate of false positives (hits that cannot be confirmed when remeasured).
Here, we used a recently developed GFP/RFP “2-color” SERCA2a (2CS) construct as a FRET biosensor (Fig. 1A),9 and a fluorescence lifetime plate reader (FLT-PR) to screen, in live cells, for small molecules that affect SERCA structure. We previously experimented with the location of the intramolecular GFP to make a construct with maximal FRET, properly folded fluorescent proteins, and a functional enzyme.9 The optimal insertion site was after residue 509 in a loop on the N domain of SERCA, where full Ca2+-transport and ATPase activity are retained.9
Figure 1.
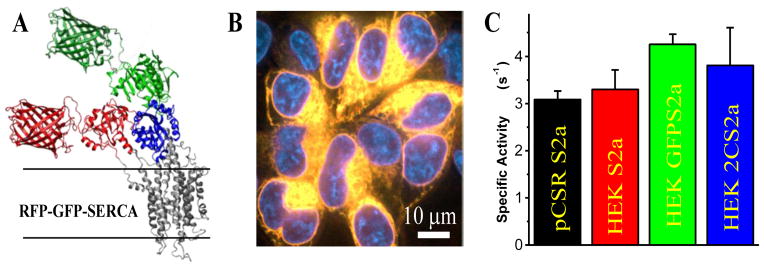
2-Color SERCA. (A) Computational model of GFP and RFP modeled on the crystal structure of SERCA1a (PDB 1IWO). (B) Confocal imaging of RFP and GFP (merged) fluorescence with DAPI nuclear stain shows 2-color SERCA localized to intracellular ER membranes. (C) ATPase activity data (mole ATP hydrolyzed per mole of SERCA per second) for pig cardiac SR (black), and stable cell lines expressing unlabeled SERCA2a (red), GFP-SERCA2a (green), and 2-color-SERCA2a (blue).
The FLT-PR is a prototype instrument constructed in collaboration with Fluorescence Innovations, Inc. (Minneapolis, MN), that uses a direct waveform recording (DWR) approach, which records an entire lifetime decay waveform on each pulse of a laser. A typical measurement averages 1000 laser pulses for a total acquisition time of about 200 ms, and a 384-well plate can be scanned in under 3 minutes, enabling high throughput screening. This is much faster than the conventional time-correlated single photon counting (TCSPC) method which requires a minimum of several seconds and sometimes minutes to acquire a single decay waveform. The precision of the lifetime measurement is critical for removing the intensity based errors associated with cell density and pipetting errors.10 Furthermore, the ability to obtain high-quality lifetime data from recombinantly labeled proteins in living cells eliminates many variables in potentially complicated FRET measurements, including labeling inefficiencies, variable well volume, and light scattering from cellular debris.
2CS is both active and properly localized to intracellular membranes when stably expressed in HEK cells (Fig. 1B,C). GFP-RFP FRET was shown to be sensitive to both [Ca2+] and thapsigargin (TG), a potent SERCA inhibitor, making it an ideal tool to screen for small molecule effectors of SERCA.9 We evaluated 2CS FRET in intact live cells in the presence of a variety of previously identified SERCA activators and inhibitors,8 and found that live-cell 2CS FRET is sensitive to SERCA effectors in a dose-responsive manner. We then performed a pilot screen of the 1280-compound Library of Pharmacologically Active Compounds (denoted LOPAC; Sigma-Aldrich) using the live-cell 2CS FRET assay. This screen yielded a few hits, i.e., compounds that significantly changed FRET. Secondary assays, measuring the Ca-ATPase activity, revealed that several hits from the FRET assay also altered SERCA function. This in-cell FRET assay represents a novel, highly sensitive tool to detect both activators and inhibitors of SERCA. The high-precision afforded by direct waveform recording technology implemented in the fluorescence lifetime plate reader (FLT-PR) enables in-cell high-throughput screening (HTS) using 2CS.
Materials and Methods
Molecular biology, cell culture, and localization
Using recombinant DNA technology, the gene for TagRFP was fused to the N-terminus of canine SERCA2a. This fusion position is in the A-domain of the pump. EGFP was fused as an intrasequence tag before residue 509 in the N-domain. The structural model presented in Fig. 1A corresponds to the isoform SERCA1a, which is 84% homologous to SERCA2a. Hou et al. evaluated several intrasequence fusion positions chosen using x-ray crystal structures of SERCA1a to identify unstructured loops and predict large relative distance changes without affecting SERCA function.9 The 509 site was chosen because the labeled construct had normal SERCA activity and its intramolecular FRET signal was sensitive to SERCA structural state, as shown below.
Cells were transiently transfected using 293fectin (Invitrogen), and stable cell lines were generated by G418 (Sigma) selection. Surviving clones expressing GFP-SERCA2a or 2CS were further selected by fluorescence microscopy and flow cytometry. Cell lines with the smallest population of non-expressing cells were selected and have been grown continuously while stably expressing 2CS or GFP-SERCA2a for over a year.
Fluorescence microscopy of HEK-GnTI− cells (ATCC, Manassas, VA) expressing 2CS was performed in glass-bottom chambered coverslips (Matek Corporation, Ashland, MA) several weeks after establishing a stable cell line. Confocal microscopy was performed using a Zeiss cell observer SD spinning disk confocal microscope equipped with a 0.55 N.A. 63 X oil immersion objective. Cells were stained 20 min before imaging with Hoechst 33342 NucBlue counterstain (Invitrogen) Excitation was accomplished with laser illumination at 405 nm for NucBlue, 488 nm for GFP, and 561 nm for RFP.
Homogenization of Cells for Activity Assays
Cells were homogenized as previously described.11 Briefly, cells were pelleted by spinning at 500g and washed 3X with PBS. Pellets were resuspended in 1–2 mL homogenization buffer (0.5 mM MgCl2, 10 mM Tris-HCl, pH 7.5, 4°C) and homogenized with 30 strokes in a Potter-Elvehjem homogenizer. Sucrose buffer (500mM sucrose, 100mM MOPS, pH 7.0, 4°C) was added at a 1:1 ratio and homogenate was assayed for protein concentration (BCA kit from Thermo Scientific, Rockford, IL) before flash freezing and storing at −80°C.
Cardiac SR and skeletal SR preparation
Cardiac sarcoplasmic reticulum (SR) vesicles were prepared from the ventricular tissue of swine hearts by adapting a previous method.12 The heart was placed in a solution (10 mM NaHCO3, 10mM Tris-HCl, 0.1 mg/mL Aprotinin, 0.1 mg/mL Leupeptin, 80 mM Benzamidine, 100 mM PMSF, 0.1 mg/mL Pepstatin A, pH 7.2, 4°C) immediately after being sacrificed, and kept on ice. All following procedures were performed at 4°C. The atria, fat and connective tissue were removed from the heart. The ventricular muscles were minced into 1 cm3 pieces and homogenized in 500 mL of SR buffer (100 mM KCl, 20 mM MOPS, pH 7.0, 4°C) using a blender (Waring, Torrington, CT). The homogenates were centrifuged at 5000 rpm (Sorvall, GSA rotor) for 20 minutes. The pellets were collected and treated with homogenization and centrifugation like before. Two supernatants were combined, filtered through 6 layers of cheesecloth, and spun at 8500 rpm (Sorvall, GSA rotor) for 20 min. KCl was added to the filtrate to a final 600 mM, and stirred for 15 minutes. The filtrate was spun at 12000 rpm for 1 hour, and the pellet was homogenized in 100 mL sucrose buffer (0.1 M sucrose, 1 mM NaN3, 20 mM MOPS, pH 7.0 4°C), and spun at 30000 rpm (Beckman Ti45 rotor) for 45 minutes. The pellet was homogenized in 15 mL sucrose buffer with a Potter-Elvehjem homogenizer. Total SERCA by weight in these vesicles was 30 ± 5% as determined by densitometry analysis of SDS-PAGE. Rabbit light skeletal SR vesicles were prepared using a method previously reported.13 Rabbit muscles were harvested from the hind leg of New Zealand White rabbits and purified using a sucrose gradient. The result light skeletal SR contains 80% SERCA.
Enzymatic activity of 2CS and compound effects on pig cardiac SR
An enzyme-coupled, NADH-linked ATPase assay was used to measure SERCA ATPase activity14 in 96-well microplates. Each well contained 50 mM MOPS (pH 7.0), 100 mM KCl, 5 mM MgCl2, 1 mM EGTA, 0.2 mM NADH, 1 mM phosphoenol pyruvate, 10 IU/mL of pyruvate kinase, 10 IU/mL of lactate dehydrogenase, 3.5 μg/mL of the calcium ionophore A23187, and CaCl2 added to set free [Ca2+] to the desired values.15 2.5 μg of cardiac SR or 25 μg of HEK cell homogenate were used in each well to correct for the difference in SERCA content. The assay was started upon the addition of ATP at a final concentration of 5 mM and read in a SpectraMax Plus microplate spectrophotometer (Molecular Devices, Sunnyvale, CA), bring the total volume to 200 μL. Results were normalized to SERCA content determined from immunoblotting.
Effects of known activators and LOPAC hits on SERCA ATPase activity was evaluated in pig cardiac SR. Compounds were dissolved in DMSO, and adjusted to 40 times the concentrations used in the final assay wells. Compounds were prediluted and 4 μL was added to each well to keep the final [DMSO] at 2% (v/v).
Western blot to quantify SERCA content
Cell homogenates and cardiac SR were subjected to SDS-PAGE on 4–20% Tris-HCl gels (Criterion, Biorad) at 5 μg total protein, transferred to Immobilon-FL membranes (Millipore), and blocked by dipping in methanol and air drying. The membrane was incubated with SERCA2a primary antibody diluted 1:1000 (2A7-A1, Abcam) for 3 h, washed, and visualized by 1 h incubation with goat-anti-mouse 800 nm IR secondary antibody (LI-COR Biosciences). Blots were quantified on the Odyssey scanner (LI-COR Biosciences).
Compound plating and fluorescence lifetime measurements in plate reader
The 96-well plate format LOPAC library was reformatted into four 384-well polystyrene mother plates (Corning, Corning, NY) using a Biomek FX liquid handler (Beckman Coulter, Brea, CA), and diluted to 500 μM using DMSO. Column 1, 22, 23 and 24 were loaded with DMSO for in-plate no-compound controls. The LOPAC compounds were distributed in column 2 through 21. Each plate contains 64 wells of DMSO controls, and 320 wells of compounds from the LOPAC library. Black well, high-quality-glass bottom Greiner 384-well microplates (PN 781892) were selected as the assay plates for their optical clarity, low autofluorescence, and low inter-well cross-talks. Compounds (1 μL) were transferred from the mother plates into assay plates using a Mosquito HV liquid handler (TTP Labtech Ltd, UK). The plates were sealed and stored at −20 °C until use. On the day of screening, the plates were equilibrated to room temperature (25 °C). Stable GFP-SERCA2a (donor only control) or 2CS cells were lifted from a 225 cm2 flask by incubating with TrypLE (Invitrogen) for 5 min. Cells were collected and pelleted for 5 min at 500g and resuspended in 10mL PBS, then analyzed on a Countess cell counter (Invitrogen) and diluted to 1x106 cells/mL. Cells (49 μL) were plated on top of the compounds by a FlexDrop IV reagent dispenser (PerkinElmer, Waltham, MA). Assay plates were spun for 1 min at 200 g and allowed to incubate at RT for 20 min before reading on a prototype fluorescence lifetime plate reader (constructed in collaboration with Fluorescence Innovations, Inc., Minneapolis, MN). GFP fluorescence was excited with a 473 nm microchip laser from Concepts Research Corporation (Belgium, WI) and emission was filtered with 490 nm long pass and 520/35 nm band pass filters from Semrock (Rochester, NY).
HTS data analysis
Time-resolved fluorescence waveforms for each well were fitted to single-exponential decays using least-squares minimization global analysis software (Fluorescence Innovations, Inc.). Each plate contained 64 control wells with only DMSO, and a hit was defined as a compound that changed the 2CS donor lifetime by more than three times the standard deviation (SD) relative to the controls. Fluorescent compounds that caused the intensity of both untransfected HEK cells and 2CS cells to be more than 3SD outside the mean of the 64 controls on a plate were excluded from the hits as likely false positives.
Results
Ca-ATPase Activity of 2CS
We have previously reported that a 2CS construct with an N-terminal cerulean and internal yellow fluorescent protein, transiently expressed in HEK cells, actively transports Ca2+ and has normal Ca2+-dependent ATPase activity9. Here, we show that SERCA ATPase activity is not significantly affected by the RFP or GFP probes in stable cell lines (Fig. 1C). Both unlabeled SERCA2a and 2CS stably expressed in HEK cells have Ca2+-dependent ATPase specific activity comparable to pig cardiac SR, which expresses SERCA2a at a high level (20–30% of total protein).
GFP-RFP FRET in FLT Plate Reader
The FLT-PR allowed us to rapidly obtain precise fluorescence waveforms from 96- or 384-well plates in under two minutes per plate (Fig. 2A). We previously showed that this plate reader could measure lifetimes from purified proteins in the same amount of time that a conventional plate reader measures intensity, but with a much better coefficient of variation (CV)8. This was true to an even greater extent for cells stably expressing GFP-SERCA2a or 2CS (Fig. 2B). The 30-fold improvement in precision means that a very small change in lifetime can be detected reliably, creating an excellent high-throughput screen (Fig. 2C). We were initially concerned about optical interference from concentrated cell suspensions (up to ~2.5 x 106 cells/mL), but found that both GFP-SERCA and 2CS stable cell lines were >30 times more fluorescent in the plate reader than untransfected cells at the same concentration. In contrast, our conventional intensity-mode fluorescence plate reader barely distinguishes GFP fluorescence from the background (CV ~ 1). Cell density was optimized to minimize CV, and we found that a wide range of densities gave the same lifetime with high precision for both donor and donor-acceptor cells. A cell density of 1x106 cells/mL gave CV values of 0.2–0.4% consistently for the GFP lifetime in both cell lines and this concentration was used for further experiments, however, we were able to obtain CVs as low as 0.34% with <5x104 cells/mL (data not shown). This level of precision enables reliable detection of changes in lifetime on the order of 10 picoseconds (Fig. 2C). The donor-only lifetime on separate days was consistently 2.5 ± 0.1 ns and the 2CS lifetime was 2.2 ± 0.1 ns, giving a basal FRET efficiency of 0.12 ± 0.05.
Figure 2.
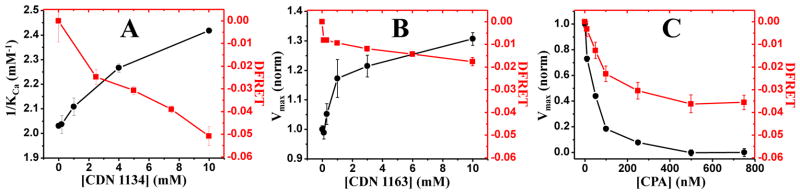
FLT-PR performance. (A) Waveforms from a high-throughput FRET assay performed in FLT-PR, on live cells expressing 2-color SERCA (identical control samples, no compounds added). (B) Lifetime measurement yields a 30-fold decrease in CV compared with intensity detection. This increased precision greatly decreases the probability of false negatives and positives, thus improving the z′ parameter (C) that defines the quality of HTS data 8,16. Thus even a 1% change in FRET produces excellent HTS quality.
We previously carried out a screen based on FRET, (measured by fluorescence intensity), from dye-labeled SERCA to dye-labeled PLB in reconstituted membranes, resulting in several SERCA activators8. CDN1134 and CDN1163 represent two distinct classes of SERCA activators, with CDN1134 increasing the apparent Ca2+ affinity of SERCA (1/KCa), and CDN1163 increasing the activity at saturating [Ca2+] (Vmax). We found that these SERCA activators both reduce FRET between GFP and RFP in 2CS, with a dose dependence similar to that of SERCA activation (Fig. 3A,B). FRET in 2CS was also reduced by the known SERCA inhibitor cyclopiazonic acid (CPA), with a concentration dependence similar to that of inhibition (Fig. 3C). The FRET and functional data both yielded apparent Kd values of about 40 nM, consistent with previous reports of the IC50 for inhibition of SERCA by CPA.16
Figure 3.
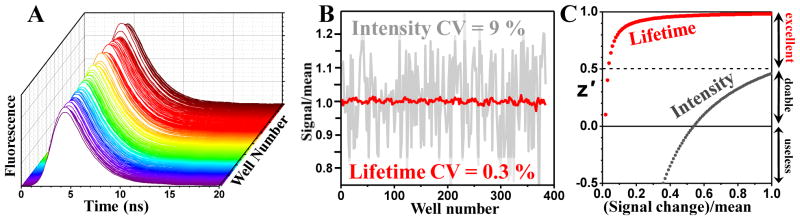
Dose-response effects of known activators and inhibitors on 2CS FRET. Small-molecule SERCA activators developed from our previous SERCA-PLB fluorescence intensity screen both reduce FRET (A and B), and the inhibitor CPA also reduces FRET (C). In all three cases, the concentration dependence of FRET and function are similar
LOPAC Library Screen
The 1280-compound LOPAC library, arranged in 384-well plates, was scanned in the FLT plate reader. Compounds that caused the 2CS GFP lifetime to be outside the 3SD window from the mean (established in controls with DMSO only) were defined as hits. Assays on ten hit compounds were repeated to determine whether they were reproducible, and we found that seven of the hits were false positives. A 3SD cutoff predicts ~four random false positives for a screen of this size. We also eliminated compounds that caused fluorescence intensity to be more than 3SD outside the range of the 64 DMSO controls on each plate (7.1% of the library). Fig. 4 shows results from one of the LOPAC plates that contained two of the three reproducible hits. Both hits were clearly distinct from the other compounds and controls on the plate when the single-lifetime fits of the fluorescence decays were analyzed (Fig. 4A) In contrast, when analyzed by the conventional fluorescence intensity method, the hits were not distinguishable from the other compounds or controls (Fig. 4B). Intensity mode analysis has a much greater window to identify hits due to the much greater CV, and many false positives would be detected before either of the two true hits would be identified.
Figure 4.
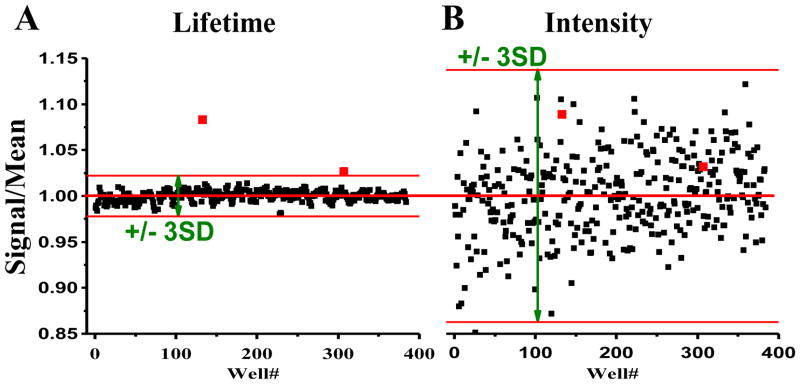
Results from one of the four 384-well plates containing 320 compounds of the 1280-compound LOPAC library. Lifetime fitting of waveforms (A) identifies two hits (red symbols) that are SERCA effectors (TG and XCT 790), both of which are missed by intensity-based screening (B). Red bars delimit the 3SD hit selection window.
The three reproducible hits were thapsigargin (TG, Sigma Cat. No. T 9033), diphenyline iodonium chloride (DPI, Sigma Cat. No. D 2926), and XCT 790 (Sigma Cat. No. X 4752). TG is a well-known SERCA inhibitor that we did not know was present in the library,16 and DIC has also been shown to inhibit SERCA activity.17 The third compound, XCT 790, is a well-known antagonist of the estrogen-like receptor α,18 but has not been previously identified as an effector of SERCA activity.
TG was the most potent FRET effector in both the LOPAC screen and subsequent dose-response assays, reducing FRET by ~33% at only 7.5 nM, when ATPase activity is nearly ablated (Fig. 5A). This result is consistent with previous steady-state FRET results of 2CS, which showed TG promoting a more open, low-FRET headpiece conformation.9
Figure 5.
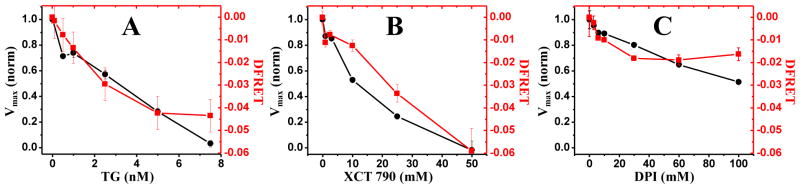
FRET and functional effects of LOPAC compounds identified in the screen. All three compounds reduce FRET and inhibit ATPase activity in a dose-response manner with similar dose dependence. TG (A) and XCT 790 (B) completely inhibit ATPase activity at the highest concentrations tested, while DPI inhibits <50% of ATPase activity at 100 μM (C).
XCT 790 was also a strong FRET effector, reducing FRET by ~42% while inhibiting ATPase activity with similar concentration dependence (Fig. 5B). XCT 790 is yellow in color when dissolved in DMSO, but it did not contribute any fluorescence intensity to plate reader measurements and had no effect on the lifetime of GFP-SERCA2a alone.
The third hit, DPI, was the weakest FRET effector, maximally reducing FRET from 0.12 to 0.10 (~18%). However, the FRET change was extremely reproducible and the concentration dependence of DPI was again similar to that of ATPase inhibition (Fig. 5C). The ATPase dependence is very similar to a previous report showing 18% inhibition of Ca2+ uptake into microsomes with 30 μM DPI and 32% inhibition at 100 μM DPI.17
Discussion
SERCA activation is a sought-after therapeutic goal to treat a wide variety of diseases including HF,19 muscular dystrophy,20,21 and diabetes,22 and SERCA inhibitors are sought for cancer therapeutics.23 Many approaches have been taken to achieve the desired aim of increasing SERCA activity and reducing cytosolic [Ca2+]. In this respect, gene therapy to overexpress SERCA2a in the heart is quite effective as a means to enhance Ca-transport capacity. Phase II clinical trials showed improvement in hemodynamic parameters and quality of life when patients were treated with an adeno-associated viral vector expressing SERCA2a in the heart.3 These results show that normalizing Ca2+ cycling in failing hearts mitigates disease symptoms and improves muscle function. However, only about 50% of the HF patients qualify for the gene therapy approach, due to the prevalence of AAV neutralizing antibodies in the general population. Small-molecule SERCA activators are designed to restore Ca2+ cycling by activating existing SERCA pumps in the SR, and are expected to provide a more mainstream pharmacological therapy for HF patients without the limitations currently associated with gene therapy.
We previously conducted a FRET-based high-throughput screen to detect small molecules that disrupt the interaction between SERCA and PLB.8 Several compounds were identified in that screen as SERCA activators, but none were specific for the SERCA-PLB interaction. Those compounds were apparently allosteric SERCA activators that directly perturbed the structure of SERCA and altered SERCA-PLB FRET as a result. Here, by placing both donor and acceptor probes within SERCA, we have searched exclusively for compounds that directly affect SERCA.
Crystal structures of SERCA1a show large rearrangements of the cytoplasmic headpiece during the enzymatic cycle, with large (~3 nm) changes in distance between the N and A domain, making them ideal locations for FRET probes to find functional effectors. Previous studies showed that a CFP-SERCA fusion protein actively pumps Ca2+ and is properly localized to intracellular membranes, and that FRET is sensitive to both TG inhibition and Ca2+ activation.9 Here we have confirmed the result that TG promotes a more open SERCA headpiece and that stably expressed 2CS in HEK cells is sensitive to allosteric regulators, thus suitable to use in screens for small molecules that perturb SERCA structure.
Using the fluorescence lifetime plate reader, we have developed the first high-throughput fluorescence lifetime assay in living cells. The precision of the lifetime measurement (CV = 0.3%) allowed us to detect very small changes in FRET between GFP and RFP, and we showed that this FRET signal is sensitive to both activators and inhibitors of SERCA function. Compounds tested showed similar concentration dependence for FRET and ATPase assays, indicating a tight link between structural and functional perturbations.
Our pilot-scale screen of the LOPAC library did not uncover any new activators, but we did identify two known inhibitors and a new inhibitor, XCT 790. Each of these hits showed similar FRET and ATPase concentration dependence, supporting a strong connection between the measured structural perturbations and enzyme function. DPI is also interesting because it was identified in a 2006 screen of the LOPAC library as a compound that inhibited growth of the malarial parasite plasmodium falciparum.24 The SERCA-like pump in this parasite is a target for the development of new drugs to treat drug-resistant strains of malaria.25 The IC50 for inhibition of parasite growth was submicromolar, while both the ATPase data here and Ca2+ uptake assays reported previously agree that there is very little functional inhibition below 10μM.17 While DPI may prove to be an effective treatment for drug resistant malaria, the connection between SERCA inhibition, structural perturbation, and restriction of plasmodium falciparum growth remains unclear.
Structures obtained from crystals grown in the presence of TG have shown a compact (closed) headpiece conformation,26 but our results with 2CS suggest that TG opens the headpiece in living cells (Fig. 5A), consistent with previous results.9
The assay presented here shows promise for large-scale HTS campaigns to identify new SERCA activators or inhibitors. The FLT-PR enables detection of very small changes with high precision, and here we show that the instrument is capable of measuring lifetimes precisely in live cells. Two-color constructs could be made with other SERCA isoforms or different proteins that undergo conformational changes associated with function. Increased activity (through overexpression) of the SERCA1a isoform in skeletal muscle has shown promise in treating muscular dystrophy,21,27 and overexpression of SERCA2b in non-muscle cells slows the progression of diabetes in mouse models.28 Thus the identification of new SERCA activators, whether they are isoform-specific or not, could generate new drugs that treat several of the most common and costly diseases.
Acknowledgments
The LOPAC library was provided by Courtney Aldrich. Joseph M. Muretta and J. Michael Autry provided helpful discussions, and Octavian Cornea helped prepare the manuscript for publication. Fluorescence microscopy was performed at the UMN Imaging Center and spectroscopy was performed at the UMN Biophysical Spectroscopy Center.
Funding
This work was supported by grants to D.D.T. (NIH GM27906, AR57220, AG42996; MDA218102) and R.L.C. (NIH HL92097). S.J.G. was supported by a predoctoral fellowship from the American Heart Association (Midwest Affiliate 13PRE13230005).
References
- 1.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415(6868):198. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 2.MacLennan DH, Kranias EG. Phospholamban: a crucial regulator of cardiac contractility. Nature Reviews. 2003;4:666. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- 3.Jessup M, et al. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. 2011;124(3):304. doi: 10.1161/CIRCULATIONAHA.111.022889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gruber SJ, Haydon S, Thomas DD. Phospholamban mutants compete with wild type for SERCA binding in living cells. Biochem Biophys Res Commun. 2012;420(2):236. doi: 10.1016/j.bbrc.2012.02.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banerjee Ranajit K, Datta Asoke G. Proteoliposome as the model for the study of membrane-bound enzymes and transport proteins. Molecular and Cellular Biochemistry. 1983;50(1):3. doi: 10.1007/BF00225276. [DOI] [PubMed] [Google Scholar]
- 6.Johnson RG, Jr, Kranias EG. Cardiac sarcoplasmic reticulum function and regulation of contractility. Introduction. Ann N Y Acad Sci. 1998;853:xi. [PubMed] [Google Scholar]
- 7.Johnson RG., Jr Pharmacology of the cardiac sarcoplasmic reticulum calcium ATPase-phospholamban interaction. Ann N Y Acad Sci. 1998;853:380. doi: 10.1111/j.1749-6632.1998.tb08305.x. [DOI] [PubMed] [Google Scholar]
- 8.Cornea RL, et al. High-throughput FRET assay yields allosteric SERCA activators. J Biomol Screen. 2013;18(1):97. doi: 10.1177/1087057112456878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hou Z, et al. 2-Color calcium pump reveals closure of the cytoplasmic headpiece with calcium binding. PLoS ONE. 2012;7(7):e40369. doi: 10.1371/journal.pone.0040369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muretta JM, et al. High-performance time-resolved fluorescence by direct waveform recording. Rev Sci Instrum. 2010;81(10):103101. doi: 10.1063/1.3480647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maruyama K, MacLennan DH. Mutation of aspartic acid-351, lysine-352, and lysine-515 alters the Ca2+ transport activity of the Ca2+-ATPase expressed in COS-1 cells. Proc Natl Acad Sci U S A. 1988;85(10):3314. doi: 10.1073/pnas.85.10.3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feher JJ, Briggs FN. Determinants of calcium loading at steady state in sarcoplasmic reticulum. Biochim Biophys Acta. 1983;727(2):389. doi: 10.1016/0005-2736(83)90424-8. [DOI] [PubMed] [Google Scholar]
- 13.Mueller B, et al. SERCA structural dynamics induced by ATP and calcium. Biochemistry. 2004;43(40):12846. doi: 10.1021/bi0489457. [DOI] [PubMed] [Google Scholar]
- 14.Stergiopoulos V, et al. Moving from rhetoric to reality: adapting Housing First for homeless individuals with mental illness from ethno-racial groups. BMC Health Serv Res. 2012;12:345. doi: 10.1186/1472-6963-12-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mueller B, et al. Direct detection of phospholamban and sarcoplasmic reticulum Ca-ATPase interaction in membranes using fluorescence resonance energy transfer. Biochemistry. 2004;43(27):8754. doi: 10.1021/bi049732k. [DOI] [PubMed] [Google Scholar]
- 16.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4(2):67. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 17.Michelangeli F, East JM. A diversity of SERCA Ca2+ pump inhibitors. Biochem Soc Trans. 2011;39(3):789. doi: 10.1042/BST0390789. [DOI] [PubMed] [Google Scholar]
- 18.Tazzeo T, Worek F, Janssen Lj. The NADPH oxidase inhibitor diphenyleneiodonium is also a potent inhibitor of cholinesterases and the internal Ca(2+) pump. Br J Pharmacol. 2009;158(3):790. doi: 10.1111/j.1476-5381.2009.00394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ariazi EA, Jordan VC. Estrogen-related receptors as emerging targets in cancer and metabolic disorders. Curr Top Med Chem. 2006;6(3):203. doi: 10.2174/1568026610606030203. [DOI] [PubMed] [Google Scholar]
- 20.Inesi G, Prasad AM, Pilankatta R. The Ca2+ ATPase of cardiac sarcoplasmic reticulum: Physiological role and relevance to diseases. Biochem Biophys Res Commun. 2008;369(1):182. doi: 10.1016/j.bbrc.2007.11.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gehrig SM, et al. Hsp72 preserves muscle function and slows progression of severe muscular dystrophy. Nature. 2012;484(7394):394. doi: 10.1038/nature10980. [DOI] [PubMed] [Google Scholar]
- 22.Goonasekera SA, et al. Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J Clin Invest. 2011;121(3):1044. doi: 10.1172/JCI43844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu S, et al. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011;473(7348):528. doi: 10.1038/nature09968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hermanson D, et al. Dual mechanisms of sHA 14-1 in inducing cell death through endoplasmic reticulum and mitochondria. Mol Pharmacol. 2009;76(3):667. doi: 10.1124/mol.109.055830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan J, et al. Genetic mapping of targets mediating differential chemical phenotypes in Plasmodium falciparum. Nat Chem Biol. 2009;5(10):765. doi: 10.1038/nchembio.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arnou B, et al. The Plasmodium falciparum Ca(2+)-ATPase PfATP6: insensitive to artemisinin, but a potential drug target. Biochem Soc Trans. 2011;39(3):823. doi: 10.1042/BST0390823. [DOI] [PubMed] [Google Scholar]
- 27.Inesi G, et al. Concerted conformational effects of Ca2+ and ATP are required for activation of sequential reactions in the Ca2+ ATPase (SERCA) catalytic cycle. Biochemistry. 2006;45(46):13769. doi: 10.1021/bi061255d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morine KJ, Sleeper MM, Barton ER, Sweeney HL. Overexpression of SERCA1a in the mdx diaphragm reduces susceptibility to contraction-induced damage. Hum Gene Ther. 2010;21(12):1735. doi: 10.1089/hum.2010.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park SW, Zhou Y, Lee J, Ozcan U. Sarco(endo)plasmic reticulum Ca2+-ATPase 2b is a major regulator of endoplasmic reticulum stress and glucose homeostasis in obesity. Proc Natl Acad Sci U S A. 2010;107(45):19320. doi: 10.1073/pnas.1012044107. [DOI] [PMC free article] [PubMed] [Google Scholar]