

Abstract
Nucleotide excision repair (NER) excises bulky DNA lesions induced by mutagens and carcinogens. The repair process includes recognition of DNA damage, excision of a short patch of nucleotides containing the damaged base, re-synthesis of a new DNA strand and ligation of the nicks to restore the sequence integrity. Mutation or aberrant transcription of NER genes reduces repair efficiency and results in the accumulation of mutations that is associated with the development of cancer. Here we present a rapid, sensitive and quantitative assay to measure NER activity in human cells, which we term the Oligonucleotide Retrieval Assay (ORA). We used oligonucleotide constructs containing the UV-damaged adduct, cyclobutane pyrimidine dimer (CPD), to transfect human cells, and retrieved the oligonucleotides for quantification of the repaired, CPD-free DNA by real-time quantitative PCR. We demonstrate that ORA can quantify the extent of NER in diverse cell types, including immortalized, primary and stem-like cells.
Cells employ nucleotide excision repair (NER) to remove bulky DNA adducts and restore the canonic nucleotide sequence1,2. This repair process comprises sequential steps including damage recognition, strand incision/excision, repair synthesis and ligation. The NER pathway can be divided into two processes, one maintaining the integrity of the whole genome via global genome repair (GGR) and the other sustaining the function of active gene expression via transcription-coupled repair (TCR)3,4,5. The proteins that are involved in the core reaction, i.e. excision, synthesis and ligation, are the same for both processes, and include: XPA, XPB and XPD for unwinding and stabilization of a 30 nucleotide (nt) bubble encompassing the adduct; ERCC1/XPF and XPG for strand-incision on both ends of the bubble; RFC/PCNA and polymerase δ/ε for synthesis of a new DNA strand; and XRCC1/ligase III for ligation. The key difference between GGR and TCR is damage recognition. In the GGR pathway, UV-DDB and the XPC/RAD23/CETN2 complex recognize and bind to the DNA adduct or the helical distortion. In TCR, however, a stalled RNA polymerase II recruits CSB ATPase and the CSA complex including DDB1, Cullin 4A, ROC1 and other proteins, for recognition and binding of the DNA adduct on the transcribed strand. Although many NER proteins have been identified and functionally characterized, new proteins that participate in these processes are continually being discovered6,7. The increasing complexity of the NER pathway consequently makes it difficult to ascertain the exact causal factor of NER deficiency that leads to mutation accumulation and cancer8,9,10.
The causal association of mutations in NER genes with inherited human diseases was first documented in xeroderma pigmentosum (XP), an autosomal recessive genetic disorder in which repair of DNA damage caused by UV light is compromised11. Patients with XP are sensitive to light and often develop skin cancers. The complementation groups of XP, termed alphabetically from XP-A to –G, form the basic components of the NER pathway. The effects of polymorphic variants and altered levels of gene expression of the NER protein components have been implicated in the pathogenesis of breast cancer and other cancers of gynecological origin including ovarian and cervical cancers, and deregulated NER is thought to result in the accumulation of mutations12,13,14. Epidemiological mapping of single nucleotide polymorphisms (SNPs) has also identified candidate protein variants of NER that are associated with different types of cancer15,16. Aberrant gene expression of NER proteins, mostly measured at the mRNA level or by immunoblotting, is also proposed to be a causal factor in several types of cancer17,18. Importantly, however, the relative repair efficiencies of individuals in these reports are unknown because of the lack of a simple and efficient assay to quantify NER activity in human cells.
We have developed a versatile method, using oligonucleotide fragments to construct DNA substrates that can be easily transfected into and retrieved from human cells, to rapidly evaluate repair efficiency and other DNA transaction activities. We term this method Oligonucleotide Retrieval Assay (ORA). In this study, we have used oligonucleotides containing a cyclobutane pyrimidine dimer (CPD) to create an oligonucleotide construct that serves as a substrate for NER. This construct can be transfected into cells with high efficiency. We demonstrated that depending on cell type, up to 10,000 molecules of oligonucleotide could be introduced into and retrieved from a single cell. As an assay of NER efficiency, ORA employs real-time quantitative PCR (qPCR) for the rapid and quantitative assessment of the proportion of oligonucleotides repaired by NER processes. We show that ORA can be applied to various types of human cells, including immortalized cell lines of assorted origins, primary fibroblasts and a primary human breast epithelial cell model that includes adult stem-like cells and non-stem cells. This method will be of value in the screening and diagnosis of compromised repair efficiencies due to mutations or polymorphic variants of NER proteins in the human population. Its versatility should facilitate epidemiological studies to establish associations between deficits in DNA repair and disease. Owing to the flexibility and simplicity of this methodology, it also should be feasible to automate this assay for high throughput analyses.
Results
Outline of the Oligonucleotide Retrieval Assay (ORA)
We first assembled three synthetic oligonucleotides to form a 5′-biotinylated duplex DNA that contains a CPD adduct (T-T dimer) to serve as a substrate for nucleotide excision repair. After transient transfection and incubation, cells were harvested for DNA extraction and the oligonucleotides retrieved using streptavidin-biotin binding from either whole cell extracts or isolated nuclei. The repair efficiency was determined by employing real-time quantitative PCR (qPCR) to quantify the percentage of DNA lesions that were repaired in the cells (Fig. 1).
Figure 1. Overview of the procedure for the Oligonucleotide Retrieval Assay (ORA).
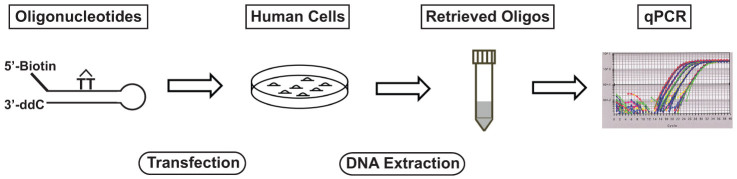
Synthetic 5′-biotinylated oligonucleotide containing a CPD adduct (TT dimer) was used to transfect human cells. After incubation, the oligonucleotides were retrieved from cells by streptavidin capture. Real-time quantitative PCR was employed for detection of the repaired oligonucleotides.
Measuring NER activity by qPCR quantification of the repaired CPD-containing oligonucleotides
To measure the activity of NER in vivo, we constructed an oligonucleotide substrate harboring a site-specific cyclobutane pyrimidine dimer (CPD), a gold standard lesion in the study of UV-induced DNA damage, in a hairpin duplex structure (Fig. 2A). The double-stranded region is 77-nt long and the CPD is located in the middle of the DNA duplex, providing sufficient length for excision by NER. A 4-nt hairpin structure is utilized to enhance ligation efficiency and to prevent exonucleolytic degradation from the 3′-end. At the 5′-end of the construct, a terminal biotin residue is positioned for retrieval of the oligonucleotide by streptavidin bead binding; a single-stranded DNA region with a unique sequence specifying the 5′-forward primer is used for qPCR amplification. The 3′-end of the construct was capped with a dideoxycytidine residue to prevent 3′ degradation by non-specific exonuclease activities present in cells.
Figure 2. Design of ORA oligonucleotide construct for NER.

(A) Construction of the ORA oligonucleotide that contains a CPD lesion. The 30-nt synthetic ssDNA fragment containing a CPD lesion (marked as a T-T dimer) was hybridized with a 5′-biotinylated (49 nt) and a 3′-ddC protected (98 nt) fragment to form a hairpin dsDNA construct after ligation. The resulting construct contains a central 77-bp dsDNA region with a 19-nt ssDNA in the 5′-terminal region and a 4-nt hairpin at the end of the 3′ arm. The CPD residue locates approximately at the center of the dsDNA stretch, 33 bp from the 3′ end and 42 bp from the 5′ end of the dsDNA region. Including the 19-nt ssDNA sequence, the 5′ arm of the CPD-containing construct is 61 nt/bp in length. Primers for qPCR quantification are shown in arrow lines, where amplification by primers a/b represents the total amount of the oligonucleotide and by a/c the amount of the repaired oligonucleotides. The percentages of oligonucleotide repaired were calculated by the following equation: % Repaired = (2−ΔCt) × 100, ΔCt = Ct Control (a/b) − Ct Test (a/c). (B–E) Calibration of CPD-containing oligonucleotide constructs. qPCR was carried out using 1 μl of 5 nM ligated normal TT- (B) or CPD-containing construct (C) as template. PCR amplifications with primers a and b serve as the internal control for the total number of substrate molecules, whereas amplifications with primers a and c quantify the amount of the repaired products. Based on the ΔCt value (ac-ab) of the TT construct, the ligation efficiency (% ligation = 1/2ΔCt × 100) was 1.5% (B). Since CPD adducts block the activity of Taq polymerase (C), the ΔCt value (ac-ab) of the CPD construct is greater than that of the normal TT control (D), yielding a ΔΔCt value (CPD-TT) of 10.71 that converts to a 1,670-fold blockage by CPD (Fold Difference = 2ΔΔCt) (E). Using this 2ΔΔCt method, we were able to obtain differential repair efficiencies of the CPD construct in cells with altered genetic backgrounds. Data are presented as mean ± S.D. from three independent experiments.
To quantify the repaired oligonucleotides, we confirmed the finding that a CPD adduct blocks Taq polymerase activity in qPCR amplification19 (Fig. 2C), allowing only the repaired DNA that lacks the lesion to be amplified. We used primers flanking DNA sequence 5′ of CPD (primers a and b, Fig. 2A) to measure the total amount of construct and employed primers spanning the CPD site (primers a and c, Fig. 2A) to quantify repaired DNA. The ratio of repaired DNA to total DNA thus represents the extent to which the CPD lesion was removed and the canonical DNA sequence restored.
Since we produce the hairpin construct by ligation of three oligonucleotides, the quantity of the complete product is dependent on the efficiency of ligation in the preparation of the construct. As CPD adducts block the activity of Taq polymerase during amplification, we are not able to directly determine ligation efficiency. We therefore assembled a reference construct identical to the CPD-containing construct, except that the oligonucleotide with the CPD adduct is replaced with an oligonucleotide with TT bases at the same positions. This reference construct allowed us to measure: (1) the quantity of the input oligonucleotides by using qPCR with primers a and b (Fig. 2A and B); and (2) the quantity of the completely ligated product, using PCR amplification with primers a and c (Fig. 2A and B). The ΔCt value derived from subtraction of the Ct value of primers a/c from that of primers a/b, as shown in Fig. 2B (ΔCt = 6.05 ± 0.24), represents the efficiency of complete ligation of the three oligonucleotides. In this experiment, the ligation efficiency is ~1.5%, as calculated by equation: % ligation = 2−ΔCt × 100. We obtained an average ligation efficiency of ~4% in all preparations, confirmed by both qPCR and gel electrophoresis. The ligation efficiency of this construct is expected to be similar to that of the oligonucleotide containing CPD, as both reactions are carried out side-by-side under the same conditions.
The blockage by CPD is evident in Fig. 2C, where the Ct value of a/c is much greater than that for the TT construct, resulting in ΔCt = 16.75 ± 0.37. The ΔCt value for CPD thus represents the background of detection, and that for the TT reference construct is illustrative of conditions in which 100% of the CPD adducts are restored to the normal TT nucleotides (Fig. 2D). The difference of the ΔCts between TT and CPD (ΔΔCt) therefore reveals the detection limits of this assay, between the baseline and the state of fully repaired, a fold difference of 1670, as computed by 2ΔΔCt (Fig. 2E). The detection baseline, which likely represents the subtle activity of Taq polymerase to bypass CPD19, is calculated as 0.06%, by the equation: % repaired = 2−ΔΔCt × 100 (Fig. 2E). Therefore, the quantitative range of this assay for measuring the repair of CPD lesion is between 0.06% and 100%.
In ORA, the amplification with primers a/b serves as a control for the total number of the retrieved oligonucleotides, whereas amplification with primers a/c represents the number of the repaired DNA. Thus, the repair efficiency, defined as % repaired, can be calculated from the ratio of the number of the repaired DNA over the number of total oligonucleotides retrieved. We have used the calculated ligation efficiency to adjust the total number of the retrieved oligonucleotides and normalized the number of % repaired by the detection base line (see detail in Methods).
Quantification of NER activities in cell lysates using CPD-containing oligonucleotides
Cell-free extracts have been used for measurement of NER activities in vitro20. Here we isolated cell lysates from 293T cells to validate our CPD construct in assaying NER activities. Since XPA is a key component in the NER machinery, for validation of the assay's specificity we used a monoclonal antibody against XPA in the reaction to suppress the NER activities. We incubated 0.075 nM of oligonucleotides with 80 μg of cell lysates in 30 μl of the reaction solution at 30°C for 2 h. After incubation, the oligonucleotide constructs were isolated by streptavidin-bead binding and eluted in deionized water. A fraction of the reaction solution, which contains 150 million of the input oligonucleotides, was used for qPCR quantification. As shown in Fig. 3, the background signals amplified by qPCR for the construct alone without incubation with lysates were 140 molecules. By incubation with 293T cell lysates, the number of the repaired oligonucleotides increased to 12,000. Addition of anti-XPA antibodies decreased the number to 5,000, an approximately two-fold reduction of the NER activities. This result demonstrated that the oligonucleotide assay specifically and quantitatively measures the NER activity.
Figure 3. Validation of oligonucleotide construct for measuring NER activity by using cell-free lysates.
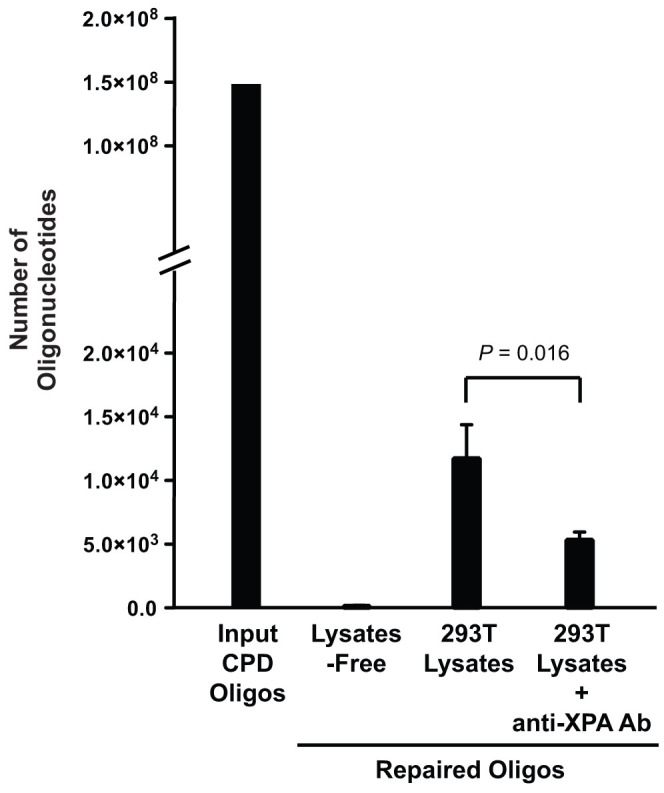
CPD-containing oligonucleotides (150 million molecules) were incubated in the solutions of three reaction conditions: alone, with 293T cell lysates or with 293T lysates including anti-XPA antibodies at 30°C for 2 h. Quantification of the PCR-amplifiable, repaired oligonucleotides shows the background number of the detected oligonucleotides is 135. Incubation with cell lysates increased the number to 12,000 and addition of anti-XPA antibodies decreased the number to 5,000, suggesting that the assay is specific to the NER activity. Data were presented as mean ± S.D. from three independent experiments and P-value was calculated by t-test.
Measuring NER efficiency by Oligonucleotide Retrieval Assay (ORA) in immortalized cell culture
We transfected 5 × 105 HEK293T cells with 0.02 nM of the CPD-containing oligonucleotide construct. After incubation, the cells were harvested at increasing time intervals for up to 24 h. Whole-cell DNA was extracted and the transfected oligonucleotides were recovered by streptavidin bead capture. The repaired oligonucleotides, unlike the input CPD-containing constructs, are amplifiable and detected by qPCR. Relative to the amplification of a control sequence on chromosome 17p13, we calculated by qPCR that we retrieved approximately 100–300 oligonucleotides per cell at each time interval (Fig. 4B); the quantity of the repaired oligonucleotides steadily increased in a time-dependent manner (Fig. 4B). NER activity was detected as early as 2 h after transfection. At 16 h, about 20% of the retrieved CPD constructs per cell were restored to their canonical, qPCR-amplifiable sequence; at 24 h, 40% were restored (Fig. 4A).
Figure 4. NER activities of human embryonic kidney cell 293T and human colon cancer cell SW480 calibrated by ORA.

(A) NER activities in a single HEK293T cell are presented as % CPD-containing oligonucleotide repaired at incremental time points. Data are presented as mean ± S.D. from three independent experiments. (B) Repair of CPD-containing oligonucleotides in HEK 293T cells. HEK293T cells (5 × 105) were transfected with 0.02 nM of CPD oligonucleotide and incubated at 37°C. Cells were harvested at increasing incubation times, followed by oligonucleotide retrieval and qPCR analysis. Data are shown as the number of oligonucleotides retrieved per cell and the number of oligonucleotides repaired per cell at different time points from three independent experiments (mean ± S.D.). (C) Comparison of NER activities measured by ORA and ELISA between 293T and SW480 cells. The CPD repair efficiencies measured by ORA or the CPD removal efficiencies measured by ELISA are obtained 24 h after oligonucleotide transfection or 24 h after UV-C irradiation, respectively, and are presented as % repaired determined from three independent experiments (mean ± S.D.). (D) The survival of 293T and SW480 cells after UV-C irradiation. Each data point is presented as mean ± S.D. from three independent experiments.
Calibrating NER efficiency in a colon cancer cell line
To verify the feasibility of ORA in different types of cells, we next measured the NER efficiency of the colon cancer cell line SW480. We transfected 2 × 105 cells with 0.02 nM of the CPD-containing oligonucleotides using calcium phosphate; incubated the transfected cells at 37°C for 24 h and 48 h. The oligonucleotides were retrieved from whole cell extracts after incubation and quantified by qPCR for repair efficiency. In contrast to the embryonic kidney cell 293T in which 44% of the retrieved oligonucleotides were repaired within a 24 h incubation, the colon cancer cell SW480 only repaired 0.4% of the retrieved oligonucleotides from whole cell extracts (Table 1). The repair efficiency stayed at 0.5% after incubation for 48 h. The oligonucleotides retrieved from nuclei were repaired more effectively, while after 24 h or 48 h incubation 1% was restored to the canonical sequence. The transfection/retrieval efficiencies of the embryonic kidney- and colon tumor-derived cells are: ~80 copies per cell for 293T and ~400 copies per cell for SW480 when retrieved from whole cell extracts after 24 h incubation; and ~20 molecules per cell for 293T and ~70 molecules per cell for SW480 when retrieved from nuclei after 48 h incubation, suggesting that both epithelial cells have similar DNA internalization capability and/or that the oligonucleotide stability inside both cells is similar. The disparity in NER efficiencies is thus cell-type specific (Table 1). We further confirmed this repair discrepancy by ELISA that monitors CPD levels in UV irradiated cells. As shown in Fig. 4C, the 293T cells removed 75% of the UV-induced CPD lesions whereas the SW480 cells repaired only 33% of the lesions, 24 h after UV irradiation. In comparison with ORA, using 293T 44% of the CPD lesions were repaired; only 0.5% were repaired using SW480. ELISA scores more CPD removals since the assay measures both TCR and GGR activities, while ORA only measures the GGR activities on the non-transcribing oligonucleotides. The NER disparity of the two cell lines is also shown in UV-irradiated cell survival. Using colony-forming assay we found that the proliferation of SW480 cells is more sensitive to UV damages than the 293T cells (Fig. 4D).
Table 1. NER efficiency measured by ORA in live cells.
| Cells | Number of Cells for Transfection | Incubation Time | Whole Cells or Nuclei Oligo Retrieval | Oligos Retrieved (per Cell) | NER Efficiency (% Repaired) |
|---|---|---|---|---|---|
| 293T | 5 × 105 | 24 h | Whole Cells | 84 | 43.7 |
| 2 × 105 | 48 h | Nuclei | 23 | 92.2 | |
| SW480 | 2 × 105 | 24 h | Whole Cells | 445 | 0.4 |
| 2 × 105 | 24 h | Nuclei | 404 | 1.1 | |
| 2 × 105 | 48 h | Whole cells | 162 | 0.5 | |
| 2 × 105 | 48 h | Nuclei | 68 | 1.0 | |
| GM0637 (normal) | 2 × 105 | 48 h | Nuclei | 31 | 2.6 |
| GM04429 (XPA) | 2 × 105 | 48 h | Nuclei | 40 | 0.7 |
| AG01440 (normal) | 4 × 105 | 48 h | Nuclei | 27 | 0.7 |
| 4 × 105 | 96 h | Nuclei | 0.72 | 13.2 | |
| AG06971 (XPA) | 3 × 105 | 48 h | Nuclei | 386 | 0.4 |
| 3 × 105 | 96 h | Nuclei | 9.65 | 1.5 | |
| Breast Non-Stem Cells (1) | 5 × 105 | 48 h | Whole Cells | 3,160 | 4.6 |
| 5 × 105 | 48 h | Nuclei | 7,240 | 2.1 | |
| Breast Stem-Like Cells (1) | 5 × 105 | 48 h | Whole Cells | 331 | 1.7 |
| 5 × 105 | 48 h | Nuclei | 255 | 1.2 | |
| Breast Non-Stem Cells (2) | 5 × 105 | 48 h | Whole Cells | 5,830 | 2.4 |
| 5 × 105 | 48 h | Nuclei | 10,000 | 1.4 | |
| Breast Stem-Like Cells (2) | 5 × 105 | 48 h | Whole Cells | 434 | 0.5 |
| 5 × 105 | 48 h | Nuclei | 60 | 0.4 | |
| Breast Non-Stem Cells (3) | 5 × 105 | 48 h | Whole Cells | 15,200 | 2.7 |
| 5 × 105 | 48 h | Nuclei | 14,100 | 1.6 |
All data are presented as the mean of three independent experiments, except for the breast stem-like cells (2) that are from two independent experiments.
The NER efficiencies of human immortalized and primary fibroblasts with XPA defects
To validate that ORA can specifically measure NER efficiency, we compared the repair efficiency of SV40-immortalized human fibroblast cell lines derived from a normal individual (GM00637) to those from a xeroderma pigmentosum (XP) patient with mutant XPA alleles (GM04429). We transfected 2 × 105 cells with 0.02 nM of the CPD-containing oligonucleotide. After 48 h incubation, nuclei were isolated and CPD-containing oligonucleotides were retrieved from the purified nuclei for quantification. Approximately 30 and 40 oligonucleotides per cell were retrieved from GM00637 and GM04429, respectively (Table 1). As shown in Fig. 5A, the XPA cells are about 80% less efficient in repairing the thymine dimers than the normal cells, verifying that the assay is capable of detecting the NER deficiency in XP cells.
Figure 5. NER deficiency in human XPA fibroblasts validated by ORA.

(A) NER efficiencies of human SV40-transformed, normal (GM00637) versus XPA (GM04429) fibroblast cells. Cells were transfected with CPD-containing oligonucleotides. After 48 h incubation at 37°C, nuclei were isolated and oligonucleotides were retrieved from the purified nuclei for quantification. Data are presented as mean ± S.D. (n = 3) and the P value was calculated by Student's t-test. (B) NER efficiencies of human primary fibroblasts. Human primary fibroblast cells, normal (AG01440) versus XPA (AG06971), were transfected with CPD-containing oligonucleotide and incubated at 37°C for 48 h and 96 h prior to retrieval. Data derived from qPCR analyses of the retrieved oligonucleotides from three independent experiments are presented as % repaired of the CPD-containing constructs per cell (mean ± S.D., n = 3). The P value was obtained by Student's t-test. The absolute values likely reflect differences in kinetics of repair between primary and immortalized cells. (C) Validation of NER deficiencies by CPD-specific ELISA. After irradiating the cells with 25 J/m2 of UV-C, CPD lesions were quantified by ELISA at 0 h, 24 h, 48 h and 96 h for the normal fibroblasts and only at 0 h, 24 h and 48 h for the XPA cells since XPA fibroblasts were dying at 96 h. The results are shown as % of remaining CPD relative to 0 h after irradiation and data are presented as mean ± S.D. (n = 3).
Similarly, using primary human fibroblasts AG01440, derived from a healthy individual, and AG06971, obtained from an individual with XPA deficiency, we transfected 4 × 105 cells of AG01440 and 3 × 105 cells of AG06971 with 0.02 nM of the CPD-containing oligonucleotide, and retrieved oligonucleotides from isolated nuclei followed periods of incubation. We observed that after 48 h incubation, the normal fibroblasts repaired 0.7% of the retrieved oligonucleotides, compared to 0.4% for the XPA cells (Table 1 and Fig. 5B). However after 96 h, the normal fibroblasts had repaired 13% of the input CPD constructs, whereas the XPA fibroblasts had repaired only 1.5% of the constructs - 90% less efficient than that of the normal cells (Table 1 and Fig. 5B). This result confirms the efficacy of this assay in assessing NER efficiencies in a variety of cells including human primary cells.
We further confirmed the NER deficiency of the primary fibroblasts by CPD-specific ELISA. As shown in Fig. 5C, the normal fibroblasts AG01440 repaired about 20% of the UV-induced CPD lesions 96 h after irradiation, while no detectable removal of CPD could be observed in the XPA fibroblasts AG06971 for up to 48 h following UV exposure; there was measurable cell death after 96 h. This result is consistent with the measurements by ORA. Although ELISA could not detect any repair in the XPA cells, ORA however, could detect subtle repair activities, i.e. 0.4% at 48 h and 1.5% at 96 h (Table 1), highlighting the sensitivity of ORA.
The NER efficiencies of epithelial stem-like cells versus non-stem cells derived from human breast tissues
Adult stem cells in tissue are the source of terminally differentiated cells and are responsible for repopulating the tissue. Whether these cells possess different repair activities than somatic cells is controversial21,22. To demonstrate that ORA can be applied to stem cell research, we obtained human breast epithelial stem-like cells from individuals undergoing reduction mammoplasty, as well as breast epithelial non-stem cells from the same individuals. The breast stem-like cells were characterized by Oct4 expression23, and the abilities to differentiate into other cell types and to form budding/ductal structures on matrigel24,25. We transfected 5 × 105 cells in 60 mm dishes with 0.02 nM of the CPD-containing oligonucleotide, and incubated the cells at 37°C for 48 h, prior to retrieval of the oligonucleotides either from whole cells or from isolated nuclei. We retrieved on average about 8,000 oligonucleotides from the whole cell extract of a single epithelial non-stem cell, and about 10,000 oligonucleotides per single isolated nucleus (Table 1). For the stem-like cells, the number of oligonucleotides retrieved is in the range of 300 oligonucleotides per cell for whole cell extraction and about 150 oligonucleotides per cell for isolated nuclei (Table 1). Based on assays of whole cells of two individuals, we observed a 3- to 5-fold greater NER activity of non-stem cells than that of stem-like cells (Fig. 6A). The results parallel the NER efficiencies in isolated nuclei, in which non-stem cells are 2- to 4-fold greater than stem-like cells (Fig. 6B).
Figure 6. Breast epithelial stem-like cells possess lower NER activities than breast epithelial non-stem cells.
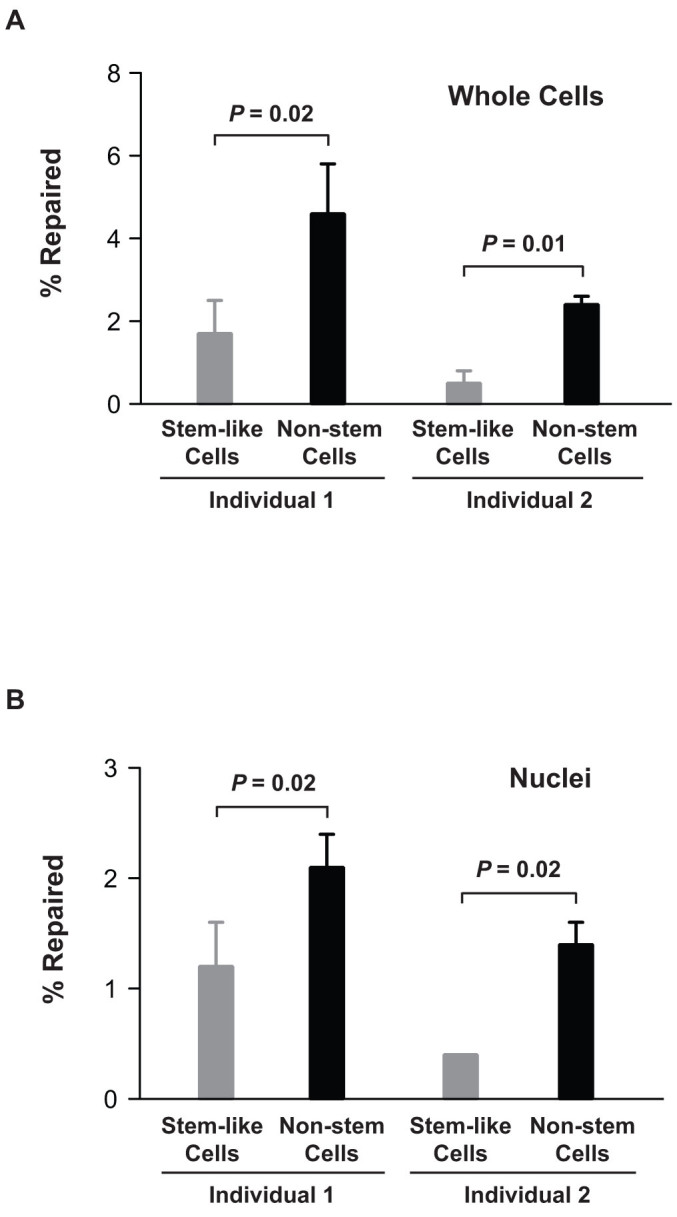
(A) NER activities calibrated from whole cells. Human breast stem-like cells and breast epithelial non-stem cells were isolated from two individuals. Both types of cells (5 × 105) were transfected with CPD-containing oligonucleotide (0.02 nM) mediated by calcium phosphate and incubated at 37°C for 48 h. After incubation, oligonucleotides were retrieved from whole cells and qPCR was employed for calibration of repair efficiencies. (B) NER activities calibrated from nuclei. The cells were treated under the same condition as described in (A). Oligonucleotides were retrieved from isolated nuclei for qPCR analyses. Data are presented as % repaired of the CPD-containing oligonucleotides per cell (mean ± S.D., n = 3 for both types of cells of individual 1 and for non-stem cells of individual 2; n = 2 for stem-like cells of individual 2).
Discussion
We have developed a simple and cost-effective method, which we term the Oligonucleotide Retrieval Assay (ORA), for in vivo calibration of DNA repair activities in eukaryotic cells. Using synthetic oligonucleotides, we were able to construct versatile DNA substrates for DNA repair processes. Here we demonstrate that an oligonucleotide construct containing a UV-damaged product CPD, a known target of NER, is repaired and restored to the canonical DNA sequence when delivered into live human cells.
The CPD-containing DNA substrate used in this study is comprised of three synthetic oligonucleotides adjoined together by ligation at 1:1:1 ratio to form a hairpin-like duplex with a 4-nt turn (Fig. 2). The central piece of the oligonucleotides (30 nt) in the construct possesses a CPD at the 13th and 14th nucleotide position 5′ from the terminus. The CPD-containing oligonucleotide was not 5′-phosphorylated when originally synthesized. To make ligation possible, we first phosphorylated the oligonucleotide by T4 polynucleotide kinase with ATP. This is the first ligation site that adjoins the 5′-terminus of the central CPD-containing oligonucleotide to the oligonucleotide that contains a 5′-terminal biotin for streptavidin bead capture and a unique 19-nt, single-stranded identity sequence for strand-specific PCR amplification. The second ligation site adheres the 3′ terminus of the CPD-containing oligonucleotide to the 5′-phosphorylated terminus of the 5′-fold-back oligonucleotide that forms a 5′-partial duplex structure with a 4-nt turn (hairpin) and consists of an exonuclease-resistant nucleotide 2′,3′-dideooxycytidine at the 3′-terminus. By avoiding the exposure of the terminal nucleotides on one side and blocking the terminal residues on the other side, this design protects the duplex DNA fragment from exonucleolytic degradation when introduced into cells. As demonstrated by transfection of primary fibroblast, this DNA construct remained intact after 96 h-incubation in cells, following which approximately 1–10 molecules were retrieved per nucleus (see AG01440 and AG06971 in Table 1), sufficient for PCR quantification for the repaired oligonucleotides.
The lengths of the synthetic oligonucleotides used in this study have been optimized to provide effective length of substrate for NER and to minimize cost. The effective substrate length for strand incision by NER has been reported in experiments carried out in cell-free extracts derived from HeLa cells by using restricted DNA fragments containing a CPD with various lengths as substrates for the incision reactions26. By electrophoretic resolution of the reaction products resulting from a 90-min incubation, the conclusion for the minimum distance from the CPD lesion required for detectable strand incision is 28 bp for the 3′-flanking sequence and 50 bp for the 5′-arm. As shown in Fig. 2, the duplex DNA used herein is 77-bp long, where the CPD residue resides in the middle, with a 3′-arm of 33 bp and a 5′-arm of 42 bp (or 61 nt including the 5′ single-stranded sequence) in length.
Here we show that this CPD-containing construct can be used to detect NER activities in both cell-free extracts (Fig. 3) and living cells (Fig. 4 and Table 1). The addition of an anti-XPA antibody into the cell lysate in order to suppress XPA function validated that ORA specifically and quantitatively measures NER activity (Fig. 3). We also transfected the substrate into cells; allowed endogenous NER activities to excise the lesion and repair the substrate in vivo. Depending on cell type, NER activities could be quantified within 2 h to 96 h (Fig. 4 and Table 1).
We determined the NER efficiency of several human cell lines, including immortalized and primary cells by ORA. For human embryonic kidney cells 293T, more than 40% of the input oligonucleotides (~80 copies per cell retrieved) were repaired in a 24 h incubation, while with human colon cancer cells SW480 under the same condition only 0.4% of the input oligonucleotides (approximately 400 copies per cell retrieved) were repaired (Table 1). The differential repair efficiencies of those two cell lines may be due to tissue-specific repair responses or differences resulting from immortalization. In addition, as ORA uses an exogenous DNA substrate to cumulatively measure NER activities in cells, the mechanism(s) involving damage surveillance of these substrates might be limited to extra-chromosomal constructs and may not necessarily reflect the realities of DNA repair in chromatin. Also, since ORA measures basal NER activity in cells, the status of NER components in cells will likely be critical. For example, in the relatively dormant primary fibroblasts, which have a population doubling time of about 6 days, it took a 96 h incubation before appreciable repair was detected. In comparison, in rapidly dividing cells such as 293T, with a population doubling time of 18 h, repair is more rapidly detected than in the slowly dividing SW480 cells, which divide every 36 h.
ORA can be applied to monitor repair events in nuclei. We were able to retrieve approximately 20 CPD-containing oligonucleotides per nucleus from 293T cells after 48 h incubation, of which more than 90% were repaired. This is consistent with the retrieval of about 80 oligonucleotides per cell from the whole cell extracts of 293T following 24 h of incubation when ~40% of constructs were repaired (Table 1). These results also suggest that the termini-protected oligonucleotides are stable against non-specific nucleotide degradation and indicate the feasibility of longer incubations of these constructs in cells.
To further validate the assay, we used immortalized and primary fibroblasts derived from patients deficient for XPA and calculated the repair efficiencies of these cells and their normal counterparts. We demonstrated that NER in XPA-deficient fibroblasts is less efficient than in normal fibroblasts, where it was ~4-fold less in immortalized fibroblasts and ~9-fold less in primary cells (Fig. 5 and Table 1). These results suggest that ORA specifically measured NER activities. We also demonstrated that even primary cells could be efficiently transfected with these oligonucleotide constructs, and incubated for up to 96 h allowing the DNA constructs to enter the nucleus and the repaired constructs to be subsequently retrieved from isolated nuclei (e.g., ~400 constructs/nucleus at 48 h and ~10 constructs/nucleus at 96 h for AG06971 cells) (Fig. 5B and Table 1).
Adult stem-like cells derived from human breast tissues have been isolated and characterized for stem cell properties23,24,25,27. Here we employed human breast epithelial stem-like cells for ORA to evaluate their NER activities, in comparison with those of breast epithelial non-stem cells derived from the same individuals. We first demonstrated that ORA could be applied to stem cells and primary epithelial cells. For breast stem-like cells, we were able to retrieve 300–400 oligonucleotides per cell from whole cells of two individuals after 48 h incubation, while 60–250 oligonucleotides per cell were retrieved from nuclei of the same individuals under the same experimental conditions (Table 1). The transfection efficiencies of these stem-like cells are in fact greater than any other cell types that were tested in this study (Table 1). For breast epithelial non-stem cells, the transfection efficiencies are comparable to those for the stem-like cells and ORA can be used to quantify the repair activities of both (Fig. 6). We retrieved 3,000–15,000 oligonucleotides per whole cell and 7,000–14,000 oligonucleotides per isolated nucleus of three individuals after 48 h incubation (Table 1). Since we only measured the basal NER activity in the absence of UV- or any other DNA damaging agent stimulation, it cannot be ruled out that the network of DDR (DNA damage responses) pathways may enhance NER activities in response to exogenous damage in stem-like cells. It will also be important to examine whether breast stem cells are more prone to apoptosis in response to DNA damage.
Direct measurements of catalytic activities of specific DNA repair enzymes in cell lysates have been used to identify repair deficient phenotypes20,28,29. These in vitro biochemical assays require the isolation of cell-free extracts that contains NER activities, for example, nucleotide excision for measuring the efficiency of strand incision of a DNA substrate by gel electrophoresis. In vivo methods such as the Comet assay30,31, Unscheduled DNA Synthesis (UDS)12,32 and Host Cell Reactivation assay (HCR)33,34,35 have been established for monitoring NER. Much effort has been made to simplify these assays. For example, several systematic methods have been used to modify the Comet assay36,37 and UDS has also been updated to a high throughput analysis by using an in-cell-analyzer to monitor repair DNA synthesis through fluorescent signals, based on the principle of coupling fluorescent agents to the incorporated nucleotides such as 5-ethynyl-2′-deoxyuridine (EdU)38,39. Other methods also commonly used include the immuno-slot-blotting assay21 and the gene-specific qPCR assay40,41.
Because of the large amount of oligonucleotide that can be introduced into a single cell, as well as the stability of the oligonucleotide once inside the cells, we were able to monitor DNA repair for as long as 96 hours using a small number of cells (Fig. 5B). ORA can also be performed with or without UV-induction, providing data of both basal and UV-induced NER activities. Unlike the Comet or UDS assay that requires microscopy, immuno-slot-blotting that requires adduct-specific antibodies or HCR that needs bacterial transformation, ORA analysis of DNA repair can be quantified simply by qPCR (the 2−ΔΔCt method). Furthermore, ORA can be tailored to perform multiplex qPCR repair assays. One can use a variety of oligonucleotide constructs to simultaneously determine the repair efficiencies of different DNA damaged products in the same cell by assigning a unique identifier sequence to each oligonucleotide. For example, DNA photoproduct 6-4 PPs can be synthesized in an oligonucleotide42 similar to the CPD one used here and both oligonucleotides can be co-transfected into cells for monitoring the repair of the two DNA lesions simultaneously.
Methods
Oligonucleotides
The 30-nt oligonucleotide containing a CPD was a generous gift from Dr. S. Iwai (Osaka University, Osaka, Japan). PCR primers and other oligodeoxynucleotides used to construct the CPD-containing DNA substrate were purchased from IDT. The sequence of the 30-nt CPD-containing oligonucleotide is: 5′-CTCGTCAGCATCTTCATCATACAGTCAGTG-3′, where CPD is marked as TT in bold and underlined. The other two oligonucleotides used to construct the CPD-containing DNA are: JS140, 5′-P-GTCCGCTCGAGACACCGAAAACGGTGTCTCGAGCGGACCACTGACTGTATGATGAAGATGCTGACGAGGTCTGGATCCGGCCTTGCCGCAGCTGGTAC-ddC-3′ and JS141, 5′-biotin-GCACGTCAGGCACGGCGTCGGTACCAGCTGCGGCAAGGCCGGATCCAGAC-3′. The PCR primers used to quantify the unrepaired and repaired CPD-containing construct are: JS012, 5′-GCACGTCAGGCACGGCGTC-3′; JS004, 5′-GTCTGGATCCGGCCTTGCC-3′ and JS012; JS144, 5′-TTCGGTGTCTCGAGCGGAC-3′, respectively.
Construction of oligonucleotide containing a CPD adduct
The DNA construct containing a CPD was created by ligation of three synthetic oligonucleotides: a 5′-biotinylated oligomer (JS141), a 30 nt ssDNA containing a CPD dimer in the center of the sequence, and a fold-back oligonucleotide (JS140) which forms a hairpin and also serves as a complementary strand for the other two oligomers to form a complete dsDNA construct (Fig. 1). The 5′-termini of the hairpin oligonucleotides were chemically phosphorylated to facilitate ligation and the 3′-termini were blocked by addition of a dideoxycytosine (ddC) residue to prevent the DNA from exonuclease degradation. Since the CPD oligomers do not contain a 5′-phosphate, 5′-phosphorylation of the oligonucleotides was carried out by T4 polynucleotide kinase in the presence of ATP. The three oligonucleotides (the 5′-phosphorylated 30-nt CPD oligomer, JS140 and JS141) were hybridized together at equivalent molarities by boiling and re-annealing in a water bath. T4 DNA ligase was used to seal the nicks between the oligomers and complete the hairpin-like dsDNA construct.
Cell lines and cell culture
Cell lines used here include human embryonic kidney cell HEK293T, human colon cancer cell SW480, SV40-transformed normal human fibroblasts GM00637, SV40-transformed human XPA fibroblasts GM04429 (courtesy of Dr. Junko Oshima, University of Washington), normal human primary fibroblasts AG01440 and human XPA primary fibroblasts AG06971 (Coriell Institute). HEK293T, SW480, GM00637 and GM04429 were propagated in Dulbecco's Modification of Eagle's Medium (DMEM, Mediatech) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin G, 0.1 mg/ml streptomycin and 2 mM L-glutamine. AG01440 and AG06971 were grown in the same medium except supplemented with 15% FBS. Cultures were maintained in a humidified incubator at 37°C with 95% air and 5% CO2.
Breast tissues of healthy women were obtained during reduction mammoplasty in accordance with institutional guidelines at a hospital in Lansing, Michigan. The establishment and culturing of epithelial stem cells (stem-like cells) and epithelial cells without stem cell characteristics (non-stem cells) from the breast tissues have been described previously27. This primary cell model of human breast epithelial cells was a generous gift from Dr. C. C. Chang (Michigan State University).
Transfection
All transfections were performed with calcium phosphate. Oligonucleotides were first prepared in 250 μl of H2O containing 200 mM CaCl2; the solution was then added drop-by-drop with gentle mixing to 250 μl of 2× HBS buffer (50 mM Hepes, pH 7.04, 10 mM KCl, 12 mM Dextrose, 280 mM NaCl and 1.5 mM Na2HPO4). The mixed solution (500 μl) was incubated at room temperature (23°C) for 20 min, allowing formation of DNA-calcium phosphate precipitates. Transfection was carried out by adding the DNA-calcium phosphate solution (500 μl) to designated cell cultures in 60 mm dishes containing 3 ml of medium. The transfected cell culture was maintained in a humidified incubator at 37°C with 95% air and 5% CO2. The transfection apparently does not affect cell viability, as all cells used in this report continue proliferating after transfection.
Isolation of nuclei
Cell nuclei were isolated by treating the cells with hypotonic buffer; followed by gentle homogenization and subcellular separation by centrifugation. Cells were harvested in pellets and resuspended in hypotonic buffer (10 mM Hepes, pH 7.5, 2 mM MgCl2, 25 mM KCl, 1 mM DTT, 1 mM PMSF and 1:200 (v/v) Protease Inhibitor cocktail III, CalBiochem). Depending on cell types, the cellular suspension was incubated on ice for different periods of time, prior to homogenization using a Dounce Homogenizer with pestle B. Immediately after homogenization, 200 mM sucrose was added to the cell lysates and mixed, and centrifugation was carried out to spin down the nuclei from other cellular debris. The isolated nuclei were immediately processed or stored in −80°C for later use.
Oligonucleotide retrieval
To retrieve oligonucleotides from the transfected cells, we used the DNA extraction kit from Stratagene to isolate total DNA from the cells or from the isolated nuclei. With a biotin molecule at the 5′-end of the oligonucleotide construct, we were able to purify and enrich the constructs by streptavidin bead binding (Invitrogen). After several washes following the manufacturer's protocol, the oligonucleotides were released from the beads by incubation at 80°C in deionized water.
Real-time quantitative PCR analysis
DNA Engine Opticon 2 (MJ Research) was used to carry out real-time quantitative PCR (qPCR) reactions. DNA and primers were mixed with Brilliant III Ultra-Fast SYBR Green qPCR Master Mix (Agilent) following manufacturer's instruction and PCR cycling was typically run with the following protocol: hot-start, 95°C, 3 min; cycling, 95°C, 5 sec and 60°C, 15 sec, 50 cycles; completion, 72°C, 1 min; melting curve, 60°C to 95°C, recording 0.2°C/sec.
Calibration of nucleotide excision repair
The hairpin-like construct of dsDNA, containing a CPD, was used as an oligonucleotide substrate for assaying the efficiency of NER in cells. This construct contains a CPD lesion in the middle of the dsDNA stretch, allowing the cellular NER protein complex to excise and remove a long patch of nucleotides flanking the lesion, synthesize a new patch of DNA to fill in the gap, and rejoin the strands together by ligation (Fig. 2A). If not repaired, CPD will prevent the polymerase from copying DNA past the lesion in PCR reactions (Fig. 2C). Using primers flanking the CPD site, the number of repaired oligonucleotides can be quantified based on amplification across the restored sequence. A single-stranded region 5′ of the oligonucleotide was utilized as a unique forward primer sequence, labeled (a) in Fig. 2A, to specify the CPD bearing strand for PCR amplification with the reverse primer sequence located before (b) or after (c) the CPD. The amplification from the forward primer (a) with the reverse primer (b), located 5′ of the lesion, serves as a control for the total number of the retrieved oligonucleotides (Ctcontrol), whereas amplification using the same forward primer (a) but with the reverse primer (c), which anneals 3′ to the CPD site, represents the number of the repaired DNA (Cttest). To obtain the number of oligonucleotides retrieved per cell, a control sequence on chromosome 17p13 is amplified (Ctchr) and the numbers of the total and the repaired oligonucleotides per cell (for example, in diploid cells) are calculated by equations: total oligonucleotides per cell = (2ΔCt) × % ligation × 2, where ΔCt = Ctchr − Ctcontrol, and repaired oligonucleotides per cell = (2ΔCt) × 2, where ΔCt = Ctchr − Cttest, respectively. Thus, the repair efficiency, defined as % repaired, can be calculated from the ratio of the number of the repaired DNA over the number of total oligonucleotides retrieved. To perform this assay therefore, cells were transfected with the 5′-biotinylated CPD construct. After a designated time of incubation, which permits the construct to enter the cells and get repaired, the oligonucleotides were retrieved by streptavidin bead capture from either the whole cell extract or the nuclei and the repair efficiency in the cells quantified by qPCR.
Measuring nucleotide excision repair activities in cell-free extracts
The cell-free lysates were extracted from HEK293T cells by incubating 2 × 107 cells on ice in 500 μl lysis buffer (20 mM Hepes-KOH, pH 7.4, 155 mM KCl, 1.5 mM MgCl2, 0.5% Triton X-100, 2 mM DTT, and 10 ug/ml each of pepstatin, leupeptin, aprotinin, and 0.1 mM PMSF) for 10 min. After incubation, the lysis solutions were centrifuged at 2,000× g for 10 min at 4°C and the supernatants were saved as cell lysates. To measure NER activities, 0.075 nM of the CPD-containing oligonucleotides was incubated in 30 μl of the reaction buffer (40 mM Hepes-KOH, pH 7.5, 70 mM KCl, 8 mM MgCl2, 2 mM ATP, 20 μ dNTP, 1.2 mM DTT, 0.3 mM EDTA, 10% glycerol and 100 μg/ml BSA) with 80 μg of cell lysates at 30°C for 2 h. Mouse monoclonal antibody against human XPA (5 μl) (Clone 12F5, Thermo Scientific) was used in reactions to inhibit NER activities. The reaction was terminated by inactivating the enzymes at 75°C for 20 min, and the oligonucleotides were isolated for calibration of NER activities from 20 μl of the reaction solution by streptavidin beads binding (Invitrogen). The streptavidin-bound oligonucleotides were eluted in 30 μl H2O at 80°C and 5 μl of the eluted solution was used for qPCR analyses.
ELISA and colony-forming assay
The ELISA CPD-removal (Cell Biolabs) and the colony-forming assay were performed following the manufacturer's protocol or using standard protocols described elsewhere43, respectively.
Author Contributions
J.C.S. and L.A.L. designed research; J.C.S. and E.J.F. performed research; E.H.A. prepared primary breast epithelial cells; J.C.S. and L.A.L. wrote the paper.
Acknowledgments
We thank Dr. Chia-Cheng Chang for providing primary human breast epithelial cells, and are grateful to Drs. Philip C. Hanawalt, Graciela Spivak, and Ashwini Kamath-Loeb for suggestions and comments; Kate Bayliss and Jessica Kuong for critical reading of the manuscript. Research reported in this publication was supported by grants from National Cancer Institute of the National Institute of Health to L.A.L. under award numbers: R01CA102029, R01CA115802, and P01CA077852, and by a grant from National Institute on Aging of the National Institute of Health to L.A.L. under award number: P01AG001751.
References
- Cleaver J. E., Lam E. T. & Revet I. Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat. Rev. Genet. 10, 756–768 (2009). [DOI] [PubMed] [Google Scholar]
- Friedberg E. C. et al. DNA Repair and Mutagenesis. Second edn, (ASM Press, 2005). [Google Scholar]
- Kamileri I., Karakasilioti I. & Garinis G. A. Nucleotide excision repair: new tricks with old bricks. Trends Genet. 28, 566–573 (2012). [DOI] [PubMed] [Google Scholar]
- Hanawalt P. C., Ford J. M. & Lloyd D. R. Functional characterization of global genomic DNA repair and its implications for cancer. Mut. Res. 544, 107–114 (2003). [DOI] [PubMed] [Google Scholar]
- Hanawalt P. C. & Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 9, 958–970 (2008). [DOI] [PubMed] [Google Scholar]
- Nouspikel T. Multiple roles of ubiquitination in the control of nucleotide excision repair. Mech. Ageing Dev. 132, 355–365 (2011). [DOI] [PubMed] [Google Scholar]
- Kang T. H., Reardon J. T. & Sancar A. Regulation of nucleotide excision repair activity by transcriptional and post-transcriptional control of the XPA protein. Nucl. Acids Res. 39, 3176–3187 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleaver J. E. Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat. Rev. Cancer 5, 564–573 (2005). [DOI] [PubMed] [Google Scholar]
- Barnes D. E. & Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu. Rev. Genet. 38, 445–476 (2004). [DOI] [PubMed] [Google Scholar]
- Loeb L. A. Human cancers express mutator phenotypes: origin, consequences and targeting. Nat. Rev. Cancer 11, 450–457 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleaver J. E., Greene A. E., Coriell L. & Riccardi V. M. An autosomal dominant inheritance for multiple sunlight-induced malignancy in a patient without abnormalities in DNA repair or replication. Repository identification No. GM2881. Cytogenet. Cell Genet. 29, 122–124 (1981). [DOI] [PubMed] [Google Scholar]
- Latimer J. J. et al. Nucleotide excision repair deficiency is intrinsic in sporadic stage I breast cancer. Proc. Natl. Acad. Sci. U.S.A. 107, 21725–21730 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg E. C. How nucleotide excision repair protects against cancer. Nat. Rev. Cancer 1, 22–33 (2001). [DOI] [PubMed] [Google Scholar]
- Saldivar J. S., Wu X., Follen M. & Gershenson D. Nucleotide excision repair pathway review I: implications in ovarian cancer and platinum sensitivity. Gynecol. Oncol. 107 (2007). [DOI] [PubMed] [Google Scholar]
- Crew K. D. et al. Polymorphisms in nucleotide excision repair genes, polycyclic aromatic hydrocarbon-DNA adducts, and breast cancer risk. Cancer Epidem. Biomark. 16, 2033–2041 (2007). [DOI] [PubMed] [Google Scholar]
- Benhamou S. & Sarasin A. ERCC2/XPD gene polymorphisms and cancer risk. Mutagenesis 17, 463–469 (2002). [DOI] [PubMed] [Google Scholar]
- Slyskova J. et al. Differences in nucleotide excision repair capacity between newly diagnosed colorectal cancer patients and healthy controls. Mutagenesis 27, 519–522 (2012). [DOI] [PubMed] [Google Scholar]
- Koberle B. et al. ERCC1 and XPF expression in human testicular germ cell tumors. Oncol. Rep. 23, 223–227 (2010). [PubMed] [Google Scholar]
- Wellinger R. E. & Thoma F. Taq DNA polymerase blockage at pyrimidine dimers. Nucl. Acids Res. 24, 1578–1579 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood R. D., Robins P. & Lindahl T. Complementation of the xeroderma pigmentosum DNA repair defect in cell-free extracts. Cell 53, 97–106 (1988). [DOI] [PubMed] [Google Scholar]
- Karbaschi M., Brady N. J., Evans M. D. & Cooke M. S. Immuno-slot blot assay for detection of UVR-mediated DNA damage. Methods Mol. Biol. 920, 163–175 (2012). [DOI] [PubMed] [Google Scholar]
- Zhang Y., Wu X., Guo D., Rechkoblit O. & Wang Z. Activities of human DNA polymerase kappa in response to the major benzo[a]pyrene DNA adduct: error-free lesion bypass and extension synthesis from opposite the lesion. DNA Repair 1, 559–569 (2002). [DOI] [PubMed] [Google Scholar]
- Tai M. H. et al. Oct4 expression in adult human stem cells: evidence in support of the stem cell theory of carcinogenesis. Carcinogenesis 26, 495–502 (2005). [DOI] [PubMed] [Google Scholar]
- Chang C. C. et al. A human breast epithelial cell type with stem cell characteristics as target cells for carcinogenesis. Radiat. Res. 155, 201–207 (2001). [DOI] [PubMed] [Google Scholar]
- Sun W., Kang K. S., Morita I., Trosko J. E. & Chang C. C. High susceptibility of a human breast epithelial cell type with stem cell characteristics to telomerase activation and immortalization. Cancer Res. 59, 6118–6123 (1999). [PubMed] [Google Scholar]
- Huang J. C. & Sancar A. Determination of minimum substrate size for human excinuclease. J. Biol. Chem. 269, 19034–19040 (1994). [PubMed] [Google Scholar]
- Kao C. Y., Nomata K., Oakley C. S., Welsch C. W. & Chang C. C. Two types of normal human breast epithelial cells derived from reduction mammoplasty: phenotypic characterization and response to SV40 transfection. Carcinogenesis 16, 531–538 (1995). [DOI] [PubMed] [Google Scholar]
- Feng Z., Hu W. & Tang M. S. Trans-4-hydroxy-2-nonenal inhibits nucleotide excision repair in human cells: a possible mechanism for lipid peroxidation-induced carcinogenesis. Proc. Natl. Acad. Sci. U.S.A. 101, 8598–8602 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Elizur T. et al. Reduced repair of the oxidative 8-oxoguanine DNA damage and risk of head and neck cancer. Cancer Res. 66, 11683–11689 (2006). [DOI] [PubMed] [Google Scholar]
- Collins A. R. The comet assay for DNA damage and repair: principles, applications, and limitations. Mol. Biotechnol. 26, 249–261 (2004). [DOI] [PubMed] [Google Scholar]
- Wu X. et al. Cell cycle checkpoints, DNA damage/repair, and lung cancer risk. Cancer Res. 65, 349–357 (2005). [PubMed] [Google Scholar]
- Thomas J. C. G. Measurement of unscheduled synthesis by autoradiography. DNA Repair, A Laboratory Manual of Research Procedures 1, 277–287 (1981). [Google Scholar]
- Hu J. J. et al. Deficient nucleotide excision repair capacity enhances human prostate cancer risk. Cancer Res. 64, 1197–1201 (2004). [DOI] [PubMed] [Google Scholar]
- Spivak G. & Hanawalt P. C. Host cell reactivation of plasmids containing oxidative DNA lesions is defective in Cockayne syndrome but normal in UV-sensitive syndrome fibroblasts. DNA Repair 5, 13–22 (2006). [DOI] [PubMed] [Google Scholar]
- Wang H. T., Choi B. & Tang M. S. Melanocytes are deficient in repair of oxidative DNA damage and UV-induced photoproducts. Proc. Natl. Acad. Sci. U.S.A. 107, 12180–12185 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiskinis E., Suter W. & Hartmann A. High throughput Comet assay using 96-well plates. Mutagenesis 17, 37–43 (2002). [DOI] [PubMed] [Google Scholar]
- Rosenberger A. et al. Validation of a fully automated COMET assay: 1.75 million single cells measured over a 5 year period. DNA Repair 10, 322–337 (2011). [DOI] [PubMed] [Google Scholar]
- Limsirichaikul S. et al. A rapid non-radioactive technique for measurement of repair synthesis in primary human fibroblasts by incorporation of ethynyl deoxyuridine (EdU). Nucl. Acids Res. 37, e31, 10.1093/nar/gkp023 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa Y., Yamashita S., Lehmann A. R. & Ogi T. A semi-automated non-radioactive system for measuring recovery of RNA synthesis and unscheduled DNA synthesis using ethynyluracil derivatives. DNA Repair 9, 506–516 (2010). [DOI] [PubMed] [Google Scholar]
- Furda A. M., Bess A. S., Meyer J. N. & Van Houten B. Analysis of DNA damage and repair in nuclear and mitochondrial DNA of animal cells using quantitative PCR. Methods Mol. Biol. 920, 111–132 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bess A. S., Ryde I. T., Hinton D. E. & Meyer J. N. UVC-induced mitochondrial degradation via autophagy correlates with mtDNA damage removal in primary human fibroblasts. J. Biochem. Mol. Toxicol. 27, 28–41 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai S., Shimizu M., Kamiya H. & Ohtsuka E. Synthesis of a phosphoramidite coupling unit of the pyrimidine (6-4) pyrimidone photoproduct and its incorporation into oligodeoxynucleotides. J. Am. Chem. Soc. 118, 7642–7643 (1996). [Google Scholar]
- Franken N. A., Rodermond H. M., Stap J., Haveman J. & van Bree C. Clonogenic assay of cells in vitro. Nat. Protocols 1, 2315–2319 (2006). [DOI] [PubMed] [Google Scholar]