

ABSTRACT
Intraovarian factors play important roles in coordinating germ cell and somatic cell growth in the ovary. Prior to the onset of gonadotropin stimulation and reproductive cyclicity, follicle development is dependent upon locally produced growth factors, such as the transforming growth factor beta family members inhibin, activin, and GDF9. In the absence of inhibin in prepubertal mice (Inha−/−), there are marked alterations in preantral follicle growth, but no evidence of ovarian tumors characteristic of adult Inha-null mice. To ascertain the contribution of GDF9 to the Inha-null phenotype, we analyzed folliculogenesis in postnatal Inha Gdf9 double knockout mice. Deletion of Gdf9 from Inha−/− rescues the initial growth defects found at early follicle stages in Inha−/− ovaries, but surprisingly enhances the onset of pretumor lesions. The normalization of growth dynamics between granulosa cells and oocytes of Inha Gdf9 double knockout mice is also accompanied by a reduction in levels of the activin/inhibin beta B subunit, Inhbb, which is upregulated in Inha−/− ovaries. However, at later ages, Inha Gdf9 double knockout ovaries are similar to Inha−/− ovaries, and show upregulation of the activin/inhibin subunits and downregulation of the growth factor, kit ligand, thus resulting in a local environment that is growth-promoting for granulosa cells but growth-inhibitory for oocytes. These data suggest a sequential mechanism of action initiated by GDF9 in the Inha knockout mouse that promotes defective folliculogenesis. These studies thus provide a novel role for GDF9 in causing reproductive defects and suppressing tumor initiation in the Inha−/− mouse model.
Keywords: cancer, fertility, ovary, reproduction, TGFβ, tumor
Deletion of GDF9 from the inhibin alpha-null mouse partially rescues the defects in early folliculogenesis but enhances the onset of tumor formation.
INTRODUCTION
Factors that control the development of postnatal folliculogenesis are critical in establishing a fully functional gonad capable of steroidogenesis, gamete production, and the maternal recognition of pregnancy. A large number of these intraovarian factors belong to the transforming growth factor β (TGFβ) family, including inhibin, activin, growth and differentiation factor 9 (GDF9), anti-Müllerian hormone, and bone morphogenetic proteins. Genetically modified mouse models for ligands, receptors, intracellular signaling proteins, and antagonists of the TGFβ family have provided key insights into the in vivo roles of these growth factors for both granulosa cell and oocyte development [1, 2].
Female inhibin alpha knockout (Inha−/−) mice develop granulosa cell tumors by 4–6 wk of age and die of an activin-driven cancer cachexia before 19 weeks of age [3, 4]. Tumor foci appear in the Inha−/− ovary by 4–6 wk of age [4], but full granulosa cell tumor development is dependent on adult levels of pituitary gonadotropins [5–7]. Prior to tumor initiation, prepubertal Inha−/− ovaries display disrupted folliculogenesis by 12 days of age [8]. At this age in Inha−/− ovaries, granulosa cell proliferation is accelerated and the resultant precocious follicle growth is accompanied by aberrant expression of multiple important growth factors, including kit ligand (Kitl) and the genes encoding the two isoforms of activin, Inhba and Inhbb [8].
Because activin and GDF9 are known to have similar regulatory roles in granulosa cells (i.e., inhibition of Kitl expression and enhancement of proliferation) [9–11], we previously suggested that these growth factors may have functionally redundant roles during folliculogenesis [8]. To investigate the contribution of GDF9 to the defects in the prepubertal Inha−/− ovary, we analyzed mice genetically deficient for both inhibin and GDF9 (Inha−/− Gdf9−/−). All Inha−/− mice and Inha−/− Gdf9−/− double mutant mice die of granulosa cell tumors followed by a cachexia-related death in a similar time frame [12]. However, we hypothesize that deletion of GDF9 will rescue any reproductive abnormalities in the prepubertal Inha−/− ovary for those genes that are not redundant with activin, and thus provide insight into functional differences between activin and GDF9 during early follicle development.
MATERIALS AND METHODS
Experimental Mice
All the mouse lines used in the present study were maintained on a mixed C57BL/6/129S7/SvEv genetic background and manipulated according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved animal protocols at Baylor College of Medicine. Ovaries were collected from mice at Postnatal Day (PND) 3, 6, and 12, with the day of birth considered as PND 0. Generation of Inha and Gdf9 single mutant and Inha Gdf9 double mutant mice was previously described [4, 12, 13]. PCR analysis of genomic tail DNA was used to genotype all the mice. Control and experimental mice of each genotype were collected from the Inha Gdf9 colony to reduce potential effects due to differences in genetic background. To increase genotype frequency, most crosses were set up between Inha+/− Gdf9+/− females with Inha+/− Gdf9−/− males, which are fertile. This results in all mice being either heterozygous or homozygous for Gdf9−/−, and where we used the Inha+/− Gdf9+/− as controls. For ease of reading, throughout the manuscript, double heterozygous mice (Inha+/− Gdf9+/−) are referred to as controls; Inha−/− Gdf9+/− are referred to as Inha−/−; Inha+/− Gdf9−/− are referred to as Gdf9−/−; and Inha−/− Gdf9−/− are referred to as Inha−/− Gdf9−/− unless indicated otherwise. Morphometric analysis and images in Figure 2 were performed using Inha−/− Gdf9+/+females for Inha−/− (i.e., not heterozygous for Gdf9). These mice were also derived from the Inha−/− Gdf9−/− colony, except they came from double heterozygous crosses.
FIG. 2.
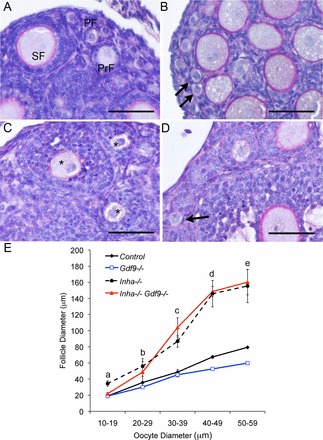
Loss of Gdf9 partly reduces the prevalence of abnormally small oocytes in multilayered follicles present in Inha−/− ovaries. A) At PND 12, primordial (PF) and primary (PrF) and growing secondary follicles (SF) are detected in control ovaries. B) In Gdf9−/− ovaries, primordial follicles (arrows) are comparable in size to control ovaries. C) Inha−/− ovaries exhibit many secondary follicles with multiple layers of granulosa cells enclosing oocytes that are equivalent in size to oocytes in control primordial and early stage (3a) primary follicles (asterisks). D) Unlike Inha−/−, the Inha−/− Gdf9−/− double mutant mice show more small oocytes in primordial follicles surrounded by a single layer of granulosa cells (arrow). E) Morphometric analysis of oocyte and follicle dynamics illustrates a GDF9-sensitive window during the primordial to primary follicle stages (10–29 μm sized oocytes) in Inha−/− mice. The Inha−/− Gdf9−/− ovary (red line) did not display abnormal germ and somatic cell dynamics to the same extent as Inha−/− ovaries (black dashed line) until the oocytes were beyond 30 μm. Bars = 50 μm (A–D). Values are mean ± SEM of a minimum of three mice of each genotype. Letters (a–e) above the lines indicate post hoc analysis of follicle diameter using Fisher LSD following ANOVA for Inha−/− Gdf9−/− oocyte size compared to control, Gdf9−/−, and Inha−/− oocytes as follows: a) the double knockout is not significant (NS) compared to control or Gdf9−/−, but P < 0.0001 compared to Inha−/−; b) the double knockout is NS compared to control and NS compared to Inha−/−, but P < 0.05 compared to Gdf9−/−; c) the double knockout is P < 0.01 compared to control and Gdf9−/−, but NS compared to Inha−/−; and d and e) the double knockout is P < 0.001 compared to control and Gdf9−/−, but NS compared to Inha−/−.
Tissue Collection and Histologic Analysis
All the mice were anesthetized by isoflurane inhalation (Abbott Laboratories) and either euthanized by decapitation (prepubertal mice) or by cervical dislocation (adult mice). Ovaries for histological and immunohistological analyses were collected in either Bouin solution (Sigma-Aldrich) or 10% neutral buffered formalin (Electron Microscopy Sciences). Processing and embedding were performed at the Pathology Core Histology Facility at Baylor College of Medicine. Ovaries for gene expression studies were stored in RNAlater (Ambion) at −80°C prior to processing for RNA.
For histological assessment, ovaries were serially sectioned into 4–6 μm sections and stained with periodic acid/Schiff reagent and hematoxylin. Morphometric analysis was performed similar to previous studies [8].
Quantitative RT-PCR
RNA was isolated using the QIAGEN RNeasy Microkit. RNA concentration was quantified using a NanoDrop SpectrophotometerND-1000 (NanoDrop Technologies). Complementary DNA was reverse transcribed from 200 ng of total RNA in a 20 μl reaction using High Capacity RNA-to-cDNA Master Mix (Applied Biosystems). Real-time PCR was performed using the Applied Biosystems StepOne Plus Sequence Detection System. Quantitative PCRs (qPCRs) of Inhba and Inhbb were performed using Taqman Master Mix and Gene Expression Assays and the following Taqman Assays: Inhba, Mm00434338_ml; Inhbb, Mm03023992_ml; and mouse glyceraldehyde-3-phosphate dehydrogenase (Gapdh) as an endogenous control [8]. Kitl1 and Kitl2 qPCR were performed using Fast Sybr Green Master Mix (Applied Biosystem) based on previously published primer sequences [14] that were specifically designed to amplified Kitl1 (forward: 5′-TGCAGCCAGCTCCCTTAGGA; reverse: 5′-TGTAGGCCCGAGTCTTCA) and Kitl2 (forward: 5′-TTATGGTGGCATCTGACACTAGT; reverse: 5′-TGTAGGCCCGAGTCTTCA). All the qPCR data was analyzed by the ΔΔ cycle threshold (CT) method using the ABI StepOne Plus Software and normalized to the endogenous reference (Gapdh). The mean ± SEM were calculated, and the results were plotted using the relative expression of each target gene with each sample compared to the mean of the calibrator (Control, PND 12).
Statistical Analyses
Statistical analysis was carried out using SPSSX version 20 (IBM). One-way ANOVA followed by the Fisher least significance differences (LSD) post hoc test were used for multiple comparisons, unless otherwise indicated. A minimum of three independent experiments were carried out for all the experiments, and P < 0.05 was considered statistically significant.
RESULTS
Loss of Both Inha and Gdf9 Results in Enlarged Ovaries and Precocious Follicle Development at PND 12
To determine if GDF9 contributes to the dysregulated postnatal folliculogenesis of Inha−/− ovaries, we genetically ablated Gdf9 from Inha−/− mice by generating Inha−/− Gdf9−/− double knockout mice, similar to a previous study that analyzed tumor development in adult mice [12]. For this study, we analyzed follicle development in the prepubertal postnatal ovary. Control and Gdf9−/− ovaries were approximately half the size of Inha−/− and Inha−/− Gdf9−/− double mutant ovaries at PND 12 (Fig. 1). Similar to adult Gdf9−/− ovaries, PND 12 Gdf9−/−ovaries contain follicles that do not progress beyond the primary stage of development [13]; however, unlike ovaries from adult mice, at PND 12, Gdf9−/− oocytes are not bigger than 50 μm (Fig. 1D). As previously reported [8], Inha−/− ovaries display a precocious follicular phenotype with large multilayered follicles, some of which exhibit small fluid filled cavities (Fig. 1, E and F). Gdf9 levels prior to development of the Inha−/− follicle defect (i.e., at PND 3) [8] are similar to the controls (Supplemental Fig. S1; all the Supplemental Data are available online at www.biolreprod.org). Abnormal growth of the postnatal ovaries in Inha−/− mice occurs in the absence of significant changes in serum follicle-stimulating hormone (FSH) [8]. Serum FSH levels also do not change in postnatal Inha−/− Gdf9−/− mice compared to controls or Inha−/− mice (Supplemental Fig. S2 and Supplemental Materials and Methods). The combinational loss of Inha and Gdf9 results in a morphologically similar phenotype to Inha−/− ovaries, with large multilayered follicles not evident in the age-matched controls (Fig. 1, G and H).
FIG. 1.
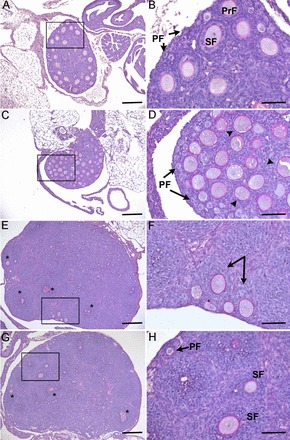
Genetic deletion of Inha and Gdf9 alters folliculogenesis at PND 12. Twelve-day-old control (A, B) ovaries contain primordial (PF) and primary (PrF) and secondary follicles (SF), while 12-day-old Gdf9−/− (C, D) ovaries block at the primary stage with a single layer of granulosa cells (arrowheads). E, F) Inha−/− ovaries are much larger at PND 12 and contain large multilayer follicles (* in E) and abnormal asymmetric secondary follicles (arrows in F). G, H) Twelve-day-old Inha−/− Gdf9−/− ovaries have a morphologically similar phenotype to Inha−/− ovaries with large multilayered follicles (* in G) that are not observed in age-matched controls or Gdf9−/− alone. Boxed regions in the left panels are shown at higher magnification in the right panels. Bars = 200 μm (A, C, E, G) and 50 μm (B, D, F, H).
The Growth Dynamics Between Germ and Somatic Cell Components Are Partly Rescued by Ablation of Gdf9 from Inha−/− Ovaries but Only at the Earliest Stages
The synchronized growth between the germ cell and somatic cells of the ovarian follicle at the early stages of growth is dependent upon locally produced growth factors such as GDF9 and KITL. For example, in the absence of Gdf9, primary follicles of adult ovaries contain large oocytes (up to 80 μm) that are likely due to the upregulation of Kitl in the Gdf9−/− ovaries [13, 15]. In contrast, loss of the inhibin alpha subunit (and thus, a gain of activin production) results in very small oocytes (20–30 μm) and a loss of Kitl expression [8]. The inverse nature of these phenotypes led us to investigate the respective contributions of GDF9, KITL, and activin to the loss of synchrony between germ and somatic cell components. Morphometric analysis of oocyte versus follicle diameter showed that removal of Gdf9 from Inha−/− recovered the normal oocyte to follicle diameter relationship when oocytes were less than 20 μm in size (i.e., corresponding to primordial follicles; Fig. 2). At an oocyte size of 20–29 μm, follicle diameter measurements for Inha−/− Gdf9−/− were midway between control and Inha−/− ovaries, and not statistically different from either genotype. However, when oocyte diameters were over 30 μm, Inha−/− and Inha−/− Gdf9−/− had similar follicle sizes that were abnormally enlarged compared to the controls (Fig. 2). These data suggest that a GDF9-sensitive window exists prior to the early primary follicle stage of development (i.e., when oocytes are <20 μm) in Inha−/− ovaries.
Intraovarian Activin/Inhibin Beta B Subunit Expression Is Increased in Inha−/− Ovaries at PND 3 and Reduced by Loss of Gdf9
Activin is an integral regulator of granulosa cell proliferation. Subunits encoding both activin isoforms (Inhba and Inhbb) are upregulated in Inha−/− ovaries at PND 12 [8]. To investigate at what time point activin subunit expression is aberrantly expressed, Inhba and Inhbb were measured at PND 3, 6, and 12 in Inha−/− ovaries with and without Gdf9 (Fig. 3). These days were chosen to represent the different developing populations of follicles in the initial follicular wave (i.e., PND 3 corresponds to the initial appearance of primary follicles, while PND 6 corresponds to initial appearance of secondary follicles). Inhba was barely detectable in all the genotypes at PND 3 (Fig. 3A). However, by PND 6, Inha−/− and Inha−/− Gdf9−/− ovaries had a marked and similar expression of Inhba (Fig. 3C). Expression levels of Inhba subunit continued to rise at PND 12 in all the genotypes except Gdf9−/−, where it remained low though not statistically different from the control (P = 0.057) (Fig. 3E). At PND 12, Inhba expression was statistically higher in Inha−/− Gdf9−/− ovaries than in Inha−/− ovaries, and both were greater than control (Fig. 3E).
FIG. 3.
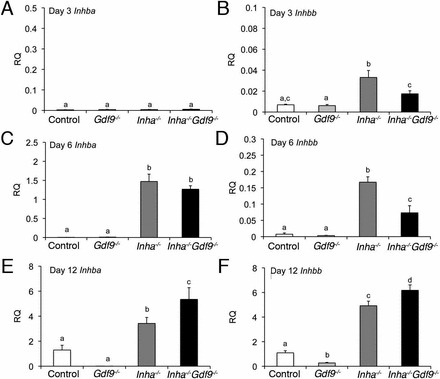
Inhbb levels are reduced in Inha−/− Gdf9−/− mutant ovaries at PND 3 and 6 compared to the pathologically higher levels in Inha−/− single mutants. Relative quantities (RQ) of mRNA for Inhba (A, C, E) or Inhbb (B, D, F) at PND 3, 6, and 12. A) Inhba is not detectable at PND 3, but increases significantly in Inha−/− and Inha−/− Gdf9−/− ovaries at PND 6 (C) and PND 12 (E). B) In contrast, Inhbb is significantly increased in Inha−/− ovaries compared to all other genotypes at PND 3 and 6. Inha−/− Gdf9−/− have lower levels of Inhbb at PND 3 (B) and PND 6 (F). Quantitative PCR results are expressed as RQ to PND 12 control ovaries of the respective gene. Values are mean ± SEM (n = 5–6 each genotype). Statistical significance of each time point as determined by ANOVA and Fisher LSD post hoc analysis are shown as letters above the bars. Different letters indicate statistically different means (i.e., “a” has a significantly different mean from “c” but not “a, c”).
In contrast to Inhba, expression of the activin/inhibin βB subunit (Inhbb) was significantly increased at PND 3 in Inha−/− mice (Fig. 3B). Removal of Gdf9 from Inha−/− significantly lowered Inhbb levels in Inha−/− Gdf9−/− compared to Inha−/− (Fig. 3B). By PND 6, levels of Inhbb were increased in Inha−/− Gdf9−/− as compared to the controls, but were still reduced compared to Inha−/− (Fig. 3D). In contrast, by PND 12, both Inha−/− and Inha−/− Gdf9−/− had higher levels of Inhbb than control or Gdf9−/− ovaries (Fig. 3F). These data suggest that precocious expression of Inhbb in Inha−/− as seen at PND 3 and PND 6 depend on GDF9, and that Inhbb levels may be a key contributor to proliferation of granulosa cells seen in the earliest stages of follicle growth in Inha−/− ovaries (Fig. 2E)
We additionally measured expression of Kitl in the four genotypes at PND 3, 6, and 12 because both activin and GDF9 have been shown to suppress Kitl expression [9–11, 16]. The isoforms of Kitl show little difference at PND3 (Fig. 4, A and B). However, both isoforms are significantly suppressed by PND 12 (Fig. 4, E and F). However, the levels of Kitl1 and Kitl2 are similar at all ages in Inha−/− and Inha−/− Gdf9−/− ovaries. Therefore, suppression of Kitl most closely reflects the overexpression pattern of Inhba (Fig. 3, A, C, and E).
FIG. 4.
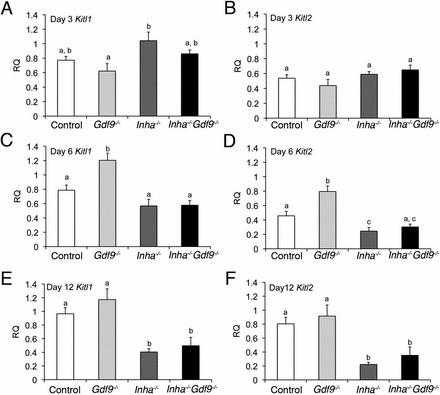
Kitl isoform expression similarly suppressed in Inha−/− and Inha−/− Gdf9−/− ovaries at PND 6 and 12. Relative quantities (RQ) of mRNA for the Kitl isoforms, Kitl1 (A, C, E) or Kitl2 (B, D, F) at PND 3, 6, and 12. Expression levels for either isoform do not significantly change between genotypes until PND 6 (C, D), where they increase in Gdf9−/− ovaries (E, F). PND 12 Kitl1 (E) and Kitl2 (F) are suppressed in Inha−/− and Inha−/− Gdf9−/− ovaries. Quantitative PCR results are expressed as relative quantity to PND 12 control ovaries of the respective gene. Values are mean ± SEM (n = 5–6 each genotype). Statistical significance of each time point as determined by ANOVA and Fisher LSD post hoc analysis are shown as letters above the bars. Different letters indicate statistically different means (i.e., “a” has a significantly different mean from “b” but not “a, b”).
The Loss of Gdf9 Enhances the Onset of Tumor Foci in Inha−/− Ovaries
Ovarian tumors are microscopically evident by 4 wk of age in Inha−/− mice as foci of nodular proliferation [4], but never observed in PND 12 Inha−/− ovaries (Fig. 5A) [8]. In contrast, we readily identified areas of microscopic foci of early forming tumors at PND 12 in Inha−/− Gdf9−/− ovaries (Fig. 5, C and D) that resembled the tumor foci found in 4-wk-old Inha−/− ovaries (Fig. 5B) [4]. Tumors are also visible in Inha−/− Gdf9−/− ovaries at 3 wk of age [12]. These data indicate that the loss of Gdf9 from Inha−/− ovaries enhanced the onset of the tumor development, which is typically observed approximately 2 wk later in Inha−/− ovaries.
FIG. 5.
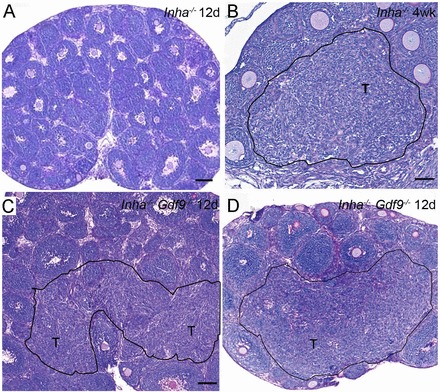
Loss of Gdf9 enhances the onset of microscopic tumor foci in Inha−/− ovaries. A) PND 12 Inha−/− ovaries do not display evidence of tumor formation until 4 wk of age. B) Black outlined region denoted as T shows tumor formation in 4-wk-old Inha−/− ovaries. C, D) Twelve-day-old Inha−/− Gdf9−/− double mutants shows regions of hyperplasia of various sizes that resemble the pretumor foci (T) observed in older Inha−/− ovaries (B). Bars = 100 μm (A, C, D) and 50 μm (B).
DISCUSSION
Activins, inhibins, and GDF9 are key players in mammalian folliculogenesis. Alone, or in conjunction with other signaling pathways, these growth factors play important roles in maintaining oocyte and granulosa cell integrity. In the present study, we describe novel data that highlight the interrelationship between activin, inhibin, and GDF9 during early folliculogenesis and focus on how misregulation of these factors during postnatal life leads to adult ovarian dysfunction, including cancer development. In the current study, our aim was to extend our previous investigations of the aberrant folliculogenesis in the prepubertal Inha−/− mouse and to dissect the distinctive roles of activin and GDF9. Using single and double knockouts for Inha and Gdf9, we were able to further identify the mechanisms into how misregulation of these growth factors affects pre- and postpubertal folliculogenesis, eventually resulting in granulosa cell tumor development.
The interrelationship between activin, inhibin, and GDF9 is highlighted by the contributions of each of these growth factors in their respective knockout mouse model. A summary of these changes is shown in Table 1. A critical role for GDF9 during early follicular development is supported by the striking appearance of Gdf9−/− ovaries, which in adult mice exhibit large oocytes that arrest during the one-layer primary stage (3b stage) of development [12, 13, 17]. Arrested primary follicles in Gdf9−/− ovaries have elevated inhibin α [12, 13], which inhibits granulosa cell development via loss of activin production and/or signaling. Removing Inha from Gdf9−/− allows follicles to progress further, presumably by increasing levels of activin [12].
TABLE 1.
Summary of phenotype and gene expression changes in 12-day-old Gdf9−/−, Inha−/−, and Inha−/− Gdf9−/− mutant ovaries.

GC, granulosa cell.
Plus sign (+) denotes expression in the wild type to which all others are compared, the number of arrows indicates relative increase or decrease in expression in the mutant mice, equal sign (=) denotes similar expression to the wild type, and 0 denotes no expression.
Measured at PND 3.
In the present study, we discovered a stage-specific role for Gdf9 in the Inha−/− ovary prior to the development of stage 3b follicles. Primordial and primary follicles are characterized by a small oocyte (less than 30 μm in diameter) surrounded by unilaminar pregranulosa or granulosa cells, respectively. In Inha−/− ovaries, we observed small oocytes encapsulated by multiple layers of granulosa cells, confirming a dysregulation between germ and somatic cell components [8]. Genetic ablation of Gdf9 from Inha−/− ovaries restores the follicle diameter back to appropriately sized oocytes in unilaminar follicles when oocytes are less than 20 μm. This suggests that it is GDF9 that is ultimately responsible for the initial desynchronization of oocyte and granulosa cell growth in Inha−/− ovaries. The initial alteration in follicle growth also appears to hinge on premature Inhbb expression in Inha−/− at the time of primary follicle formation (i.e., PND 3). Thus, in Inha−/− ovaries, GDF9 causes an upregulation (either directly or indirectly) of Inhbb and, subsequently, an improper initiation of granulosa cell growth. A direct regulation is supported by the demonstration that recombinant mGDF9 upregulates Inhbb in both control and Inha−/− granulosa cells (Supplemental Figure S3). Other studies have shown that recombinant GDF9 upregulates INHBB mRNA in cultures of human granulosa-luteal cells [18]. In addition, at 12 days of age, Gdf9−/− mice have reduced levels of Inhbb, as do adult Gdf9−/− ovaries [17].
We have previously reported that PND 12 Inha−/− ovaries contain large multilayered follicles greater than 90 μm in diameter, reminiscent of follicles observed in 3-wk-old wild-type mice [8]. In this study, we found that this phenotype was observed irrespective of the presence or absence of Gdf9. Because of the significant increases in Inhba and Inhbb after PND 6, as well as the morphologic similarity between Inha−/− and Inha−/− Gdf9−/−, we propose that it is primarily excessive activin production that is responsible for the more advanced follicular structures. In addition, our previous model hypothesized that in the absence of inhibin, activin and/or GDF9 suppressed Kitl, resulting in the loss of a key trophic signal required for oocyte growth [8]. Therefore, we examined the expression of Kitl in the single and double knockout mice to identify differences between GDF9 and activin regulation of Kitl. At no time did we detect an expression difference between Inha−/− and Inha−/− Gdf9−/−. By PND 6, at the time of a dramatic increase in Inhba and Inhbb in both Inha−/− and Inha−/− Gdf9−/− mutant ovaries, the Kitl2 isoform, but not the Kitl1 isoform, is suppressed in Inha−/− ovaries. By PND 12, both Kitl isoforms are downregulated in Inha−/− and Inha−/− Gdf9−/− ovaries, and at this time, levels of Inhba and Inhbb are much higher than at PND 6, particularly Inhbb. The latter data support studies that show that activin may preferentially suppress the Kitl2 isoform and therefore juxtacrine signaling (Kitl2 encodes the membrane-bound form of KITL [19]). However, based on our data, we further suggest that activin's effect on Kitl might be dose-dependent, with higher levels of activin able to suppress both forms of Kitl.
In addition to regulating the growth dynamics between germ and somatic cells, intraovarian factors also play significant roles in maintaining the differentiation state of granulosa cells. Granulosa cells have a regulated program of differentiation that involves marked cellular and molecular changes [20, 21]. In the Inha−/− ovary, either with or without GDF9, granulosa cells show excessive proliferation and eventually form granulosa cell tumors [3, 4, 12], while deletion of Gdf9 by itself does not. To our surprise, deletion of Gdf9 from Inha−/− mice enhanced the onset of granulosa cell tumor foci comparable to those observed in ovaries from 4–6 wk-old Inha−/− mice, a time correlating with sexual maturity and surge of pituitary gonadotropins [4, 6]. Pituitary gonadotrophins are known modifiers of the tumor phenotype of the Inha−/−ovary, and remarkably only ablation of both gonadotropins is able to prevent full tumor formation, though not the onset of tumor lesions [5–7]. We therefore hypothesize that oocyte-derived GDF9 is a key paracrine factor for maintaining Inha−/− granulosa cells in a nontumor phenotype, and we suggest this protects granulosa cells from other paracrine interactions that cause atypical differentiation leading to tumor formation. In a wild-type ovary, stimulation of the gonadotropins results in ovulation and formation of corpora lutea. However, Inha−/− granulosa cells respond to gonadotropin stimulation with an altered gene expression pattern [22]. We hypothesize that in ovaries of sexually mature Inha−/− mice, stimulation by the gonadotropins increases both granulosa cell proliferation and net activin production, which contribute to excessive granulosa cell proliferation and ultimately oocyte loss. Loss of oocytes in the Inha−/− ovary would thus phenocopy deletion of Gdf9 and allow tumor foci to form. It is interesting to note that although the onset of the tumor lesions is much earlier in Inha−/− Gdf9−/− mice (PND 12 vs. 4 wk of age), they eventually die of cancer cachexia-induced death at the same postnatal age as Inha−/− mice [12]. This suggests that regardless of the initial protective effects of GDF9, the progression of the disease in Inha−/− mice is primarily driven by gonadotropins and activin.
In summary, our findings demonstrate that Inha−/− Gdf9−/− ovaries also exhibit a precocious follicle growth phenotype evident within the first 2 wk after birth, similar to Inha−/− ovaries. However, deletion of Gdf9 from Inha−/− ovaries reduces a subset of multilayer follicles containing abnormally small oocytes and restores aspects of the normal coupling of oocyte and granulosa cell growth at the earliest stages of folliculogenesis. In part, this is likely due to a reduction in pathologically higher Inhbb expression levels that is found in Inha−/− ovaries. Surprisingly, removal of Gdf9 from Inha−/− also advanced the onset of the tumor foci within the ovary to 12 days of age. Together, our data reveal a sequential role for GDF9 and activin during early folliculogenesis and a novel role for GDF9 in preventing tumorgenesis in the Inha−/− ovary. These are previously uncharacterized roles for GDF9 in the ovary and highlight the interrelationship of members of the TGFB family during early follicle development to maintain granulosa cells in the appropriate state of differentiation.
Supplementary Material
ACKNOWLEDGMENT
We would like to thank Dr. Martin Matzuk (Baylor College of Medicine) for the Inha and Gdf9 lines, recombinant mGDF9, and the αSMA antibody. We thank The University of Virginia Ligand Center for Research in Reproduction, Ligand Assay and Analysis Core (Eunice Kennedy Shriver NICHD/NIH, SCCPIR, Grant U54-HD28934) for the FSH measurements and the Baylor College of Medicine Pathology Core (P30-CA125123). We additionally thank Alysse Ligon, Robert Cook, and Brooke Middlebrook (BCM) for technical assistance.
Footnotes
This work was supported by a Burroughs Wellcome Career Award in the Biomedical Sciences and a National Institute of Health R01 CA138628 (to S.A.P.).
These authors contributed equally to this manuscript.
REFERENCES
- Myers M, Pangas SA. Regulatory roles of transforming growth factor beta family members in folliculogenesis. Wiley Interdiscip Rev Syst Biol Med 2010; 2: 117 125. [DOI] [PubMed] [Google Scholar]
- Edson MA, Nagaraja AK, Matzuk MM. The mammalian ovary from genesis to revelation. Endocr Rev 2009; 30: 624 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzuk MM, Finegold MJ, Mather JP, Krummen L, Lu H, Bradley A. Development of cancer cachexia-like syndrome and adrenal tumors in inhibin-deficient mice. Proc Natl Acad Sci U S A 1994; 91: 8817 8821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzuk MM, Finegold MJ, Su JG, Hsueh AJ, Bradley A. Alpha-inhibin is a tumour-suppressor gene with gonadal specificity in mice. Nature 1992; 360: 313 319. [DOI] [PubMed] [Google Scholar]
- Kumar TR, Palapattu G, Wang P, Woodruff TK, Boime I, Byrne MC, Matzuk MM. Transgenic models to study gonadotropin function: the role of follicle-stimulating hormone in gonadal growth and tumorigenesis. Mol Endocrinol 1999; 13: 851 865. [DOI] [PubMed] [Google Scholar]
- Kumar TR, Wang Y, Matzuk MM. Gonadotropins are essential modifier factors for gonadal tumor development in inhibin-deficient mice. Endocrinology 1996; 137: 4210 4216. [DOI] [PubMed] [Google Scholar]
- Nagaraja AK, Agno JE, Kumar TR, Matzuk MM. Luteinizing hormone promotes gonadal tumorigenesis in inhibin-deficient mice. Mol Cell Endocrinol 2008; 294: 19 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers M, Middlebrook BS, Matzuk MM, Pangas SA. Loss of inhibin alpha uncouples oocyte-granulosa cell dynamics and disrupts postnatal folliculogenesis. Dev Biol 2009; 334: 458 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutts SM, Childs AJ, Fulton N, Collins C, Bayne RA, McNeilly AS, Anderson RA. Activin signals via SMAD2/3 between germ and somatic cells in the human fetal ovary and regulates kit ligand expression. Dev Biol 2008; 314: 189 199. [DOI] [PubMed] [Google Scholar]
- Pangas SA, Jorgez CJ, Tran M, Agno J, Li X, Brown CW, Kumar TR, Matzuk MM. Intraovarian activins are required for female fertility. Mol Endocrinol 2007; 21: 2458 2471. [DOI] [PubMed] [Google Scholar]
- Joyce IM, Clark AT, Pendola FL, Eppig JJ. Comparison of recombinant growth differentiation factor-9 and oocyte regulation of KIT ligand messenger ribonucleic acid expression in mouse ovarian follicles. Biol Reprod 2000; 63: 1669 1675. [DOI] [PubMed] [Google Scholar]
- Wu X, Chen L, Brown CA, Yan C, Matzuk MM. Interrelationship of growth differentiation factor 9 and inhibin in early folliculogenesis and ovarian tumorigenesis in mice. Mol Endocrinol 2004; 18: 1509 1519. [DOI] [PubMed] [Google Scholar]
- Dong J, Albertini DF, Nishimori K, Kumar TR, Lu N, Matzuk MM. Growth differentiation factor-9 is required during early ovarian folliculogenesis. Nature 1996; 383: 531 535. [DOI] [PubMed] [Google Scholar]
- Thomas FH, Ismail RS, Jiang JY, Vanderhyden BC. Kit ligand 2 promotes murine oocyte growth in vitro. Biol Reprod 2008; 78: 167 175. [DOI] [PubMed] [Google Scholar]
- Carabatsos MJ, Elvin J, Matzuk MM, Albertini DF. Characterization of oocyte and follicle development in growth differentiation factor-9-deficient mice. Dev Biol 1998; 204: 373 384. [DOI] [PubMed] [Google Scholar]
- Elvin JA, Clark AT, Wang P, Wolfman NM, Matzuk MM. Paracrine actions of growth differentiation factor-9 in the mammalian ovary. Mol Endocrinol 1999; 13: 1035 1048. [DOI] [PubMed] [Google Scholar]
- Elvin JA, Yan C, Wang P, Nishimori K, Matzuk MM. Molecular characterization of the follicle defects in the growth differentiation factor 9-deficient ovary. Mol Endocrinol 1999; 13: 1018 1034. [DOI] [PubMed] [Google Scholar]
- Kaivo-Oja N, Bondestam J, Kamarainen M, Koskimies J, Vitt U, Cranfield M, Vuojolainen K, Kallio JP, Olkkonen VM, Hayashi M, Moustakas A, Groome NP, et al. Growth differentiation factor-9 induces Smad2 activation and inhibin B production in cultured human granulosa-luteal cells. J Clin Endocrinol Metab 2003; 88: 755 762. [DOI] [PubMed] [Google Scholar]
- Childs AJ, Anderson RA. Activin A selectively represses expression of the membrane-bound isoform of Kit ligand in human fetal ovary. Fertil Steril 2009; 92: 1416 1419. [DOI] [PubMed] [Google Scholar]
- Myers M, van den Driesche S, McNeilly A, Duncan W. Activin A reduces luteinisation of human luteinised granulosa cells and has opposing effects to human chorionic gonadotropin (hCG) in vitro. J Endocrinol 2008; 199: 201 212. [DOI] [PubMed] [Google Scholar]
- Stocco C, Telleria C, Gibori G. The molecular control of corpus luteum formation, function, and regression. Endocr Rev 2007; 28: 117 149. [DOI] [PubMed] [Google Scholar]
- Nagaraja AK, Middlebrook BS, Rajanahally S, Myers M, Li Q, Matzuk MM, Pangas SA. Defective gonadotropin-dependent ovarian folliculogenesis and granulosa cell gene expression in inhibin-deficient mice. Endocrinology 2010; 151: 4994 5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay VL, Midgley AR, Jr,, Niswender GD. Patterns of gonadotrophin secretion associated with ovulation. Fed Proc 1970; 29: 1880 1887. [PubMed] [Google Scholar]
- Pangas SA, Jorgez CJ, Matzuk MM. Growth differentiation factor 9 regulates expression of the bone morphogenetic protein antagonist gremlin. J Biol Chem 2004; 279: 32281 32286. [DOI] [PubMed] [Google Scholar]
- Li Q, Agno JE, Edson MA, Nagaraja AK, Nagashima T, Matzuk MM. Transforming growth factor beta receptor type 1 is essential for female reproductive tract integrity and function. PLoS Genet 2011; 7: e1002320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.