

SUMMARY
When unfolded proteins accumulate to irremediably high levels within the endoplasmic reticulum (ER), intracellular signaling pathways called the unfolded protein response (UPR) become hyperactivated to cause programmed cell death. We discovered that thioredoxin-interacting protein (TXNIP) is a critical node in this “Terminal UPR.” TXNIP becomes rapidly induced by IRE1α, an ER bifunctional kinase/endoribonuclease (RNase). Hyperactivated IRE1α increases TXNIP mRNA stability by reducing levels of a TXNIP destabilizing micro-RNA, miR-17. In turn, elevated TXNIP protein activates the NLRP3 inflammasome, causing Caspase-1 cleavage and interleukin 1β (IL-1β) secretion. Txnip gene deletion reduces pancreatic β-cell death during ER stress, and suppresses diabetes caused by proinsulin misfolding in the Akita mouse. Finally, small molecule IRE1α RNase inhibitors suppress TXNIP production to block IL-1β secretion. In summary, the IRE1α-TXNIP pathway is used in the terminal UPR to promote sterile inflammation and programmed cell death, and may be targeted to develop effective treatments for cell degenerative diseases.
INTRODUCTION
The endoplasmic reticulum (ER) is the first organelle that proteins of the secretory pathway encounter as they mature structurally and fold to their native conformations (Gething and Sambrook, 1990). Cells specialized for secretion, such as insulin-producing pancreatic islet β-cells, accommodate a high rate of cargo proteins transiting through the ER (Scheuner and Kaufman, 2008). But when conditions demand that these cells further increase protein secretion, the secretory pathway can quickly become overwhelmed. Inability to properly fold large secretory loads causes accumulation of unfolded proteins within the ER. In cells experiencing such “ER stress,” intracellular signaling pathways termed the unfolded protein response (UPR) become activated. Upon detecting unfolded proteins, three ER transmembrane sensors—IRE1α, PERK, and ATF6—initiate the UPR pathways (Harding et al., 1999; Tirasophon et al., 1998; Yoshida et al., 1998). Combinatorial signals from the three sensors increase transcription of target genes encoding ER chaperones and enzymatic activities, thus enhancing folding and maturation of secretory proteins. UPR targets also allow unfolded proteins to be extracted from the ER, and subsequently degraded in the cytosol (a process called ER-associated degradation) (Vembar and Brodsky, 2008). Additionally, a transient reduction in translation relieves ER protein load (Harding et al., 2001). If these adaptive UPR outputs are successful, the decline in unfolded proteins causes UPR signaling to wane as homeostasis is restored (Merksamer et al., 2008).
Alternatively, cells may experience ER stress at levels that are high—or prolonged—enough to overwhelm adaptive responses. Such irremediable ER stress can result from genetic mutations causing improper folding or modification of encoded secretory proteins. A well-studied example is the unoxidizable mutant form of murine proinsulin—called Akita—that cannot form an intramolecular disulfide bond needed to fold in the ER. Buildup of Akita in β-cells triggers programmed cell death, leading to a dominantly inherited form of diabetes in the mutant mice (Oyadomari et al., 2002; Wang et al., 1999); similar diabetes-causing mutations in the proinsulin gene occur in humans (Stoy et al., 2007). Irremediable ER stress can also be caused by pharmacologically inhibiting important ER protein modification processes. Under chronic and uncorrected ER stress, a terminal UPR becomes activated to trigger programmed cell death (Merksamer and Papa, 2010; Shore et al., 2011). Multi-cellular organisms may have evolved the ability to cull irremediably-stressed cells through programmed cell death in order to prevent production of improperly-modified or misfolded proteins. However, massive cell loss, which goes unmatched by cell proliferation, can lead to cell degenerative diseases.
Programmed cell death during chronic/high ER stress is an active process, and is promoted by alternate outputs of the UPR itself, which bias cell fate away from adaptation to the opposite extreme of cell destruction (Han et al., 2009). As activation levels of IRE1α, PERK, and ATF6 reflect either an adapted ER, or the continued presence of unfolded proteins, these upstream sensors are centrally poised to participate in the switching process between adaptation and destruction. However, many other key downstream links in this switching process remain to be discovered, and their elucidation may provide inroads to treat diseases of cell loss.
To find undiscovered signaling mediators of a terminal UPR, we conducted an unbiased screen to discover mRNAs whose translation increases during irremediable ER stress. Through this strategy we identified thioredoxin-interacting protein—TXNIP—as a critical node in a chain of destruction leading from the ER to programmed cell death. Remarkably, IRE1α utilizes a micro-RNA intermediate to control induction of TXNIP mRNA. Induced TXNIP protein in turn activates the NLRP3 inflammasome to cleave Pro-Caspase 1 to its active form, thereby causing maturation and secretion of the inflammatory cytokine, IL-1β. Furthermore, we find that TXNIP action is critical for programmed cell death of pancreatic β-cells under ER stress in vivo, and development of diabetes in rodents. Finally, our work provides pharmacological insights to target this destructive UPR chain at its upstream source, IRE1α, and thereby preserve cell viability and function.
RESULTS
Thioredoxin-interacting protein (TXNIP) is rapidly induced through the UPR
To identify signaling proteins mediating UPR-induced cell destruction, we purified poly-ribosomes to enrich for mRNAs that become preferentially translated (Heiman et al., 2008) very early in response to catastrophic ER stress. A gene encoding an enhanced green fluorescent protein (EGFP) epitope target was fused to the large ribosomal subunit protein L10a, and expressed in INS-1 insulinoma cells, which are differentiated insulin-producing cells derived from rat pancreatic islets (Figure 1A). The chimeric gene (or an EGFP control) was driven from a tetracycline-inducible expression construct integrated at a chromosomal FRT docking site in the INS-1 cells. Exposed to doxycycline (Dox), the cells express the EGFP-L10a fusion, or EGFP (Figure 1B). Compared to EGFP, which localizes primarily to the cytosol, EGFP-L10a localizes to both cytosol and nucleosomes, consistent with assembly into ribosomes (Figure 1C) (Heiman et al., 2008).
Figure 1. TXNIP mRNA and protein are rapidly induced in cells undergoing endoplasmic reticulum stress.
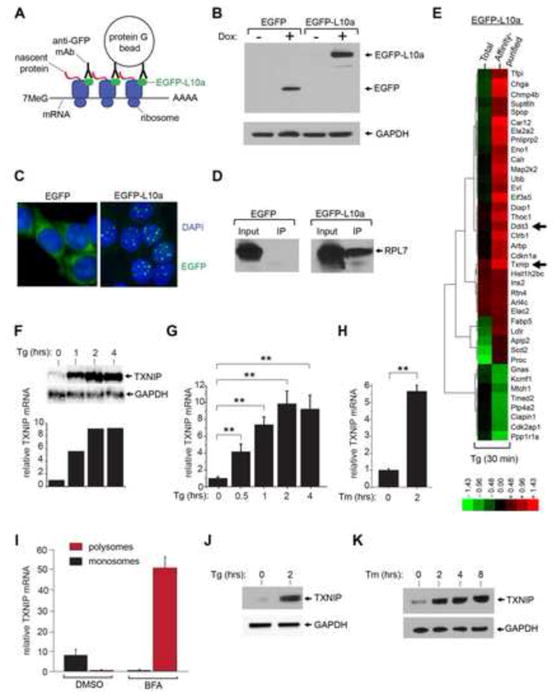
(A) Schematic of affinity purification of poly-ribosomes using a translational fusion of enhanced green fluorescent protein (EGFP) to the large ribosomal subunit protein L10a (EGFP-L10a). (B) Immunoblot analysis of whole cell extracts from 24 hour untreated and 1μg/ml doxycycline (Dox)- treated insulinoma (INS-1) cell lines expressing EGFP, or a EGFP translational fusion to the large ribosomal subunit protein L10a (EGFP-L10a) under a Dox inducible promoter. (C) Confocal images of INS-1 cells expressing EGFP or EGFP-L10a. Prior to imaging, cells were induced with 1μg/ml Dox for 24 hours, fixed with paraformaldehyde, and stained with 4′,6-diamidino-2-phenylindo (DAPI). (D) Immunoblot analysis of ribosomal protein L7 (RPL7) after anti-EGFP immunoprecipitation (i.p) confirms co-immunoprecipitation of ribosomes in INS-1 cells expressing EGFP-L10a (but not in cells expressing EGFP) following 24 hours treatment with 1μg/ml Dox. (E) Hierarchical clustering analysis of gene expression changes in INS-1 EGFP-L10a-expressing cells (Dox 1μg/ml for 24 hours) under ER stress through the use of DNA microarrays. cDNAs for hybridization were generated from total cellular mRNAs, or from mRNAs collected from anti-EGFP-L10a affinity-purified ribosomes. Indicated genes are those whose expression increased (red) or decreased (green) at least 2 fold under 1 μM thapsigargin (Tg) for 30 minutes (compared to no treatment). See Table S1 for gene identities, log2 expression changes, and statistics. (F & G) Time course analysis of TXNIP mRNA expression (normalized to GAPDH) during ER stress (1 μM Tg) in INS-1 cells by Northern blot, (F), or quantitative real-time PCR (Q-PCR), (G). (H) Analysis of TXNIP mRNA expression (normalized to GAPDH) during ER stress with 5 μg/ml tunicamycin (Tm) or 1 μM Tg in INS-1 cells by Q-PCR. (I) Poly-ribosome profiling demonstrates recruitment of TXNIP mRNA from monosomes into poly-ribosomes under treatment with 2.5 μg/ml brefeldin A (BFA) at 30 minutes. (J) Immunoblot detection of TXNIP protein in INS-1 cells during ER stress (1 μM Tg). (K) Immunoblot detection of TXNIP protein in INS-1 cells during ER stress (5 μg/ml Tm). Three independent biological samples were used for Q-PCR experiments. Data are shown as mean ± SD. **p < 0.005.
Immunoaffinity purification (using anti-EGFP antibodies) of ribosomes in cells expressing EGFP-L10a was confirmed by detecting a different ribosomal protein, L7 (Figure 1D). After inducing EGFP-L10a INS-1 cells with Dox, we treated them with the ER stress agent thapsigargin (Tg), which inhibits the SERCA (Sarcoplasmic-Endoplasmic Reticulum Calcium ATPase) pump, at a concentration (1 μM) known to trigger apoptosis in the entire population by 24 hours (Han et al., 2009). To reveal proteins translated very early under this regime of irremediable ER stress, we treated the cells with Tg for just 30 minutes before isolating mRNA from either immunoaffinity-purified poly-ribosomes, or total mRNA; both sources of RNA were used to perform comparative DNA microarray analysis (Figure 1E). Validating our approach, Ddit3, a pro-apoptotic UPR transcription factor also known as CHOP, was identified in the hit list of 38 genes with a two-fold or greater change in expression in both total and affinity-purified mRNA (Table S1). CHOP is known to be both transcriptionally and translationally upregulated in the UPR (Jousse et al., 2001; Palam et al., 2011).
Of other significantly induced genes on the list, we focused our attention on thioredoxin-interacting protein, TXNIP. TXNIP was first described as a binding partner of thioredoxin that regulates its antioxidant functions (Nishiyama et al., 1999; Patwari et al., 2006; Yamanaka et al., 2000). As TXNIP had been implicated in glucotoxicity-induced apoptosis of β-cells (Chen et al., 2008; Shalev, 2008), we reasoned that it may also mediate programmed cell death in response to ER stress; therefore we embarked on experiments to investigate the underlying mechanisms and physiology of this putative link.
TXNIP induction is evident in microarrays using both total and polysome-associated mRNA. From parent INS-1 cells exposed to 1 μM Tg, northern blots and Q-PCR show that total TXNIP mRNA increases by ~10-fold within 2 hours (Figure 1F and G). Treatment with the ER stress agent tunicamycin (Tm), an inhibitor of N-linked glycosylation, also increases TXNIP mRNA (Figure 1H). Validating our immunoaffinity purification strategy, we fractionated ribosomes according to size, and found, using yet another mediator of ER stress, the anterograde protein trafficking poison Brefeldin A (BFA), an approximately 50-fold migration of TXNIP mRNA from monosomes (in the uninduced state) to polysomes (Figure 1I).
As a consequence of strong recruitment of its mRNA to polysomes, TXNIP protein becomes rapidly and robustly translated under ER stress (Figure 1J and K). Rapid, high-level induction of TXNIP under ER stress is reminiscent of its induction under high ambient glucose (Figure S1, A–C) (Shalev et al., 2002). TXNIP was previously found to be induced by oxidant stress (e.g., H2O2) (Zhou et al., 2010), but we found that it also becomes induced upon exposure to the cell-permeable reductant dithiothreitol (DTT), which reduces disulfide bonds in the ER to cause protein misfolding (Figure S1, D, E). Taken together, these data demonstrate that diverse perturbations in ER protein folding cause robust and rapid induction of the Txnip gene, at both the mRNA and protein level.
The UPR sensors IRE1α, PERK, and ATF6 become activated as the earliest signaling events in cells experiencing ER stress. Because TXNIP is induced contemporaneously with PERK and IRE1α activation (Figure S2A), we reasoned that early UPR signaling events may mediate upregulation of TXNIP mRNA and protein. To test this, we treated mouse embryonic fibroblasts (MEFs) deficient for each UPR sensor with either Tm or Tg (Figure 2A). TXNIP mRNA induction is significantly diminished in both Ire1α−/− and Perk−/− MEFs during ER stress, but induction remains unperturbed in Atf6a −/− MEFs. Paralleling this effect, TXNIP protein induction is completely abrogated in Ire1α−/− and Perk−/− MEFs during ER stress (Figure 2B), but unaffected in Atf6a−/− MEFs (Figure S3A).
Figure 2. Robust induction of TXNIP requires activation of IRE1α’s bifunctional kinase and RNase domains.
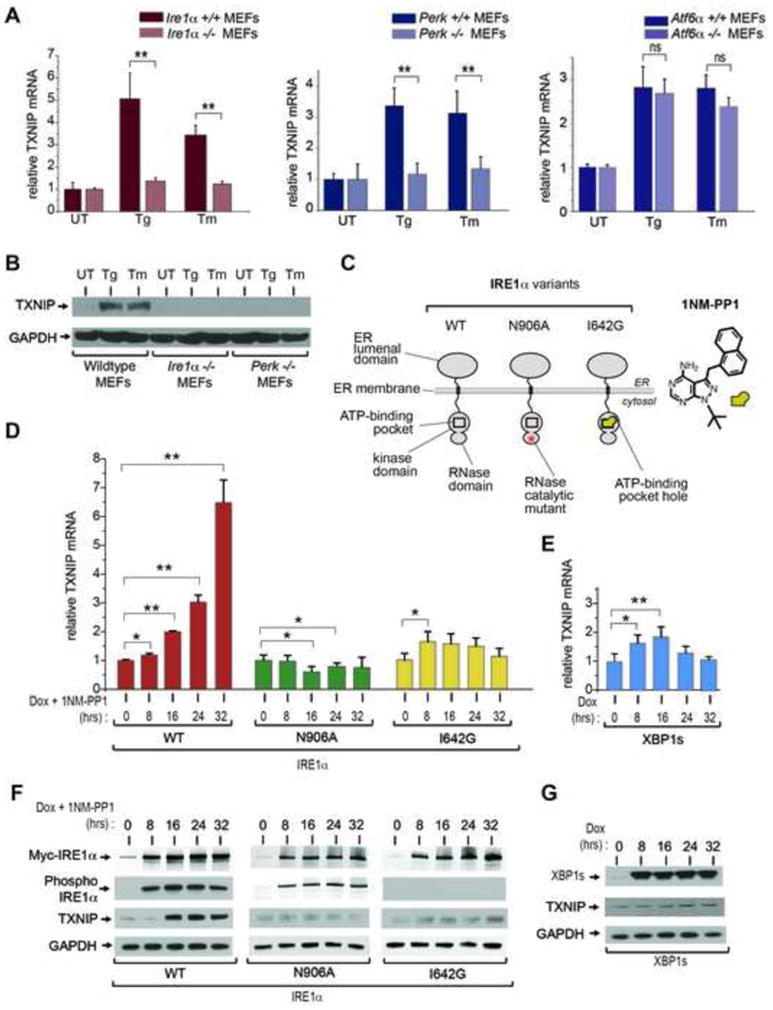
(A) Analysis of TXNIP mRNA expression (normalized to GAPDH) by Q-PCR during ER stress treatment in UPR sensor signaling mutants. Atf6α −/−, Perk−/−, and Ire1α−/− MEFs (and wild-type counterparts) were treated with 1 μM Tg, or 5 μg/ml Tm, for 6 hours. (B) Immunoblot for TXNIP protein from whole cell lysates of wild-type, Ire1α−/− and Perk−/− MEFs untreated or treated with 1 μM Tg or 5 μg/ml Tm for 3 hrs. (C) Schematic representation of IRE1α variants used in this study, and chemical structure of 1NM-PP1. (D) Time course analysis of TXNIP mRNA expression (normalized to GAPDH) by Q-PCR through ER stress-independent forcible activation of IRE1α and mutants, and forced expression of XBP1s, in INS-1 cells with 1μg/ml Dox and 5μM 1NM-PP1. (E) Time course analysis of TXNIP proteins (by immunoblot) following forced activation of IRE1α and mutants, and forced expression of XBP1s, in INS-1 cells untreated or treated with 1μg/ml Dox and 5μM 1NM-PP1. Three independent biological samples were used for Q-PCR experiments. Data are shown as mean ± SD. **p < 0.005, *p < 0.01.
IRE1α and PERK are two different ER transmembrane proteins that homo-oligomerize through an ER lumenal domain that senses unfolded proteins (Aragon et al., 2009; Credle et al., 2005; Gardner and Walter, 2011; Zhou et al., 2006). Both ER stress sensors have serine/threonine kinase activities on their cytosolic face. For both PERK and IRE1α, homo-oligomerization of ER lumenal domains juxtaposes their respective cytosolic kinase domains, and they consequently trans-autophosphorylate. For PERK, trans-autophosphorylation is a potentiating step that causes the kinase to subsequently phosphorylate the translation initiation factor, eIF2α, causing translational attenuation. We noted that forced dimerization using a chemical dimerizer of a FK506 binding protein-PERK eIF2α kinase construct is sufficient to induce TXNIP mRNA, without upstream ER stress (Figure S3B and C).
IRE1α is the more ancient of the two UPR sensors, and in addition to its kinase catalytic activity it contains an endoribonuclease (RNase) at its C-terminal end (Wang et al., 1998). For IRE1α, trans-autophosphorylation is a potentiating step that activates its RNase to initiate splicing of the mRNA encoding the XBP1 transcription factor. IRE1α-mediated splicing of XBP1 mRNA removes a 26 nucleotide intron, and alters the open reading frame (ORF); translated in the alternate ORF, spliced XBP1 mRNA encodes the XBP1s (s=spliced) transcription factor whose target genes enhance ER protein folding capacity (Figure S2B) (Calfon et al., 2002; Lee et al., 2003; Yoshida et al., 2001). Thus, by splicing XBP1 mRNA, IRE1α’s RNase promotes adaptation to ER stress. However, under irremediable ER stress, IRE1α’s RNase becomes hyperactive, and causes massive endonucleolytic degradation of ER-localized mRNAs and downstream c-Jun N-terminal kinase (JNK) phosphorylation to promote apoptosis (Han et al., 2009). Therefore, we decided to study if IRE1α uses TXNIP as an intermediary to trigger cell death under irremediable ER stress.
We previously developed tools through which we can forcibly activate IRE1α at will. Because IRE1α naturally activates through self-association in the ER membrane under ER stress, we can mimic this step by conditionally overproducing the protein from a transgene. In this situation, the transgenic IRE1α protein self-associates by mass action, without requiring upstream ER stress. Thus unlike pleiotropic ER stress-inducing agents that activate all arms of the UPR, our tools allow us to delineate the specific contribution of IRE1α to any UPR-linked physiological process. We decided to employ these tools to study the contribution of IRE1α’s catalytic activities to TXNIP induction (Figure 2C).
Expression of transgenic WT IRE1α (using doxycycline—Dox) causes the protein to spontaneously autophosphorylate as it accumulates (Figure 2E); this leads to complete conversion of cellular XBP1 mRNA to the spliced form (Figure S4C), as occurs under ER stress (Figure S2A)(Han et al., 2009). Activation of WT IRE1α through this maneuver is sufficient to induce TXNIP mRNA (Figure 2D and S4E) and protein (Figure 2F).
To dissect the effects of IRE1α’s catalytic activities on TXNIP upregulation, we tested two point mutants. The first, IRE1α (I642G), has an enlarged adenosine triphosphate (ATP)-binding pocket in its kinase domain that destroys phosphotransfer catalytic activity; the enlarged pocket can selectively bind 1NM-PP1, a cell-permeable adenosine nucleotide mimic with a bulky chemical head group (Figure 2C) (Han et al., 2008; Papa et al., 2003). Binding of 1NM-PP1 to IRE1α (I642G) allosterically activates the RNase domain, causing it to forcibly splice XBP1 mRNA, while bypassing the auto-phosphorylation requirement (Figure S4B and C). A second mutant, IRE1α (N906A), can properly autophosphorylate when expresssed, but because its RNase active site is mutated cannot splice XBP1 mRNA (Figures 2C, F and S4C). Interestingly, expression of IRE1α (N906A) leads to small, reproducible decreases in basal TXNIP mRNA (Figure 2D and Figure S4E) and protein (Figure 2F), consistent with its known dominant-negative effects against endogenous IRE1α. Furthermore, induction of either IRE1α (I642G)—or forced expression of spliced XBP1 transcription factor (XBP1s)—cause minimal elevation of TXNIP mRNA, without discernible changes in TXNIP protein (Figure 2D–G). These results argue that robust TXNIP induction requires both functional kinase and RNase catalytic activities of IRE1α, and is largely independent of XBP1 transcription factor activity. This last point was confirmed using Xbp1 −/− MEFs, in which production of TXNIP under ER stress is intact (Figure S3D). Indeed, TXNIP protein is detectable in Xbp1 −/− MEFs even under basal conditions, consistent with a previous observation that in the absence of XBP1, IRE1α becomes partially activated even without ER stress (Lee et al., 2008). In contrast, induction of TXNIP under ER stress is abrogated in Jnk1,2 −/− MEFs (Figure S3E), arguing that TXNIP regulation by IRE1α occurs downstream of JNK.
IRE1α utilizes a micro-RNA to control TXNIP levels
Transcriptional stimulation of TXNIP mRNA in response to increased glucose has been previously studied (Cha-Molstad et al., 2009; Yu and Luo, 2009). In response to elevated glucose levels, the transcription factor carbohydrate response element-binding protein (ChREBP) interacts with a consensus element in the TXNIP promoter to increase TXNIP transcription (Yu and Luo, 2009). We noted that ChREBP translocates to the nucleus, and binds TXNIP promoter elements under ER stress (Figure S5A and B). Furthermore, under ER stress, ChREBP mRNA itself increases about two-fold (Figure S5C). Luciferase reporter constructs containing variable TXNIP promoter regions, including two carbohydrate response elements (ChoREs) are activated in response to ER stress; but activation of the reporter constructs was never greater than about two-fold, in contrast to the robust induction that occurs under hyperglycemia (Figure S5D–F). Thus, while some contribution of de novo TXNIP transcription can be traced to known cis (ChORE) and trans (ChREBP) elements (Figure S5G–I), transcriptional activation is significantly weaker than under hyperglycemia, and hence insufficient to account for the robust increases in TXNIP mRNA to their new steady-states under ER stress. This implied either the existence of unidentified trans factors and/or cis elements that stimulate TXNIP transcription under ER stress, or that the induction is also due to processes other than transcription. To test this second possibility, we asked whether the rapid induction of TXNIP upon ER stress is due in part to changes in mRNA stability. By measuring mRNA half-life when transcription is arrested by Actinomycin D (ActD), we find that TXNIP mRNA is inherently labile, but that it becomes significantly stabilized (~ 3 fold) during ER stress (Figure 3A and B).
Figure 3. IRE1α Increases TXNIP mRNA Stability Through Decreasing miR-17.
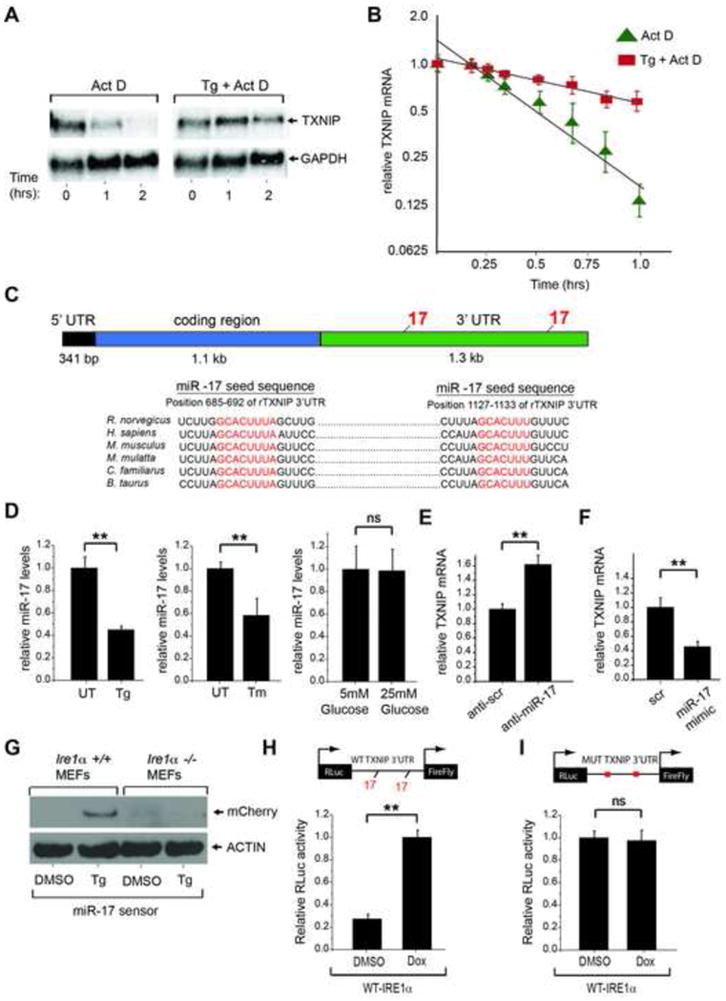
(A and B) Analysis by Northern blotting and Q-PCR shows that TXNIP mRNA is short lived, but becomes stabilized under ER stress. Total RNA extracts from INS-1 cells treated with 5 μg/ml Actinomycin D plus/minus 1 μM Tg were probed for TXNIP mRNA (or GAPDH). B) Early time course (1st hour) Q-PCR of TXNIP mRNA levels (relative to GAPDH) in INS-1 cells treated with 5 μg/ml Actinomycin D plus/minus 1 μM Tg with best fit line. (C) Schematic showing miR-17 binding sites within the 3′-UTR of TXNIP mRNA across multiple species. (D) Q-PCR of miR-17 levels from HEK293 cells untreated or treated with 1 μM Tg, or 5 μg/ml Tm, for 6 hours. (E) TXNIP mRNA levels as analyzed by Q-PCR from HEK293 cells 24 hrs post transfection with scrambled or miR-17 anti-miR. (F) TXNIP mRNA levels as analyzed by Q-PCR from HEK293 cells 24 hrs post transfection with scrambled or miR-17 mimic. (G) Immunoblot analysis of miR-17 mCherry sensor in wild-type and Ire1α−/− MEFs (36 post-transfection) following treatment with DMSO control or 1 μM Tg for 12 hours. (H) IRE1α induction of TXNIP luciferase reporter is dependent on miR-17 binding sites. Dox-inducible WT-IRE1α HEK293 cells were transfected (24 hours) with a luciferase reporter construct containing wild-type or miR-17 binding mutant TXNIP 3′-UTR. The cells were treated with DMSO control or 1μg/ml Dox for 24 hours, lysed and then analyzed for luciferase activity. Three independent biological samples were used for Q-PCR and luciferase experiments. Data are shown as mean ± SD. **p < 0.005, ns = not significant.
mRNA stability is often governed by binding of specific microRNAs to complementary sequences in the 3′-untranslated region (UTR) of gene targets (Fabian et al., 2010). Bioinformatic analysis of the TXNIP 3′-UTR identified two conserved binding sites for microRNA-17 (miR-17) (Figure 3C). This provoked the hypothesis that changes in miR-17 may regulate TXNIP mRNA stability. Consistent with this notion, we find that miR-17 levels rapidly decline under ER stress, but not under high glucose (Figure 3D). TXNIP mRNA levels can be increased by introducing anti-miR-17 into cells (Figure 3E); conversely a miR-17 mimic reduces baseline levels of TXNIP mRNA (Figure 3F).
We next used heterologous reporter systems to test the consequence of miR-17 reduction. We constructed a mCherry, red fluorescent protein (RFP) reporter that contains tandem miR-17 seed sequences in its 3′-UTR; the reporter is designed to express RFP when cellular miR-17 levels drop. Upon transfection into wild-type MEFs, the reporter becomes derepressed under ER stress to produce RFP, indicating reduction in endogenous miR-17 (Figure 3G). The reporter remains silenced in Ire1α−/− MEFs, indicating that IRE1α is necessary for reduction of miR-17 under ER stress. To further investigate whether IRE1α is sufficient for miR-17-dependent control of TXNIP, we constructed a luciferase reporter containing the entire TXNIP 3′-UTR, and a version mutated in the miR-17 seed sequences. Upon transfection of these reporters into dox-inducible WT-IRE1α cells (Figure 2C), induction with Dox increases baseline luciferase activity driven from the wild-type—but not the miR-17-mutant—TXNIP 3′-UTR reporter (Figure 3H). Together these results argue that activation of IRE1α increases TXNIP mRNA levels post-transcriptionally by reducing its inhibitory micro-RNA, miR-17. Rationalizing our results, a mathematical model (Figure S5J and K) shows that a combination of transcriptional and post-transcriptional control of TXNIP mRNA produces a sharper—and more rapid—rise to new steady-state levels upon ER stress than would occur through de novo mRNA synthesis alone.
Txnip deletion protects against ER stress-induced β-cell programmed cell death and diabetes
We next explored the physiological connection of TXNIP to ER stress-mediated cell degeneration and disease. Given that the loss of TXNIP protects against glucotoxicity, we tested whether it would similarly protect cells against ER stress-induced programmed cell death. To this end, we challenged Txnip−/− MEFs with ER stress agents and found that they are strikingly resistant to programmed cell death (Figure 4A), despite the fact that adaptive UPR outputs—XBP1 mRNA splicing and transcriptional induction of the ER chaperone BiP—are no different than in Txnip+/+ MEFs (Figure S6A and B). As with cell lines, freshly harvested pancreatic islets from wild-type C57BL/6 mice induce TXNIP mRNA under Tm (Figure 4B). However, β-cells in pancreatic islets from Txnip−/− mice are strongly protected (compared to Txnip+/+ mice) from programmed cell death under Tm (Figure 4C and D).
Figure 4. Loss of Txnip protects MEFs and pancreatic islets against ER stress-induced apoptosis.
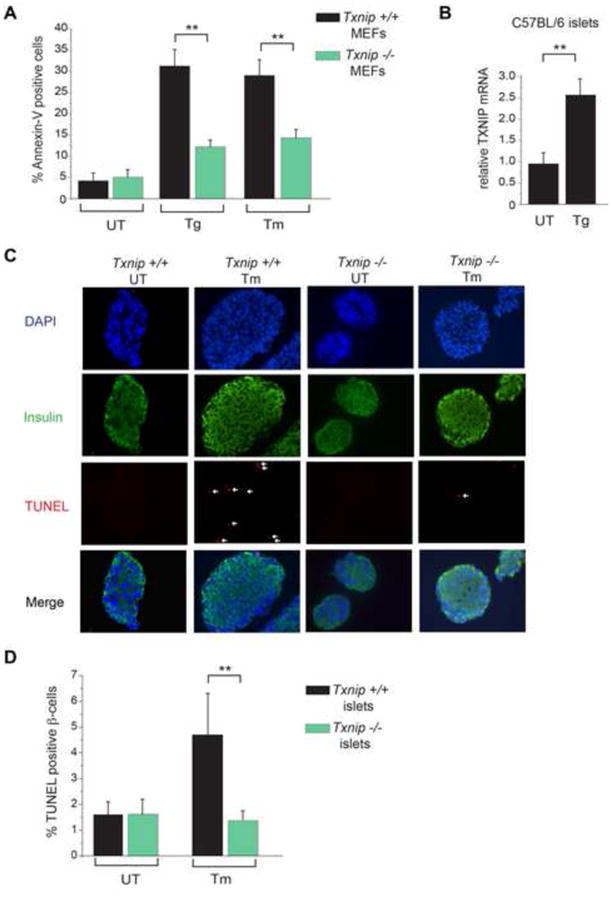
(A) Wild-type and Txnip−/− MEFs were challenged with 1 μM Tg or 5 μg/ml Tm for 24hrs and assessed for apoptosis by flow cytometry for Annexin-V binding. (B) Pancreatic islets were isolated from 6 week old C57BL/6 mice and left untreated or treated with 1 μM Tg for 6hrs. TXNIP mRNA (relative to GAPDH) was measured by Q-PCR. (C) Pancreatic islets were isolated from 6 week old Txnip+/+ and Txnip−/− mice, cultured in the absence or presence of 5 μg/ml Tm for 12hrs, and then subjected to DAPI, anti-insulin and TUNEL staining. (D) Quantification of TUNEL positive β-cells from C. Bar graphs represent three independent biological samples. All mice were on C57BL/6 genetic background. Data are shown as mean ± SD. **p < 0.005.
Considering the substantial cytoprotection enjoyed by Txnip−/− MEFs and islets against pharmacological inducers of ER stress, we next tested whether loss of TXNIP would ameliorate β-cell degeneration and development of diabetes in the Ins2WT/C96Y—“Akita”—mouse. Because INS2 (C96Y) proinsulin cannot form a critical intramolecular disulfide bond needed to fold in the ER, it accumulates as a proteotoxin that causes ER stress-induced β-cell loss and spontaneous diabetes during infancy (Oyadomari et al., 2002; Ron, 2002). Ins2WT/C96Y mice begin developing hyperglycemia at approximately 3 weeks of age, but are not frankly diabetic and can still dispose of a glucose load by glucose tolerance test (GTT) (Figure S6E). However, even at 3 weeks, islets from Ins2WT/C96Y mice display significantly elevated baseline IRE1α activation (~ two-fold increased XBP1 mRNA splicing), documenting elevated ER stress prior to development of frank diabetes (Figure 5 A). Furthermore, the IRE1α-TXNIP pathway is also activated in islets from Ins2WT/C96Y mice at 3 weeks of age, as evidenced by significantly decreased miR-17 levels and elevated TXNIP mRNA expression at baseline (Figure 5 B and C).
Figure 5. Txnip deficiency protects against β-cell loss and diabetes in the Ins2WT/C96Y mouse.
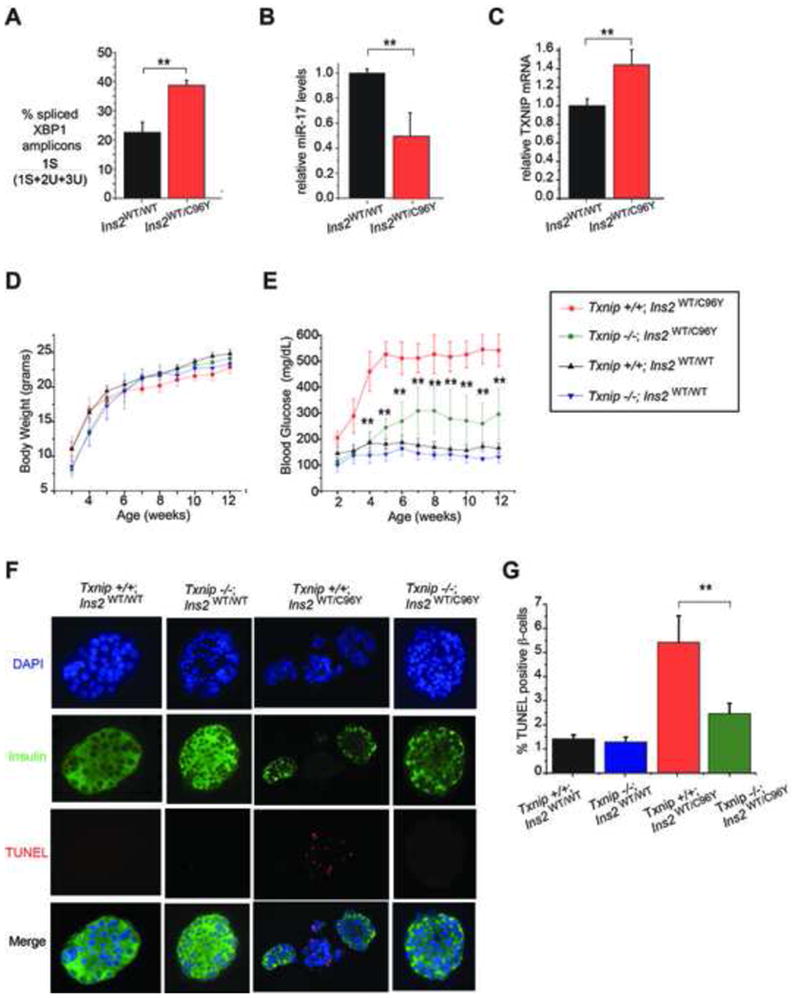
(A–C) Pancreatic islets from 3 week old Ins2WT/C96Y mice show evidence of ER stress at baseline, including increased XBP-1 splicing, decreased miR-17, and elevated TXNIP mRNA as assessed by Q-PCR. (D) Indicated genotypes showed no significant differences in body weight up to 12 weeks of age. For the 12 week timecourse, N=9 for Txnip+/+ Ins2WT/C96Y mice, N=10 for Txnip+/+Ins2WT/WT, and N=8 for both Txnip−/−Ins2WT/WT and Txnip−/−Ins2WT/C96Ymice. (E) Body glucose levels for indicated genotypes up to 12 weeks of age. Note that Txnip−/− Ins2WT/C96Ymice have significantly lower blood glucose levels compared to Txnip+/+ Ins2WT/C96Ymice at all time points. (F) Pancreatic islets were isolated from mice of indicated genotypes at 5 weeks of age and assessed by DAPI, anti-insulin and TUNEL staining. (G) Quantification of TUNEL positive β-cells from experiments in F. Bar graphs represent three independent biological samples. All mice were on C57BL/6 genetic background. Data are shown as mean ± SD. **p < 0.005.
We then crossed the Txnip−/− and Ins2WT/C96Y mice and followed β-cell apoptosis and development of diabetes in the various cohorts. While the different cohorts have no significant differences in body weight over time (Figure 5D), Txnip−/−; Ins2WT/C96Y mice are strikingly protected from hyperglycemia compared to Txnip+/+; Ins2WT/C96Y mice, for up to 12 weeks (Figure 5E). Moreover, Txnip−/−; Ins2WT/C96Y islets display significantly lower levels of β-cell apoptosis compared to islets from Txnip+/+; Ins2WT/C96Y mice (Figure 5F–G), confirming that TXNIP plays a critical role in promoting programmed β-cell death in this spontaneous ER stress model of diabetes.
Blocking TXNIP induction and IL-1β secretion through small molecule inhibition of IRE1α
We next explored the mechanistic bases of TXNIP-mediated cell death by turning our attention to the NLRP3 inflammasome. The NLRP3 inflammasome is a multi-protein complex that senses endogenous “danger” signals—also called damage associated molecular pattern molecules (DAMPs)—and leads to maturation and secretion of the pro-inflammatory cytokine, interleukin-1 β (IL-1β) (Strowig et al., 2012). TXNIP was recently discovered to bind and activate the NLRP3 inflammasome, and murine Txnip−/− islets are resistant to glucose-induced NLRP3 inflammasome activation and IL-1β secretion (Zhou et al., 2010).
Having connected ER stress to production of TXNIP, we specifically tested whether ER stress also causes production of IL-1β. Indeed, we find that Tg causes robust IL-1β secretion, as occurs during hyperglycemia in pancreatic islets (Figure 6A); or extracellular ATP, a well-known DAMP and NLRP3 inflammasome activator, in THP-1 macrophage cell lines (Figure 6B). We further tested known signaling events linking activation of the NLRP3 inflammasome to IL-1β production by DAMPs, and found that ER stress causes Caspase-1 cleavage from its zymogen form, as occurs with ATP (Figure 6C). The effects of ER stress on IL-1β appear to be largely post-transcriptional as only modest increases of IL-1β mRNA occur with Tg (Figure S7G). Furthermore, shRNA knock-down of the NLRP3 inflammasome abrogates Caspase-1 cleavage, and IL-1β production, under ER stress (Figure 6D and E), as does a specific inhibitor of Caspase-1, Z-YVAD-FMK (Figure S7H,I).
Figure 6. ER stress leads to IRE1 α-dependent TXNIP upregulation, NLRP3 inflammasome activation, Caspase-1 cleavage, and IL-1β secretion.
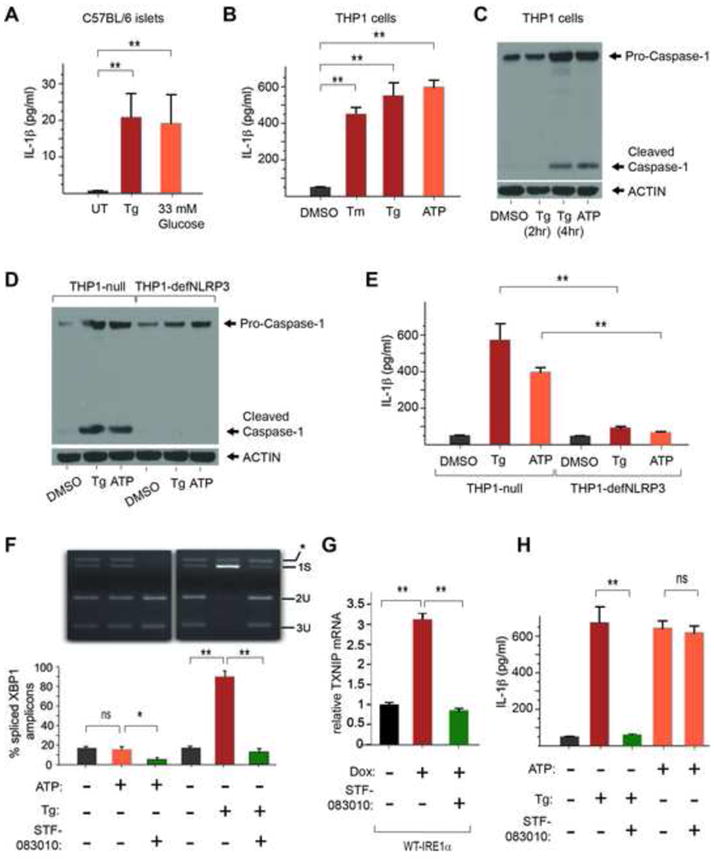
(A) IL-1β secretion from C57BL/6 murine islets exposed to 1μM Tg or 33 mM glucose. (B) IL-1β secretion from human THP-1 cells after 4hr treatment with DMSO control, 10μg/ml Tm, 1μM Tg, or 5 mM ATP as assessed by ELISA. (C) Caspase-1 cleavage from Pro-caspase-1 in THP_1 cells (detected by immunoblot) in response to ER stress 1μM Tg (at 2 hours and 4 hours), or 5 mM ATP at 4 hours. (D) Caspase-1 cleavage in response to ER stress (1μM Tg) is abrogated in THP-1 cells lacking the NLRP3 inflammasome (THP1-defNLRP3); compare to THP1-null positive control cells. Control DAMP, ATP, is at 5 mM. (E) (E) IL-1β secretion in response to ER stress (1μM Tg) is abrogated in THP-1 cells lacking the NLRP3 inflammasome (THP1-defNLRP3); compare to THP1-null positive control cells. Control DAMP ATP is at 5 mM. (F) STF-083010 blocks IRE1α RNase. Shown is an EtBr-stained agarose gel of XBP1 cDNA amplicons after induction of ER stress for 4 hrs in THP-1 cells using 1 μM Tg, with or without pre-treatment with STF-083010 at 50μM for 2 hours. The cDNA amplicon of unspliced XBP1 mRNA is cleaved by a PstI site within a 26 nucleotide intron to give 2U and 3U. IRE1α-mediated cleavage of the intron and re-ligation in vivo removes the PstI site to give the 1S (spliced) amplicon. *is a spliced/unspliced XBP1 hybrid amplicon. The ratio of spliced over (spliced + unspliced) amplicons—1S/(1S+2U+3U)—is reported as % spliced XBP1 amplicons. STF-083010 blocks: TXNIP mRNA upregulation in WT- IRE1α-overexpressing INS-1 cells (G), and IL-1β secretion from THP-1 cells (H). IL-1β secretion in response to 5mM ATP is unaffected by STF-083010. (E–F) Bar graphs represent three independent biological samples. Data are shown as mean ± SD. **p < 0.005, ns = not significant.
Finally, we reasoned that because IRE1α controls TXNIP induction, we may be able to reduce TXNIP and IL-1β by inhibiting IRE1α with small molecules. Recently, a cell-permeable small molecule—called STF-083010—capable of covalently inhibiting IRE1α RNase was described (Papandreou et al., 2011). We re-synthesized STF-083010 and tested its ability to prevent IRE1α activation. Figure 6F shows complete inhibition of IRE1α-mediated XBP1 mRNA splicing by STF-083010 when provided to cells before exposure to Tm. Note that treatment with ATP does not trigger ER stress, as evidenced by unchanged XBP1 mRNA splicing, but interestingly, STF-083010 can reduce basal levels of XBP1 mRNA splicing even in the ATP-treated cells. Strikingly, STF-083010 pre-treatment prevents production of TXNIP under forcible IRE1α activation (Figure 6G). Furthermore, provision of STF-083010 effectively shuts off secretion of IL-1β during treatment with Tg, but not ATP (Figure 6H).. This indicates that ER stress signals to the NLRP3 inflammasome can be specifically blocked by a small molecule targeting the proximal UPR sensor, IRE1α, while still allowing other DAMP signals to be relayed.
DISCUSSION
TXNIP is a signaling hub through which cells respond to irremediable ER stress
Cells expend considerable resources to maintain secretory homeostasis when ER stress levels fall within containable limits. Paradoxically when ER stress levels rise above critical thresholds, cells actively commit to programmed cell death. Robust signaling networks may force cells to make such a binary choice. We predicted the existence of signaling proteins that mediate destructive responses to catastrophic ER stress, and sought to discover such proteins using unbiased screens. Using a strategy to identify translational targets of the UPR (which was historically described and studied as a transcriptional pathway), we identified thioredoxin-interacting protein (TXNIP) as a critical mediator of cell death in response to catastrophic ER stress—a process we refer to as a terminal UPR.
TXNIP gene regulation is robustly wired into the terminal UPR. TXNIP up-regulation occurs rapidly when cells experience ER stress acutely at irremediable levels. Alternatively, chronic low-level ER stress in β-cells (e.g. due to Akita proinsulin) also increases TXNIP basal levels. While we discovered TXNIP as a translational target, its mRNA levels also climb greater than tenfold within 2 hours in a terminal UPR. Contemporaneously, TXNIP mRNA becomes loaded onto poly-ribosomes to begin translation. Intriguingly, the new steady-state level of TXNIP mRNA under ER stress is achieved through mRNA stabilization combined with de novo transcription. We found that TXNIP mRNA is inherently unstable, reminiscent of CHOP mRNA, which encodes a pro-apoptotic UPR transcription factor (Rutkowski et al., 2006), Furthermore, through a regulated event, TXNIP mRNA becomes stabilized under ER stress. Master regulatory proteins controlling switching into different cell states are often encoded by short-lived mRNAs, thus ensuring rapid interconversion of cell states. It is conceivable that other master regulators of the terminal UPR are encoded by short-lived mRNAs.
TXNIP mRNA stability during ER stress is under control of a specific micro-RNA, miR-17. miRs control gene expression at post-transcriptional levels by destabilizing target mRNAs and/or by repressing translation. Highly conserved seed sequences for miR-17 in the TXNIP 3′-UTR were found to govern post-transcriptional regulation of TXNIP mRNA under ER stress. Furthermore, steady-states levels of TXNIP mRNA could be predictably modulated: either down with a miR-17 mimic, or up with anti-miR-17. Forcible activation of IRE1α is sufficient to decrease cellular miR-17 levels, and endogenous IRE1α is necessary to decrease miR-17 under irremediable ER stress.
Opposite to its effects on miR-17, forcible activation of IRE1α is sufficient to induce TXNIP mRNA, even without ER stress, and endogenous IRE1α is necessary for TXNIP induction under irremediable ER stress. Thus, a parsimonious interpretation holds that IRE1α controls TXNIP mRNA levels—in part—post-transcriptionally by regulating levels of its repressive miR-17. We are investigating whether decreases in miR-17 proceed directly from endonucleolytic cleavage by IRE1α RNase, as we found for decay of ER-localized mRNAs (Han et al., 2009).
TXNIP was previously identified as a transcriptional target of the ChREBP transcription factor in response to elevated carbohydrate and adenosine nucleotides (Minn et al., 2005; Yu and Luo, 2009). While significant, ChREBP has a modest effect on TXNIP transcription under ER stress: ChREBP undergoes nuclear translocation and ChREBP mRNA increases slightly (Figure S5). ER stress may overlap with hyperglycemic signals that activate ChREBP; intriguingly, IRE1α is also partially activated by adenosine nucleotides or hyperglycemia (Lipson et al., 2006). An accompanying manuscript from Fumihiko Urano’s lab also explores PERK and ChREBP transcriptional control of TXNIP, which may be complementary to IRE1α’s post-transcriptional control. Combining post-transcriptional mRNA stabilization with transcriptional synthesis may allow cells to robustly and rapidly commit to self-destruction under high ER stress. Control of gene expression under ER stress through micro RNAs may be widespread, as in other biological processes.
Physiological effects of TXNIP and small molecule modulation through IRE1 α
TXNIP action has been implicated in diverse physiological and pathological contexts. TXNIP was first described as an inhibitor of thioredoxin, an anti-oxidant enzyme that catalyzes cysteine-thiol disulfide exchange (Nishiyama et al., 1999; Patwari et al., 2006; Yamanaka et al., 2000). Increased TXNIP levels renders cells susceptible to oxidative stress. Thus, we predicted, and confirmed, that increasing TXNIP levels would generate reactive oxygen species (ROS); we further predicted, and confirmed, that IRE1α hyper-activation, or irremediable ER stress, would also spontaneously generate ROS (Figure S7A–F). As ROS enhance activation of NLRP3 inflammasome, they may further amplify effects of the IRE1α-TXNIP node to increase sterile inflammation.
TXNIP levels are elevated in the muscle of diabetic humans and mice (Parikh et al., 2007), and TXNIP-deficient mice have increased adiposity while remaining insulin-sensitive (Hui et al., 2004). TXNIP is strongly induced in response to glucotoxicity, and promotes apoptosis of β-cells (Chen et al., 2008; Shalev, 2008). A recent study linked glucose toxicity and oxidative stress through TXNIP to downstream activation of the NLRP3 inflammasome and secretion of IL-1β (Zhou et al., 2010). Here we found that the loss of TXNIP prevents β-cell apoptosis and diabetes caused by ER stress in the Akita mouse. Thus through a link to the NLRP3 inflammasome, TXNIP may be well-positioned to mediate both cell autonomous and nonautonomous destructive responses to diverse DAMPs, including unfolded proteins in the ER.
IRE1α and PERK are both upstream UPR master kinases whose activation levels correlate directly with ER unfolded protein levels. Together, IRE1α and PERK may utilize the levels and duration of autophosphorylation to control homeostatic-apoptotic switching. Under low/remediable levels of ER stress, self-association of IRE1α through its lumenal domain extinguishes as adaptive UPR outputs from XBP1s re-establish homeostasis (Figure 7A). However, under irremediable ER stress, high-order oligomerization through IRE1α’s lumenal domains leads to kinase hyper-phosphorylation, and acquisition of relaxed specificity in the RNase. Endonucleolytic destruction of RNAs localizing to the ER membrane during co-translational translocation in close proximity to hyperactive IRE1αRNase occurs under irremediable ER stress, and is mimicked when overexpressing WT- IRE1α; instead, forcible activation of IRE1α (I642G) under 1NM-PP1 more closely mimics an adaptive UPR, primarily constrained to XBP1 splicing. Thus IRE1α RNase hyperactivation leading to ER-localized mRNA decay actually amplifies and promotes ER stress-mediated cell death (Han et al., 2009)(Figure S4D). Opposite to its direct effects on destabilizing ER-localized mRNAs, IRE1α RNase activation may also cleave specific miRs, and in doing so indirectly stabilize specific mRNA targets needed to promote cell death. In this scenario, adaptive outputs through XBP1 mRNA splicing may become eclipsed and irrelevant as destructive IRE1α signaling dominates in a terminal UPR (Figure 7B). A therapeutic strategy to shut down IRE1α RNase entirely should therefore reduce destructive outputs under irremediable ER stress. Consistent with this notion, the tool compound STF-083010, which selectively targets the IRE1α RNase activity (Papandreou et al., 2010), markedly reduces TXNIP induction and downstream IL-1β maturation and secretion. We interpret these results as proof-of-concept that targeting the hyperactive IRE1α RNAse can disrupt cell destructive endpoints in the terminal UPR. It is likely that the active component of STF-083010 is the salicylaldehyde that rapid hydrolysis of its sulfonylimine unmasks (Volkmann et al., 2011). Aldehydes are inherently unstable in vivo and may limit the utility of this compound class. The future development of more drug-like inhibitors will allow these concepts to be effectively explored in vivo for amelioration of ER stress disease endpoints.
Figure 7. Illustrative models of adaptive (A) and terminal (B) UPR signaling.
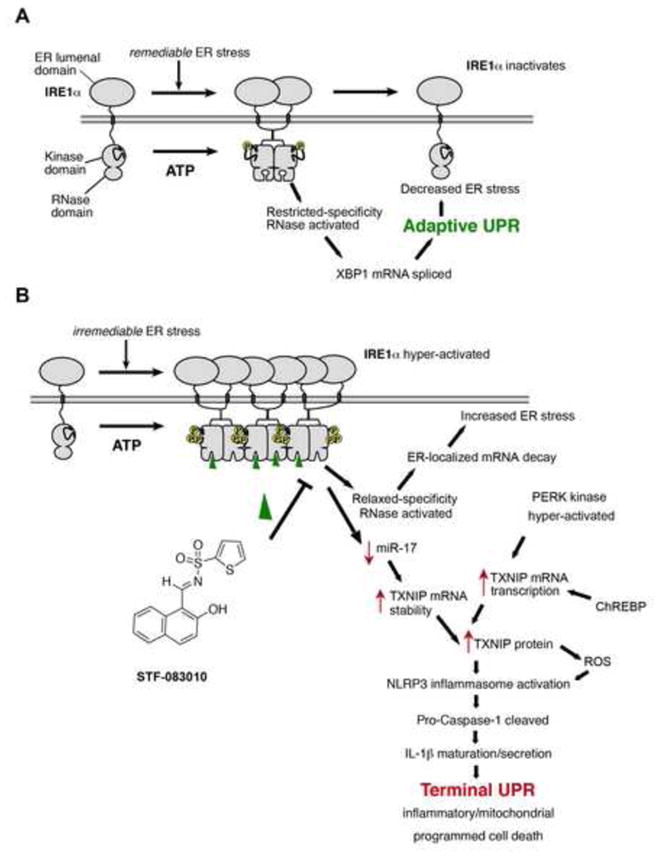
A. Under remediable levels of ER stress, adaptive UPR outputs through XBP1 mRNA splicing reduces ER stress, in turn closing negative feedback loops to shut down low-level IRE1α signaling. B. Alternatively, under irremediable levels of ER stress, hyperactivated IRE1α induces TXNIP as a potentiating step in the Terminal UPR, in part through stabilizing TXNIP mRNA by reducing levels of a repressive miR that targets TXNIP mRNA. This event combines with de novo transcription of TXNIP, through PERK kinase and ChREBP, to result in rapid elevation of TXNIP mRNA to new steady-state levels. TXNIP protein activates the NLRP3 inflammasome, which cleaves Pro-Caspase-1 to its active form, in turn causing maturation and secretion of Interleukin 1-β (IL-1β), thus promoting sterile inflammation and programmed cell death. Moreoever, ER-localized mRNA decay by hyperactivated IRE1α (requiring both a functional kinase and RNase activity) furthers—rather than corrects—ER stress, thus promoting vicious cycles of cell destruction. Also shown is the RNAse inhibitor—STF-083010—which reduces terminal UPR endpoints by inhibiting IRE1α RNAse activity.
For multi-cellular organisms, the sacrifice of irremediably stressed cells through programmed cell death is an ultimate and fail-safe method to ensure protein quality control, and thus to maintain health. Yet over-zealous cell death may cause organ failure and expose organisms to the risk of cell degenerative diseases, such as diabetes mellitus. Many cell degenerative diseases, including diabetes mellitus and some neurodegenerative diseases, are now thought to occur in part from UPR dysregulation. Drug target validation of the IRE1α-TXNIP-IL-1β chain may ultimately lead to therapeutic advances for such diseases.
EXPERIMENTAL PROCEDURES
Immunoaffinity purification of poly-ribosomal mRNA
INS-1-EGFP or INS-1-EGFP-L10a stable cell lines were induced with doxycycline (Dox), treated with cycloheximide, washed with PBS, and lysed with 20mM HEPES [pH 7.4], 150mM KCL, 2M MgCl2, 1% NP-40. Lysates were homogenized in ice-cold polysome extraction buffer and homogenates clarified. Immunoaffinity-purification of polysomal RNA used goat anti-GFP (Nathaniel Heintz, Rockefeller Univ.), which was precipitated, resuspended, and quantified for DNA microarray experiments (See Supp methods for further details).
Chemical-genetic cell lines
INS-1 cells with doxycycline inducible expression of EGFP or EGFP-L10a fusion were generated from INS-1/FRT/TO cells (Thomas et al., 2004), as were the previously described IRE1α chemical-genetic variants and XBP1s-expressing cell lines (Han et al., 2009). See Supp methods for further details on induction of transgenic proteins.
Detection of IL-1β
Human THP-1 cells were grown in RPMI-1640 media supplemented with 10% (vol/vol) FBS and 50 μM 2-mercaptoethanol (Sigma # M3148). THP-1 cells were differentiated for 2 hrs with 0.5 μM phorbol-12-myristate-13-acetate (Sigma # P8139). Differentiated THP-1 cells were primed for 18 h with ultrapure lipopolysaccharide (LPS; 1 μg/ml, Sigma #L5293). THP-1 cell culture media was changed to media without LPS and treated with ATP (5 mM, Roche, # 11162306001), or Tg (1 μM Sigma # T9033) for 4 hrs. THP-1 cells were untreated or treated with 50 μM STF-083010 for 2 hr prior to the addition of Tg (1 μM) or Tm (10 μg/ml, Sigma, # T7765) and allowed to incubate for 4 hrs. After 4 hrs the media supernatant was collected and assayed for hIL-1β by ELISA (#EH2IL1B from Thermo Scientific). Further variants of THP-1 cells used in this study were from InvivoGen: THP1-defNLRP3, deficient in NLRP3; and THP1-Null, which is a positive control line proficient for inflammasome function.
Western Blots and Antibodies
For protein analysis, cells were lyzed in 1x M-PER buffer (#78501, Pierce) plus 10U protease inhibitor (#P840 from Sigma) and 250 μM sodium fluoride (#S299-100, Fisher Scientific). The protein concentration of samples was determined using a Thermo BCA Assay. Western blots were performed using the Invitrogen XCell SureLock® Mini-Cell and XCell II™ Blot Module (#EI0002) plus NuPage 10% (#NP0315BOX) and 12% (#NP0341BOX) Bis-Tris precast gels. Gels were run using MES buffer (#NP0002) and transferred onto Immobilin-P transfer membrane (IPVH07850 from Millipore) using a XCell II™ Blot Module (#EI9051). See Supp methods for details on antibodies, dilutions, and detection.
RNA isolation, RT-qPCR, and Primers
RNA was isolated from whole cells using either the Qiagen RNeasy kit (#74104), or the ZR RNA Mini-prep kit (Zymo Research, #R1064). See Supp methods for details on primer sequences and quantitative PCR.
Flow Cytometry
For assaying apoptosis by Annexin V staining, cells were plated two days prior to FACS in 6-well plates. The day before flow cytometry, MEFs were induced with either 5μg/ml Tm or 1μM Tg. The next day, cells were trypsinized and washed in PBS and resuspended in Annexin V binding buffer with Annexin-V FITC (#K101-100, Biovision.). Flow cytometry was performed on a Becton Dickinson LSRII flow cytometer.
Animal Studies
C567BL/6 and C57BL/6 Ins2 WT/C96Y were obtained from Jackson Laboratories. Txnip−/− mice were generated as previously described (Hui et al., 2008). Txnip−/−; Ins2 WT/C96Y mice were generated by breeding Txnip−/− and Ins2 WT/C96Y mice and are both on the C57BL/6 background. All procedures described involving animals were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee at the University of California, San Francisco. Animals were maintained in a specific pathogen-free animal facility on a 12-h light–dark cycle at an ambient temperature of 21 °C. They were given free access to water and food. All experiments used age-matched male mice.
Supplementary Material
Highlights.
Under high ER stress, cells undergo programmed cell death.
The ER stress sensor IRE1α triggers cell death by inducing TXNIP.
TXNIP induction by IRE1α utilizes micro RNA control.
Inhibition of TXNIP induction under ER stress reduces diabetes.
Acknowledgments
We thank the Gladstone Histology Core and UCSF Nikon Center. We thank M. Hebrok, P. Muchowski, and members of the Papa and Oakes labs for comments. This work was supported by NIH: Director’s New Innovator Award DP2 OD001925 (F.R.P), RO1 DK080955 (F.R.P), RO1 CA136577 (S.A.O.), NIDDK/Beta Cell Biology Consortium (BCBC)— U01DK089541 (F.R.P); NIDDK P30 DK063720 (DERC Islet Core); and Research Scholar Grant RSG-12-068-01 from the American Cancer Society (S.A.O.) and Burroughs Wellcome Foundation (F.R.P.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aragon T, van Anken E, Pincus D, Serafimova IM, Korennykh AV, Rubio CA, Walter P. Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature. 2009;457:736–740. doi: 10.1038/nature07641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Cha-Molstad H, Saxena G, Chen J, Shalev A. Glucose-stimulated expression of Txnip is mediated by carbohydrate response element-binding protein, p300, and histone H4 acetylation in pancreatic beta cells. J Biol Chem. 2009;284:16898–16905. doi: 10.1074/jbc.M109.010504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Saxena G, Mungrue IN, Lusis AJ, Shalev A. Thioredoxin-interacting protein: a critical link between glucose toxicity and beta-cell apoptosis. Diabetes. 2008;57:938–944. doi: 10.2337/db07-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Credle JJ, Finer-Moore JS, Papa FR, Stroud RM, Walter P. Inaugural Article: On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2005;102:18773–18784. doi: 10.1073/pnas.0509487102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–379. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- Gardner BM, Walter P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science. 2011;333:1891–1894. doi: 10.1126/science.1209126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gething MJ, Sambrook J. Transport and assembly processes in the endoplasmic reticulum. Semin Cell Biol. 1990;1:65–72. [PubMed] [Google Scholar]
- Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, Backes BJ, Oakes SA, Papa FR. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Upton JP, Hagen A, Callahan J, Oakes SA, Papa FR. A kinase inhibitor activates the IRE1alpha RNase to confer cytoprotection against ER stress. Biochem Biophys Res Commun. 2008;365:777–783. doi: 10.1016/j.bbrc.2007.11.040. [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Bertolotti A, Zeng H, Zhang Y, Urano F, Jousse C, Ron D. Translational regulation in the cellular response to biosynthetic load on the endoplasmic reticulum. Cold Spring Harb Symp Quant Biol. 2001;66:499–508. doi: 10.1101/sqb.2001.66.499. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase [published erratum appears in Nature 1999 Mar 4;398(6722):90] [see comments] Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suarez-Farinas M, Schwarz C, Stephan DA, Surmeier DJ, et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui ST, Andres AM, Miller AK, Spann NJ, Potter DW, Post NM, Chen AZ, Sachithanantham S, Jung DY, Kim JK, et al. Txnip balances metabolic and growth signaling via PTEN disulfide reduction. Proc Natl Acad Sci U S A. 2008;105:3921–3926. doi: 10.1073/pnas.0800293105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui TY, Sheth SS, Diffley JM, Potter DW, Lusis AJ, Attie AD, Davis RA. Mice lacking thioredoxin-interacting protein provide evidence linking cellular redox state to appropriate response to nutritional signals. J Biol Chem. 2004;279:24387–24393. doi: 10.1074/jbc.M401280200. [DOI] [PubMed] [Google Scholar]
- Jousse C, Bruhat A, Carraro V, Urano F, Ferrara M, Ron D, Fafournoux P. Inhibition of CHOP translation by a peptide encoded by an open reading frame localized in the chop 5′UTR. Nucleic Acids Res. 2001;29:4341–4351. doi: 10.1093/nar/29.21.4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320:1492–1496. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson KL, Fonseca SG, Ishigaki S, Nguyen LX, Foss E, Bortell R, Rossini AA, Urano F. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab. 2006;4:245–254. doi: 10.1016/j.cmet.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Merksamer PI, Papa FR. The UPR and cell fate at a glance. J Cell Sci. 2010;123:1003–1006. doi: 10.1242/jcs.035832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merksamer PI, Trusina A, Papa FR. Real-Time Redox Measurements during Endoplasmic Reticulum Stress Reveal Interlinked Protein Folding Functions. Cell. 2008;135:933–947. doi: 10.1016/j.cell.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AH, Hafele C, Shalev A. Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces beta-cell apoptosis. Endocrinology. 2005;146:2397–2405. doi: 10.1210/en.2004-1378. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Matsui M, Iwata S, Hirota K, Masutani H, Nakamura H, Takagi Y, Sono H, Gon Y, Yodoi J. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J Biol Chem. 1999;274:21645–21650. doi: 10.1074/jbc.274.31.21645. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, Mori M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest. 2002;109:525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palam LR, Baird TD, Wek RC. Phosphorylation of eIF2 Facilitates Ribosomal Bypass of an Inhibitory Upstream ORF to Enhance CHOP Translation. J Biol Chem. 2011;286:10939–10949. doi: 10.1074/jbc.M110.216093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa FR, Zhang C, Shokat K, Walter P. Bypassing a kinase activity with an ATP-competitive drug. Science. 2003;302:1533–1537. doi: 10.1126/science.1090031. [DOI] [PubMed] [Google Scholar]
- Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, Solow-Cordero DE, Bouley DM, Offner F, Niwa M, et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood. 2010;117:1311–1314. doi: 10.1182/blood-2010-08-303099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, Solow-Cordero DE, Bouley DM, Offner F, Niwa M, et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood. 2011;117:1311–1314. doi: 10.1182/blood-2010-08-303099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh H, Carlsson E, Chutkow WA, Johansson LE, Storgaard H, Poulsen P, Saxena R, Ladd C, Schulze PC, Mazzini MJ, et al. TXNIP regulates peripheral glucose metabolism in humans. PLoS Med. 2007;4:e158. doi: 10.1371/journal.pmed.0040158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwari P, Higgins LJ, Chutkow WA, Yoshioka J, Lee RT. The interaction of thioredoxin with Txnip. Evidence for formation of a mixed disulfide by disulfide exchange. J Biol Chem. 2006;281:21884–21891. doi: 10.1074/jbc.M600427200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D. Proteotoxicity in the endoplasmic reticulum: lessons from the Akita diabetic mouse. J Clin Invest. 2002;109:443–445. doi: 10.1172/JCI15020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, Mori K, Sadighi Akha AA, Raden D, Kaufman RJ. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4:e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocr Rev. 2008;29:317–333. doi: 10.1210/er.2007-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalev A. Lack of TXNIP protects beta-cells against glucotoxicity. Biochem Soc Trans. 2008;36:963–965. doi: 10.1042/BST0360963. [DOI] [PubMed] [Google Scholar]
- Shalev A, Pise-Masison CA, Radonovich M, Hoffmann SC, Hirshberg B, Brady JN, Harlan DM. Oligonucleotide microarray analysis of intact human pancreatic islets: identification of glucose-responsive genes and a highly regulated TGFbeta signaling pathway. Endocrinology. 2002;143:3695–3698. doi: 10.1210/en.2002-220564. [DOI] [PubMed] [Google Scholar]
- Shore GC, Papa FR, Oakes SA. Signaling cell death from the endoplasmic reticulum stress response. Curr Opin Cell Biol. 2011;23:143–149. doi: 10.1016/j.ceb.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, Below JE, Hayes MG, Cox NJ, Lipkind GM, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci U S A. 2007;104:15040–15044. doi: 10.1073/pnas.0707291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481:278–286. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- Thomas H, Senkel S, Erdmann S, Arndt T, Turan G, Klein-Hitpass L, Ryffel GU. Pattern of genes influenced by conditional expression of the transcription factors HNF6, HNF4alpha and HNF1beta in a pancreatic beta-cell line. Nucleic Acids Res. 2004;32:e150. doi: 10.1093/nar/gnh144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes and Development. 1998;12:1812–1824. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9:944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmann K, Lucas JL, Vuga D, Wang X, Brumm D, Stiles C, Kriebel D, Der-Sarkissian A, Krishnan K, Schweitzer C, et al. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J Biol Chem. 2011;286:12743–12755. doi: 10.1074/jbc.M110.199737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Takeuchi T, Tanaka S, Kubo SK, Kayo T, Lu D, Takata K, Koizumi A, Izumi T. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J Clin Invest. 1999;103:27–37. doi: 10.1172/JCI4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XZ, Harding HP, Zhang Y, Jolicoeur EM, Kuroda M, Ron D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 1998;17:5708–5717. doi: 10.1093/emboj/17.19.5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka H, Maehira F, Oshiro M, Asato T, Yanagawa Y, Takei H, Nakashima Y. A possible interaction of thioredoxin with VDUP1 in HeLa cells detected in a yeast two-hybrid system. Biochem Biophys Res Commun. 2000;271:796–800. doi: 10.1006/bbrc.2000.2699. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors [published erratum appears in J Biol Chem 1999 Jan 22;274(4):2592] Journal of Biological Chemistry. 1998;273:33741–33749. doi: 10.1074/jbc.273.50.33741. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Yu FX, Luo Y. Tandem ChoRE and CCAAT motifs and associated factors regulate Txnip expression in response to glucose or adenosine-containing molecules. PLoS ONE. 2009;4:e8397. doi: 10.1371/journal.pone.0008397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Liu CY, Back SH, Clark RL, Peisach D, Xu Z, Kaufman RJ. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc Natl Acad Sci U S A. 2006;103:14343–14348. doi: 10.1073/pnas.0606480103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.