

ABSTRACT
Gestational protein restriction (PR) alters the renin-angiotensin system in uterine arteries and placentas and elevates plasma levels of angiotensin II in pregnant rats. To date, how PR increases maternal plasma levels of angiotensin II remains unknown. In this study, we hypothesize that the expression and/or the activity of angiotensin I converting enzyme (peptidyl-dipeptidase A) 1 (ACE) in lungs, but not kidneys and blood, largely contribute to elevated plasma angiotensin II levels in pregnant rats subject to gestational PR. Time-scheduled pregnant Sprague-Dawley rats were fed a normal or low-protein diet from Day 3 of pregnancy until euthanized at Day 19 or 22. Expressions of Ace and Ace2 (angiotens in I converting enzyme [peptidyl-dipeptidase A] 2) in lungs and kidneys from pregnant rats by quantitative real-time PCR and Western blotting, and the activities of these proteins in lungs, kidneys, and plasma, were measured. The mRNA levels of Ace and Ace2 in lungs were elevated by PR at both Days 19 and 22 of pregnancy. The abundance of ACE protein in lungs was increased, but ACE2 protein was decreased, by PR. The activities of ACE, but not ACE2, in lungs were increased by PR. PR did not change expressions of Ace and Ace2, the activities of both ACE and ACE2 in kidneys, and the abundance and activity of plasma ACE. These findings suggest that maternal lungs contribute to the elevated plasma levels of angiotensin II by increasing both the expression and the activity of ACE in response to gestational PR.
Keywords: angiotensin I converting enzyme, angiotensin II, gestational protein restriction, kidney, lung, pregnancy, rat
Gestational protein restriction increases plasma levels of angiotensin II by stimulating expression and enzymatic activities of ACE in lungs but not kidneys of pregnant rats.
INTRODUCTION
The renin-angiotensin system (RAS) plays an important role in regulating homeostasis and blood pressure. RAS includes multiple components with different functions that are expressed in a tissue- or organ-specific manner [1–3]. Angiotensinogin, mainly produced in liver, is the starting substrate for the production of various angiotensin peptides. Angiotensinogin is converted to angiotensin I (angiotensin 1–9) by REN (renin), an enzyme mainly produced by kidney. Angiotensin I is further converted to angiotensin II (angiotensin 1–8) by ACE (angiotensin I converting enzyme [peptidyl-dipeptidase A] 1), and angiotensin II is converted to angiotensin (1–7) by ACE2 (angiotensin I converting enzyme [peptidyl-dipeptidase A] 2). Among these angiotensin peptides, angiotensin II is considered to be the main effector of the RAS, increasing arterial pressure by its potent vasoconstrictor action and also by stimulating the release of aldosterone and consequent renal fluid retention [4].
Lung and kidney have been considered to be main sources for angiotensin hormones in blood circulation, although multiple tissues or organs possess local RAS [5]. ACE protein is predominantly localized to the surface of endothelial cells in the pulmonary circulation [6] and is a potent generator of angiotensin II [7]. It has been shown that more than 90% of conversion of angiotensin I to angiotensin II occurs in lungs [8], thus, lungs may be the primary source of circulating angiotensin II. Kidneys have the highest activity of ACE2 compared to other tissues or organs [7] and, therefore, contribute to the degradation of angiotensin II. Moreover, kidneys are the main organs producing REN, which converts angiotensinogen to angiotensin I and thus initiates the cascade of hormone production [9]. In addition, blood not only acts as a carrier for endocrine hormones and exerts their effects systemically, but also contains soluble ACE proteins that are originally derived from the transmembrance ACE of vascular endothelial cells [10].
In normal pregnancy, RAS undergoes significant changes both systemically and locally, with a progressive increase in different components of RAS [11–13]; however, normotension is maintained. In contrast, failure to adapt to the increased RAS during pregnancy may cause pregnancy complications such as preeclampsia [1, 2, 14]. To date, mechanisms for these dramatic increases in RAS during pregnancy are not fully understood. It is known that fetal RAS is susceptible to external stimuli during pregnancy, because RAS in multiple fetal organs, including the brain, kidney, lung and heart, is altered in response to gestational protein restriction (PR) [15, 16], glucocorticoid exposure [17–19], and maternal hypoxic stress [20], and these changes in fetal RAS have been suggested to underlie the mechanisms in fetal programming. However, it is unclear if there are changes in maternal RAS in different organs that might contribute to fetal programming.
Malnutrition during pregnancy is one of the causes of intrauterine growth restriction, with long-lasting detrimental effects on the health of adult offspring [21, 22]. Dietary protein insufficiency occurs in large populations of developing countries due to poverty, and in certain ethnic groups of developed countries it occurs due to varied social-economic limitations [23, 24]. It has been established that offspring from dams with maternal PR during gestation develop hypertension and cardiovascular diseases in adulthood in a gender- and time-dependent manner [25–31]; however, the underlying mechanisms have not been completely understood.
Pregnant rats fed a low-protein diet have been widely used to study the effects of fetal programming on the development of hypertension and metabolic syndromes [32]. Our previous studies have shown that, in response to maternal PR, angiotensin II type I receptor expression was increased in the uterine artery [33] and ACE2 was decreased in placental labyrinth [34]. Moreover, we also found that plasma levels of angiotensin II in the PR group were significantly higher than those in the control group at Days 19 and 22 of pregnancy, respectively. In addition, in both the PR and control groups, plasma levels of angiotensin II at Day 22 were significantly higher than those at Day 19 [33]. It is known that mild global dietary restriction reduces uterine and placental blood flow [35], and protein insufficiency itself impairs vasodilation of mesenteric artery in pregnant rats [36]. These alterations in RAS in gestational PR, together with the elevated plasma angiotensin II [33], may impair the blood flow to the fetus, resulting in intrauterine growth restriction. The fetal growth restriction is associated with cardiovascular disease, including hypertension, in adult offspring [37–40]. To date, the mechanisms of elevated plasma angiotensin II in response to gestational PR remain unknown. In the current study, we hypothesize that the expression and/or the activity of ACE in lungs, but not kidneys and blood, largely contribute to elevated plasma angiotensin II levels in pregnant rats subject to gestational PR. Ace and Ace2 gene and protein expressions, as well as activity, in lung and kidney tissue are quantitated in normal and PR dams. Additionally, plasma ACE protein and activity were measured for comparison.
MATERIALS AND METHODS
Animal and Diets
All procedures were approved by the Animal Care and Use Committee at the University of Texas Medical Branch and were in accordance with those published by the U.S. National Institutes of Health Guide for the Care and Use of Laboratory Animals. Timed pregnant Sprague-Dawley rats weighing between 175 and 225 g were purchased from Harlan (Houston, TX). At Day 3 of pregnancy, rats were randomly divided into two dietary groups, housed individually, and fed a control (CT, 20% casein) or low-protein (6% casein) diet until they were euthanized on Day 19 or 22 of pregnancy (n = 5 rats/diet/day of pregnancy). The isocaloric low-protein and normal-protein diets were purchased from Harlan Teklad (cat. TD.90016 and TD.91352, respectively; Madison, WI).
Anesthesia and Sample Procurement
At Day 19 or 22 of pregnancy, rats were anesthetized by carbon dioxide. The whole blood was collected by left ventricle puncture using a 10-ml syringe and an 18G needle partially injected into a BD Vacutainer blood-collection tube containing K2-ethylenediaminetetraacetic acid (EDTA; cat. 36643; BD, Franklin Lakes, NJ) or heparin (cat. 367874; BD) and centrifuged at 3000 × g for 15 min at 4°C. The plasma with EDTA was collected for angiotensin II enzyme immunoassay [33] and Western blotting analyses on ACE and ACE2 proteins. The plasma with heparin was used for ACE activity assay. Lungs and kidneys were collected, snap frozen in liquid nitrogen, and stored at −80°C until analysis.
Tissues were thawed on ice before analyses. The middle lobe of the right lung from each dam was trimmed off the trachea and primary bronchi and used for RNA extraction, and the upper lobe of the right lung was used for protein extraction. The right kidney from each dam was cut into two halves longitudinally, and one half was used for RNA extraction, the other half for protein extraction.
RNA Extraction and RT-PCR
Total RNA was extracted from lung and kidney tissues (n = 4–5 rats/diet/day of pregnancy) by Trizol reagent (cat. 15596-018; Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. The possible genomic DNA in total RNAs was digested with RNA-free DNase I (cat. 79254; Qiagen Inc., Valencia, CA), followed by clean-up procedures using a Qiagen RNeasy minikit (cat. 74104; Qiagen). In all these procedures the manufacturer's instructions were followed. Complementary DNA was synthesized from 1 μg of total RNA by reverse transcription in a total volume of 20 μl by using a MyCycler Thermal Cycler (cat. 170-9703; Bio-Rad Laboratories, Hercules, CA) under the following conditions: one cycle at 28°C for 15 min, 42°C for 50 min, and 95°C for 5 min.
Quantitative Real-Time PCR
Real-time PCR detection was performed on a CFX96Real-Time PCR Detection System (cat. 184-5096; Bio-Rad). Primers were designed using Primer 3 Version 4 (http://primer3.sourceforge.net/) or prepared according to the references and are shown in Table 1. Syber Green Supermix (cat. 170-8882; Bio-Rad) was used for amplification of Ace, Ace2, Ren (renin), and Rn18s. The reaction mixture was incubated at 95°C for 10 min and cycled according to the following parameters: 95°C for 30 sec and 60°C for 1 min for a total of 40 cycles. Negative control without cDNA was performed to test primer specificity. The relative gene expression was calculated by use of the threshold cycle (CT) Rn18s/CT target gene.
TABLE 1.
Quantitative real-time PCR primers.
Protein Extraction from Lung and Kidney
Lung and kidney tissues were lysed in Component C Buffer using the SensoLyte 390 ACE2 Activity Assay Kit (cat. 72086; Anaspec Inc., Fremont, CA), and total proteins were extracted and analyzed with an ACE and ACE2 activity assay and Western blotting. All these procedures were conducted by following the manufacturer's instructions with minor modifications. Briefly, tissues were homogenized in Component C Buffer containing 0.5% (v/v) Triton-X 100 with a Polytran homogenizer at 15 000 rpm for 1 min, followed by incubation for 15 min at 4°C. Tissue lysates were centrifuged for 10 min at 1000 × g at 4°C and the supernatant fractions were collected, aliquoted, and stored at −80°C until analyzed by ACE and ACE2 protein activity assays and Western blotting. Protein concentration was determined by using a Pierce BCA Protein Assay Kit (cat. 23225; Pierce Biotechnology, Rockford, IL).
Western Blotting
Aliquots of 50 μg of proteins from rat lungs and kidneys or 2 μl of rat plasma were added with 4× sample buffer (200 mM Tris [pH 6.8]; 8% [w/v] SDS; 0.005% [w/v] bromophenol blue; 20% [v/v] glycerol; 2% [v/v] β-mercaptoethanol), followed by incubation at 70°C for 10 min. The separated proteins in SDS-PAGE were transferred onto a nitrocellulose membrane at 4°C overnight. After blocking in 5% nonfat milk, a rabbit anti-ACE polyclonal IgG (cat. PT344R; Panora Biotech, Sugarland, TX) or a rabbit anti-ACE2 polyclonal IgG (cat. ab87436; Abcam Inc., Cambridge, MA) at 1:2000 dilutions was added to nitrocellulose membrane and incubated at 4°C overnight. The blots were washed and incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (cat. 1030-05; Southern Biotech, Birmingham, AL) at 1:2000 dilutions at room temperature for 1 h. ACTB (β-actin) was used as an internal control for Western blotting in this study. Primary antibody, mouse monoclonal antibody for ACTB (cat. 3700; Cell Signaling, Danvers, MA), and secondary antibody, HRP-conjugated goat antimouse IgG (cat. 1030-05; Southern Biotech, Birmingham, AL), were used at 1:10 000 dilutions. Proteins in blots were visualized with Pierce enhanced chemiluminescence detection (cat. 32209; Thermo Scientific, Rockford, IL) and Blue Lite Autorad Film (cat. F9024; BioExpress, Kaysville, UT) according to the manufacturers' recommendations. The signals in films representing the contents of the target proteins ACE and ACE2 and the internal control protein ACTB were quantified by densitometry using Fluorchem 8000 software (Cell Biosciences, Santa Clara, CA). The relative amount of target protein was expressed as a ratio to ACTB in each rat lung or kidney analyzed by Western blotting.
ACE Activity Assay
The ACE activity was measured using the substrate hippuryl-L-histidyl-L-leucine. ACE cleaves the substrate to expose a free N-terminus, which can be fluorogenically labeled with o-phthaldialdehyde. The procedures were described in detail by Hemming and Selkoe [41]. A total of 2.5 μg protein from each tissue of interest or 2.5 μl plasma was analyzed, and fluorescence was measured after 15-min reactions at Ex/Em = 355 nm/544 nm. When plasma ACE activity was measured, 2.5 μg of lung proteins from control rats at Day 19 of pregnancy was used as a positive control.
ACE2 Activity Assay
The ACE2 activity was assessed with SensoLyte 390 ACE2 Activity Assay Kit by following the manufacturer's instructions. A total of 50 μg proteins from each tissue of interest was analyzed, and fluorescence was measured after 30-min reactions at Ex/Em = 330 nm/390 nm.
Statistical Analysis
All quantitative data were subjected to least-squares ANOVA by using the general linear models procedures of the Statistical Analysis System (version 9.1; SAS Institute, Cary, NC). Data on gene expression, the relative abundance of proteins, and enzyme activity were analyzed for effects of day of pregnancy, diet treatment, and their interaction. In ANOVA, differences in treatment or day means were determined by the Student-Newman-Keuls multiple comparison test. Log transformation of variables was performed when the variance of data was not homogenous among treatment groups, as assessed by the Levene test. P ≤ 0.05 was considered significant; a P > 0.05 and P ≤ 0.10 was considered a trend toward significance. Data were presented as least-squares means with overall SEMs.
RESULTS
The mRNA Levels of Ace and Ace2 in Maternal Lung Were Increased by Gestational PR
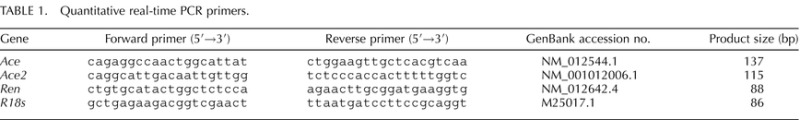
The mRNA levels of Ace were increased by 2.7- and 3.4-fold (P < 0.01) in the PR group at Days 19 and 22 of pregnancy, respectively, compared to those in the control group (Fig. 1A). The mRNA levels of Ace2 were increased by 1.7-fold (P < 0.05) in the PR group at Day 19, compared to those in the control group, while mRNA levels of Ace2 were comparable in PR and control groups at Day 22 (Fig. 1B).
FIG. 1.
Quantitative real-time PCR analysis of Ace (A) and Ace2 (B) in lungs from pregnant rats with PR at Days 19 and 22 of pregnancy. CT, control; PR, protein restriction. The error bar represents the mean ± SEM expressed as relative units of mRNA standardized against R18s (n = 4–5). *P < 0.05; **P < 0.01.
The Abundance of ACE Proteins in Maternal Lung, but Not ACE2 Proteins, Was Increased by Gestational PR
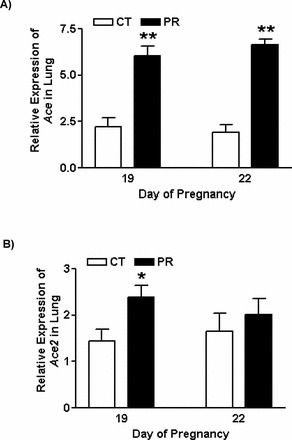
The abundance of ACE protein in lungs was increased 3.0-fold (P < 0.05) in the PR group at Day 19 of pregnancy compared to the control group; a similar trend was observed at Day 22 (P = 0.08; Fig. 2, A and B). The abundance of ACE2 protein in lungs was decreased (P < 0.05) by 1.4- and 1.6-fold in the PR group at Days 19 and 22, respectively. In both PR and control groups, the abundance of ACE2 protein was greater (P < 0.05) at Day 19 compared to Day 22 (Fig. 2, C and D).
FIG. 2.
Western blotting analysis of ACE and ACE2 proteins in lungs from pregnant rats with PR at Days 19 and 22 of pregnancy. A) ACE proteins shown as 150-kDa bands. B) Relative abundance of ACE protein. C) ACE2 proteins shown as 90-kDa bands. D) Relative abundance of ACE2 protein. ACTB, beta-actin; CT, control; PR, protein restriction. The error bar represents the mean ± SEM expressed as the ratio of density of the ACE or ACE2 band to that of ACTB (n = 4–5). *P < 0.05.
The Activity of ACE Proteins in Maternal Lung Was Increased by Gestational PR
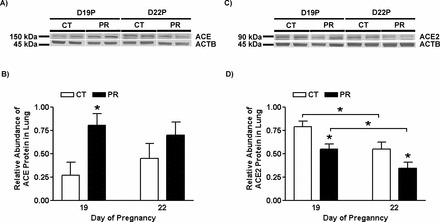
The relative activity of ACE protein in lungs was increased by 1.3- (P < 0.01) and 1.2-fold (P < 0.05) in the PR group at Days 19 and 22 of pregnancy, respectively, compared to that in the control group (Fig. 3A). The relative activity of ACE2 protein in lungs was not altered by PR at both Days 19 and 22 (Fig. 3B).
FIG. 3.
In vitro analysis of ACE (A) and ACE2 (B) activities in lungs from pregnant rats with PR. CT, control; PR, protein restriction. The error bar represents the mean ± SEM (n = 4–5). *P < 0.05; **P < 0.01.
The mRNA Levels of Ace, Ace2, and Ren in Kidney from Pregnant Rats Were Not Changed by Gestational PR
The mRNA levels of Ace (Fig. 4A), Ace2 (Fig. 4B), and Ren (Fig. 4C) in kidney in pregnant rats were not altered by gestational PR at both Days 19 and 22 of pregnancy.
FIG. 4.
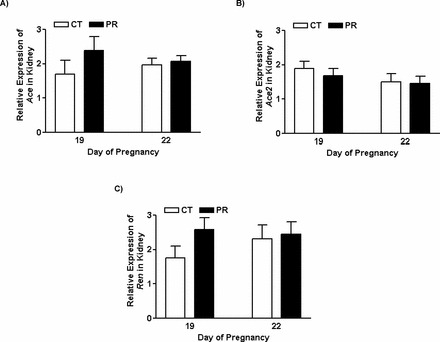
Quantitative real-time PCR analysis of Ace (A), Ace2 (B), and Ren (C) in kidneys from pregnant rats with PR at Days 19 and 22 of pregnancy. CT, control; PR, protein restriction. The error bar represents the mean ± SEM expressed as relative units of mRNA standardized against R18s (n = 4–5).
The Abundance of ACE and ACE2 Proteins in Kidney in Pregnant Rats Was Not Changed by Gestational PR
The abundance of ACE proteins in kidney in pregnant rats was not changed by PR at both Days 19 and 22 of pregnancy. In both the PR and control groups, the abundance of ACE proteins was greater (P < 0.05) at Day 19 than that at Day 22 (Fig. 5, A and B).
FIG. 5.
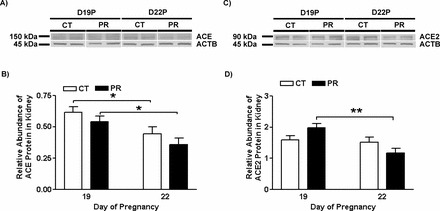
Western blotting analysis of ACE and ACE2 proteins in lungs from pregnant rats with PR at Days 19 and 22 of pregnancy. A) ACE proteins shown as 150-kDa bands. B) Relative abundance of ACE protein. C) ACE2 proteins shown as 90-kDa bands. D) Relative abundance of ACE2 protein. ACTB, beta-actin; CT, control; PR, protein restriction. The error bar represents the mean ± SEM expressed as the ratio of density of the ACE or ACE2 band to that of ACTB (n = 4–5). *P < 0.05; **P < 0.01.
The abundance of ACE2 proteins in kidney in pregnant rats was not changed by PR at both Days 19 and 22 of pregnancy. In the PR group, the abundance of ACE2 proteins was greater (P < 0.01) at Day 19 than that at Day 22 (Fig. 5, C and D).
The Activities of ACE and ACE2 Proteins in Kidney in Pregnant Rats Were Not Changed by Gestational PR
The activities of ACE (Fig. 6A) and ACE2 (Fig. 6B) proteins in kidney in pregnant rats were comparable between the PR and control groups at both Days 19 and 22 of pregnancy.
FIG. 6.
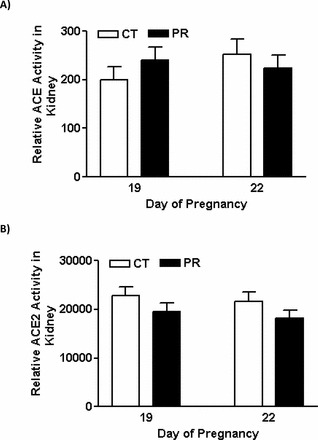
In vitro analysis of ACE (A) and ACE2 (B) activities in kidneys from pregnant rats with PR. CT, control; PR, protein restriction. The error bar represents the mean ± SEM (n = 4–5).
Plasma Levels of ACE Proteins in Pregnant Rats Were Not Changed by Gestational PR
The relative abundance of ACE proteins in maternal plasma was not altered by PR at both Days 19 and 22 of pregnancy. In both PR and control groups, the relative abundance of ACE proteins in plasma was higher (P < 0.001) at Day 22 than Day 19 (Fig. 7). In addition, ACE2 protein was not detectable by Western blotting (data not shown).
FIG. 7.
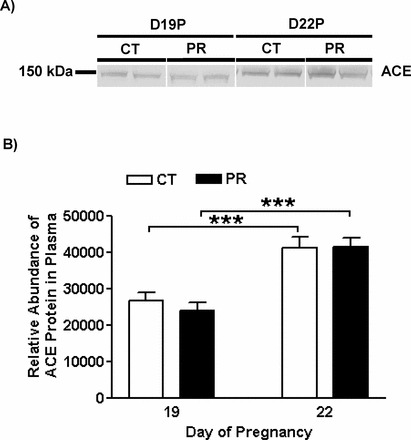
Western blotting analysis of ACE proteins in plasma from pregnant rats with PR at Days 19 and 22 of pregnancy. A) ACE proteins shown as 150-kDa bands. B) Relative abundance of ACE protein. CT, control; PR, protein restriction. The error bar represents the mean ± SEM of the density of ACE bands (n = 4–5). ***P < 0.001.
Plasma ACE Activity Was Not Changed by Gestational PR
The relative activity of ACE in maternal plasma was reduced by 1.3-fold (P < 0.05) by PR at Day 19 of pregnancy, and unchanged by PR at Day 22. Compared to Day 19, the relative activity of ACE in maternal plasma at Day 22 was decreased by 1.6- (P < 0.001) and 1.3- (P < 0.05) fold, respectively, in control and PR groups.
DISCUSSION
This study revealed for the first time that increased expression of ACE in lung tissue, but not kidneys or plasma, is likely a key contributor to elevated plasma angiotensin II in pregnant rats fed a PR diet. This conclusion is further supported by observations that Ace expression is low in uterine and mesenteric arteries and that PR does not increase Ace expression in these vessels [33, 42]. Our previous studies found that gestational PR caused elevations in the expression of angiotensin II receptor type I in uterine arteries [33] and enhanced uterine artery constriction to angiotensin II ex vivo (Gao and Yallampalli, unpublished data). Moreover, we also found that Ace2 expression in the placental labyrinth zone was reduced in PR rats [34], possibly contributing to the elevated angiotensin II locally. These two studies, together with the findings of this study, suggest that PR during pregnancy causes alterations in maternal RAS that may reduce utero-placental blood flow. In addition, it was reported that total uterine and placental blood flows in pregnant rats with global dietary restriction were reduced 60%–65% relative to the ad libitum-fed dams [35]. Therefore, findings of this study contribute to our knowledge of the RAS-mediated fetal growth restriction and associated fetal programming of hypertension.
Several lines of evidence suggest that the PR-induced elevations in plasma angiotensin II levels [33] may be derived from the increased production due to elevated expression of Ace and ACE activity in the maternal lung, and not due to the decreased degradation of angiotensin II. First, ACE proteins in rat lungs are more abundant than those in kidneys (Figs. 2A and 5A). Second, the ACE activity in lungs is 50 times higher than that in kidneys in pregnant rats (Figs. 3A and 6A), which is similar to that reported in adult nonpregnant rats [7]. Third, although the abundance of ACE2 proteins was variable in rat lungs (Fig. 2, C and D) and kidneys (Fig. 5, C and D) with gestational PR, the ACE2 activity was unchanged in both these tissues. Fourth, although ACE2 protein (Fig. 5, C and D) and activity (Fig. 6B) were relatively greater in kidneys compared to lungs, they were unchanged with PR. Furthermore, the greater ACE2 expression and activity in kidneys may limit the local net production of angiotensin II. Finally, due to the abundance of capillary numbers, density, and large capillary surface density in lungs [43], the pulmonary circulation is suited to contribute large amounts of angiotensin II to the systemic circulation. Therefore, the data presented here, together with previously published results [33], support the idea that increased pulmonary ACE expression and activity contributes to enhanced generation of angiotensin II in pregnant rats with PR, and may contribute to the elevated angiotensin II levels in maternal circulation.
Unlike in lungs, Ace and Ace2 gene expressions (Figs. 4 and 5) and enzyme activities in kidneys (Fig. 6) were not altered by gestational PR in late pregnancy. In spite of the potent activity of renal ACE2 in kidneys [7], angiotensin II degradation via renal ACE2 activity in pregnant dams was unchanged by gestational PR. This may rule out the contribution of kidneys to the elevated plasma levels of angiotensin II in response to gestational PR. Similarly, gestational PR did not alter Ren expression in kidneys (Fig. 4C). These findings suggest that maternal kidneys may not play an important role in the regulation of plasma angiotensin II in response to gestational PR. However, in offspring kidneys the expression of ACE and ACE2 depends on the type of programming insult during pregnancy. For example, in undernutrition models, there are no changes in both renal Ace and Ace2 mRNA levels in offspring [7], although nephrogenesis and renal functions are impaired [28, 44]. However, renal ACE levels are elevated with no changes in ACE2 (Yallampalli et al., unpublished) in PR models. Thus, the expression sensitivity of renal ACE and ACE2 in the offspring may be dependent upon the type of insult.
The current study contributes novel data regarding plasma ACE and angiotensin II levels investigates for the first time the relevance of plasma ACE to plasma angiotensin II levels under the setting of gestational PR. Several lines of evidence support that plasma ACE does not contribute to the elevated angiotensin II levels in plasma in response to PR. First, plasma ACE activity was decreased by PR at Day 19 but not at Day 22 of pregnancy (Fig. 8), although the abundance of ACE protein in plasma was unchanged by PR at both days (Fig. 7). Second, the reduced plasma ACE activity at Day 22 compared to that at Day 19 in the PR group (Fig. 8) obviously cannot cause the increased plasma angiotensin II levels at Day 22 compared to Day 19 [33]. Third, the contribution of other enzymes such as plasma chymases for the elevated plasma angiotensin II levels in the PR group are negligible, since 98% of total ACE activity was inhibited with ACE inhibitor and captopril (data not shown), and chymase activity was shown to be uninhibited by ACE inhibitors [45]. Fourth, the low abundance or absence of ACE2 protein in maternal plasma may also contribute to reduced degradation of angiotensin II in the systemic circulation. Overall, this study suggests that gestational PR causes elevated maternal plasma angiotensin II levels, with lung as the key contributor to its production. These elevated angiotensin II levels then act on systemic and uterine arteries, resulting in elevated blood pressure and reduced blood flows, respectively, as demonstrated in ewes infused with angiotensin II in late pregnancy [46].
FIG. 8.
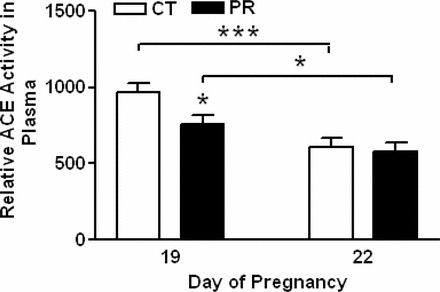
In vitro analysis of ACE activity in plasma from pregnant rats with PR. CT, control; PR, protein restriction. The error bar represents the mean ± SEM (n = 4–5). *P < 0.05; ***P < 0.001.
The functions of Ace and Ace2 in both lungs and kidneys may be regulated at multiple levels, including transcriptional, posttranscriptional, and posttranslational. For instance, mRNA levels of Ace2 in lungs of the PR group at Day 19 of pregnancy were elevated (Fig. 1B), but protein levels (Fig. 2D) and activities were not (Fig. 3B). Although the underlying mechanisms for this discrepancy remain unclear, similar observations were also reported for RAS components in other tissues, including rat uterine artery [33] and the placental labyrinth zone [34]. Recently, it has been reported that gene expressions of Ace, Ace2, and Agtr2 (angiotensin II receptor, type 2) are posttranscriptionally regulated by micro-RNAs [16, 20]. However, to date, the existence of micro-RNAs regulating expressions of ACE and ACE2 in maternal lungs and kidneys has not been reported. Moreover, the complicated glycosylation of ACE not only changes its molecular weight and band size in SDS-PAGE, but also modulates its catalytic properties, cellular localization, and affinity to substrates [47, 48]. This may partly explain the discrepancy between the protein levels and enzymatic activities in lungs, kidneys, and plasma.
The mechanisms for the elevated expression of Ace in lungs remain unknown, and this question needs to be addressed in future studies. We suggest that the increased glucocorticoids in rats fed with PR diet may contribute to the elevated activity of ACE proteins, since gestational PR has been shown to increase plasma levels of glucocorticoids [49], and glucocorticoids increases ACE activity in endothelial cells as well as in normal rat lungs in vitro [50–52]. In addition, our preliminary study found that gestational PR is associated with elevated expression of glucocorticoid receptor in maternal lungs (Gao and Yallampalli, unpublished data). Collectively, these data suggest nutrient restriction during pregnancy may increase glucocorticoids, thus impacting fetal growth and development by modulating the expression of Ace and/or the activity of ACE proteins in maternal lungs.
In summary, the current study suggests that increased expression of pulmonary ACE contributes to elevated circulating angiotensin II in pregnant rats with PR. This novel finding opens a new window for future research on mechanisms of maternal contributions to fetal programming of hypertension.
ACKNOWLEDGMENT
The authors thank Ms. Elizabeth A. Powell for editorial work on this manuscript and administrative support.
Footnotes
1Supported by National Institutes of Health grants R01HL102866 and R01HL58144.
REFERENCES
- Anton L, Brosnihan KB. Systemic and uteroplacental renin–angiotensin system in normal and pre-eclamptic pregnancies. Ther Adv Cardiovasc Dis 2008; 2: 349 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irani RA, Xia Y. Renin angiotensin signaling in normal pregnancy and preeclampsia. Semin Nephrol 2011; 31: 47 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai M, Horiuchi M. Devil and angel in the renin-angiotensin system: ACE-angiotensin II-AT1 receptor axis vs. ACE2-angiotensin-(1–7)-Mas receptor axis. Hypertens Res 2009; 32: 533 536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush JW, Aultman CD. Vascular biology of angiotensin and the impact of physical activity. Appl Physiol Nutr Metab 2008; 33: 162 172. [DOI] [PubMed] [Google Scholar]
- Paul M, Poyan MA, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev 2006; 86: 747 803. [DOI] [PubMed] [Google Scholar]
- Caldwell PR, Seegal BC, Hsu KC, Das M, Soffer RL. Angiotensin-converting enzyme: vascular endothelial localization. Science 1976; 191: 1050 1051. [DOI] [PubMed] [Google Scholar]
- Riviere G, Michaud A, Breton C, VanCamp G, Laborie C, Enache M, Lesage J, Deloof S, Corvol P, Vieau D. Angiotensin-converting enzyme 2 (ACE2) and ACE activities display tissue-specific sensitivity to undernutrition-programmed hypertension in the adult rat. Hypertension 2005; 46: 1169 1174. [DOI] [PubMed] [Google Scholar]
- Ng KK, Vane JR. Conversion of angiotensin I to angiotensin II. Nature 1967; 216: 762 766. [DOI] [PubMed] [Google Scholar]
- Mulrow PJ. The intrarenal renin-angiotensin system. Curr Opin Nephrol Hypertens 1993; 2: 41 44. [DOI] [PubMed] [Google Scholar]
- Gorin AB, Hasagawa G, Hollinger M, Sperry J, Zuckerman J. Release of angiotensin converting enzyme by the lung after Pseudomonas bacteremia in sheep. J Clin Invest 1981; 68: 163 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosnihan KB, Neves LA, Anton L, Joyner J, Valdes G, Merrill DC. Enhanced expression of Ang-(1–7) during pregnancy. Braz J Med Biol Res 2004; 37: 1255 1262. [DOI] [PubMed] [Google Scholar]
- Levy A, Yagil Y, Bursztyn M, Barkalifa R, Scharf S, Yagil C. ACE2 expression and activity are enhanced during pregnancy. Am J Physiol Regul Integr Comp Physiol 2008; 295: R1953 R1961. [DOI] [PubMed] [Google Scholar]
- Neves LA, Stovall K, Joyner J, Valdes G, Gallagher PE, Ferrario CM, Merrill DC, Brosnihan KB. ACE2 and ANG-(1–7) in the rat uterus during early and late gestation. Am J Physiol Regul Integr Comp Physiol 2008; 294: R151 R161. [DOI] [PubMed] [Google Scholar]
- Brosnihan KB, Hering L, Dechend R, Chappell MC, Herse F. Increased angiotensin II in the mesometrial triangle of a transgenic rat model of preeclampsia. Hypertension 2010; 55: 562 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal R, Galffy A, Field SA, Gheorghe CP, Mittal A, Longo LD. Maternal protein deprivation: changes in systemic renin-angiotensin system of the mouse fetus. Reprod Sci 2009; 16: 894 904. [DOI] [PubMed] [Google Scholar]
- Goyal R, Goyal D, Leitzke A, Gheorghe CP, Longo LD. Brain renin-angiotensin system: fetal epigenetic programming by maternal protein restriction during pregnancy. Reprod Sci 2010; 17: 227 238. [DOI] [PubMed] [Google Scholar]
- Connors N, Valego NK, Carey LC, Figueroa JP, Rose JC. Fetal and postnatal renin secretion in female sheep exposed to prenatal betamethasone. Reprod Sci 2010; 17: 239 246. [DOI] [PubMed] [Google Scholar]
- Forhead AJ, Fowden AL. Role of angiotensin II in the pressor response to cortisol in fetal sheep during late gestation. Exp Physiol 2004; 89: 323 329. [DOI] [PubMed] [Google Scholar]
- Dodic M, Baird R, Hantzis V, Koukoulas I, Moritz K, Peers A, Wintour EM. Organs/systems potentially involved in one model of programmed hypertension in sheep. Clin Exp Pharmacol Physiol 2001; 28: 952 956. [DOI] [PubMed] [Google Scholar]
- Goyal R, Lister R, Leitzke A, Goyal D, Gheorghe CP, Longo LD. Antenatal maternal hypoxic stress: adaptations of the placental renin-angiotensin system in the mouse. Placenta 2011; 32: 134 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Bazer FW, Cudd TA, Meininger CJ, Spencer TE. Maternal nutrition and fetal development. J Nutr 2004; 134: 2169 2172. [DOI] [PubMed] [Google Scholar]
- Wu G, Bazer FW, Wallace JM, Spencer TE. Board-invited review: intrauterine growth retardation: implications for the animal sciences. J Anim Sci 2006; 84: 2316 2337. [DOI] [PubMed] [Google Scholar]
- Muller O, Krawinkel M. Malnutrition and health in developing countries. CMAJ 2005; 173: 279 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad AS, Fitzgerald JT, Hess JW, Kaplan J, Pelen F, Dardenne M. Zinc deficiency in elderly patients. Nutrition 1993; 9: 218 224. [PubMed] [Google Scholar]
- Gangula PR, Reed L, Yallampalli C. Antihypertensive effects of flutamide in rats that are exposed to a low-protein diet in utero. Am J Obstet Gynecol 2005; 192: 952 960. [DOI] [PubMed] [Google Scholar]
- Kwong WY, Wild AE, Roberts P, Willis AC, Fleming TP. Maternal undernutrition during the preimplantation period of rat development causes blastocyst abnormalities and programming of postnatal hypertension. Development 2000; 127: 4195 4202. [DOI] [PubMed] [Google Scholar]
- Langley-Evans SC, Gardner DS, Jackson AA. Maternal protein restriction influences the programming of the rat hypothalamic-pituitary-adrenal axis. J Nutr 1996; 126: 1578 1585. [DOI] [PubMed] [Google Scholar]
- Langley-Evans SC, Welham SJ, Jackson AA. Fetal exposure to a maternal low protein diet impairs nephrogenesis and promotes hypertension in the rat. Life Sci 1999; 64: 965 974. [DOI] [PubMed] [Google Scholar]
- McMullen S, Langley-Evans SC. Sex-specific effects of prenatal low-protein and carbenoxolone exposure on renal angiotensin receptor expression in rats. Hypertension 2005; 46: 1374 1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullen S, Langley-Evans SC. Maternal low-protein diet in rat pregnancy programs blood pressure through sex-specific mechanisms. Am J Physiol Regul Integr Comp Physiol 2005; 288: R85 R90. [DOI] [PubMed] [Google Scholar]
- Watkins AJ, Wilkins A, Cunningham C, Perry VH, Seet MJ, Osmond C, Eckert JJ, Torrens C, Cagampang FR, Cleal J, Gray WP, Hanson MA. et al. Low protein diet fed exclusively during mouse oocyte maturation leads to behavioural and cardiovascular abnormalities in offspring. J Physiol 2008; 586: 2231 2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kautzky-Willer A, Handisurya A. Metabolic diseases and associated complications: sex and gender matter! Eur J Clin Invest 2009; 39: 631 648. [DOI] [PubMed] [Google Scholar]
- Gao H, Yallampalli U, Yallampalli C. Protein restriction to pregnant rats increases the plasma levels of angiotensin II and expression of angiotensin II receptors in uterine arteries. Biol Reprod 2012; 86: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Yallampalli U, Yallampalli C. Maternal protein restriction reduces expression of angiotensin I-converting enzyme 2 in rat placental labyrinth zone in late pregnancy. Biol Reprod 2012; 86: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahokas RA, Anderson GD, Lipshitz J. Effect of dietary restriction, during the last week only or throughout gestation, on cardiac output and uteroplacental blood flow in pregnant rats. J Nutr 1983; 113: 1766 1776. [DOI] [PubMed] [Google Scholar]
- Koumentaki A, Anthony F, Poston L, Wheeler T. Low-protein diet impairs vascular relaxation in virgin and pregnant rats. Clin Sci (Lond) 2002; 102: 553 560. [PubMed] [Google Scholar]
- Beinder E. [Fetal growth retardation and diseases in adult life]. Gynakol Geburtshilfliche Rundsch 2008; 48: 207 214. [DOI] [PubMed] [Google Scholar]
- Holemans K, Aerts L, Van Assche FA. Fetal growth restriction and consequences for the offspring in animal models. J Soc Gynecol Investig 2003; 10: 392 399. [DOI] [PubMed] [Google Scholar]
- Ojeda NB, Grigore D, Alexander BT. Intrauterine growth restriction: fetal programming of hypertension and kidney disease. Adv Chronic Kidney Dis 2008; 15: 101 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross MG, Beall MH. Adult sequelae of intrauterine growth restriction. Semin Perinatol 2008; 32: 213 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemming ML, Selkoe DJ. Amyloid beta-protein is degraded by cellular angiotensin-converting enzyme (ACE) and elevated by an ACE inhibitor. J Biol Chem 2005; 280: 37644 37650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathishkumar K, Balakrishnan M, Chinnathambi V, Gao H, Yallampalli C. Temporal alterations in vascular angiotensin receptors and vasomotor responses in offspring of protein-restricted rat dams. Am J Obstet Gynecol 2012; 206: 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes K, Murphy LJ, Takahashi M, Tanzawa K, Turner AJ. Localization and biochemical characterization of endothelin-converting enzyme. J Cardiovasc Pharmacol 1995; 26 (suppl 3): S37 S39. [PubMed] [Google Scholar]
- Woods LL, Ingelfinger JR, Nyengaard JR, Rasch R. Maternal protein restriction suppresses the newborn renin-angiotensin system and programs adult hypertension in rats. Pediatr Res 2001; 49: 460 467. [DOI] [PubMed] [Google Scholar]
- Wang Y, Gu Y, Lewis DF, Alexander JS, Granger DN. Elevated plasma chymotrypsin-like protease (chymase) activity in women with preeclampsia. Hypertens Pregnancy 2010; 29: 253 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld CR, Naden RP. Uterine and nonuterine vascular responses to angiotensin II in ovine pregnancy. Am J Physiol 1989; 257: H17 H24. [DOI] [PubMed] [Google Scholar]
- Baudin B, Alves N, Pilon A, Beneteau-Burnat B, Giboudeau J. Structural and biological roles of glycosylations in pulmonary angiotensin I-converting enzyme. Glycobiology 1997; 7: 565 570. [DOI] [PubMed] [Google Scholar]
- Orth T, Voronov S, Binevski P, Saenger W, Kost O. Glycosylation of bovine pulmonary angiotensin-converting enzyme modulates its catalytic properties. FEBS Lett 1998; 431: 255 258. [DOI] [PubMed] [Google Scholar]
- Zambrano E, Nathanielsz PW, McDonald TJ. Neonatal ovine adrenal cortical and medullary Cell H+ responses to ACTH and prostaglandin E(2). Biol Neonate 2002; 82: 243 249. [DOI] [PubMed] [Google Scholar]
- Ialenti A, Calignano A, Carnuccio R, Di RM. Glucocorticoid induction of angiotensin converting enzyme. Agents Actions 1986; 17: 294 295. [DOI] [PubMed] [Google Scholar]
- Michel B, Grima M, Coquard C, Welsch C, Barthelmebs M, Imbs JL. Effects of triiodothyronine and dexamethasone on plasma and tissue angiotensin converting enzyme in the rat. Fundam Clin Pharmacol 1994; 8: 366 372. [DOI] [PubMed] [Google Scholar]
- Mendelsohn FA, Lloyd CJ, Kachel C, Funder JW. Glucocorticoid induction of angiotensin converting enzyme production from bovine endothelial cells in culture and rat lung in vivo. Clin Exp Pharmacol Physiol Suppl 1982; 7: 57 62. [PubMed] [Google Scholar]