

Semiempirical quantum-chemical X-ray macromolecular refinement using the program DivCon integrated with PHENIX is described.
Keywords: X-ray crystallography, quantum-mechanics refinement, regional refinement, stereochemical restraints, ligand strain, high-throughput crystallography
Abstract
Macromolecular crystallographic refinement relies on sometimes dubious stereochemical restraints and rudimentary energy functionals to ensure the correct geometry of the model of the macromolecule and any covalently bound ligand(s). The ligand stereochemical restraint file (CIF) requires a priori understanding of the ligand geometry within the active site, and creation of the CIF is often an error-prone process owing to the great variety of potential ligand chemistry and structure. Stereochemical restraints have been replaced with more robust functionals through the integration of the linear-scaling, semiempirical quantum-mechanics (SE-QM) program DivCon with the PHENIX X-ray refinement engine. The PHENIX/DivCon package has been thoroughly validated on a population of 50 protein–ligand Protein Data Bank (PDB) structures with a range of resolutions and chemistry. The PDB structures used for the validation were originally refined utilizing various refinement packages and were published within the past five years. PHENIX/DivCon does not utilize CIF(s), link restraints and other parameters for refinement and hence it does not make as many a priori assumptions about the model. Across the entire population, the method results in reasonable ligand geometries and low ligand strains, even when the original refinement exhibited difficulties, indicating that PHENIX/DivCon is applicable to both single-structure and high-throughput crystallography.
1. Introduction
Structure-based drug discovery (SBDD) utilizes the three-dimensional structure of a drug target, such as an enzyme, complexed with one or more ligands or lead compounds in order to inform the drug-discovery process. The structure serves as the three-dimensional template for screening, docking and scoring (e.g. rank ordering) potential drug candidates, and hence the quality of the model is crucial to overall success. X-ray crystallography is the primary technique used to determine the three-dimensional structure of molecular biochemical systems, and owing to recent advances in data collection, processing, structure solution and refinement automation, X-ray crystallography has become fairly routine. The quality and correctness of the final model is dependent both on the quality of the experimental data (e.g. resolution, twinning, mosaicity etc.), as well as human factors such as the expertise and abilities of the investigator and the robustness of the refinement methodology, including the force field and the initial ligand placement (Pozharski et al., 2013 ▶). During the refinement process, the parameters of the macromolecular model, including atomic coordinates and temperature factors, are optimized to achieve the best possible agreement between the observed experimental structure factors and the chosen target function (e.g. maximum likelihood or least squares; Murshudov et al., 1997 ▶, 2011 ▶; Tronrud, 2004 ▶). Ideally, the refinement protocol should provide satisfactory structure geometry regardless of data quality.
A significant challenge in obtaining a reasonable fit during macromolecular refinement is that the model parameters are typically underdetermined in the macromolecular diffraction experiment owing to resolution limitations. At moderate and low resolutions, the observation-to-parameter ratio is low and ranges from 4 to 1, and hence a priori information about the structure such as its stereochemistry is usually required. Stereochemical restraints represent a harmonic oscillator function (1) that implies a penalty (Evans, 2007 ▶) Q P for a deviation of the geometric parameter P from its ideal value. In typical X-ray refinement, geometry parameters include bond lengths, bond angles and torsion angles as well as chirality and group planarity requirements.
Conventional refinement methods use fixed stereochemical restraints for any components included within the protein–ligand complex. For standard amino acids, these fixed stereochemical restraints are based on the ideal Engh and Huber parameters (Engh & Huber, 1991 ▶), and these restraints often promote fairly satisfactory macromolecular geometry. However, the situation becomes less certain when a small molecule (a ligand, an inhibitor and/or a metallic or non-metallic cofactor) is bound to a protein (Kleywegt, 2007 ▶). The conventional refinement approach requires a detailed description of the molecular geometry for each small molecule to be refined, and thus creating a good library or Crystallographic Information File (CIF) is extremely important to the ultimate success of the effort. Unfortunately, the creation of such a dictionary is not a trivial task because of the great variety of potential ligand chemistry and structure. When such a species is involved in the model, a correct structure requires an a priori understanding of the bond lengths and bond angles of the species within the active site, and these methods are unable to account for influences including coordination, bond making/breaking and protein-induced or environment-induced structural perturbations. Furthermore, compared with modern molecular-mechanics and quantum-mechanics methods, the functionals used in conventional refinement are rudimentary in nature and do not account for interactions such as hydrogen bonds, dispersion, electrostatics, polarization and charge transfer. In order to address these deficiencies, and since it is impossible to understand all of these influences prior to refinement, conventional refinement often requires a repeated, highly interactive, investigator-driven cycle leading to greater expense, especially in industrial laboratories. According to a recent PDB survey, the percentage of ligands having questionable geometric parameters in deposited macromolecular structures could be as high as 60% (Liebeschuetz et al., 2012 ▶; Gore et al., 2011 ▶). Overall, this survey has suggested that conventional macromolecular refinement protocols based on fixed, pre-refinement stereochemical restraints have often failed to provide a reasonable structure of small-molecule ligands. Enhancements in PDB validation tools would certainly help in detecting problematic ligands, but such tools only serve to point out problems and they do not directly improve structure. The PDB_REDO project (Joosten et al., 2012 ▶) uses conventional refinement methods to clean up these structures, but this project is still limited to the use of CIFs, other empirical parameters and a rudimentary energy functional.
To overcome the limitations associated with conventional refinement, it has been proposed (Yu et al., 2005 ▶) that a quantum-mechanical (QM) method can be substituted for the stereochemical restraint function. This proof-of-concept study clearly demonstrated that QM macromolecular refinement using QM gradients generated in real time can be successfully carried out using a combination of the QM and molecular-mechanics (MM) functionals from AMBER (Case et al., 2010 ▶) with the Crystallography & NMR System (CNS; Brünger et al., 1998 ▶) refinement platform. While this work was groundbreaking, a number of key limitations impact the accessibility of the method. In particular, in the years since this implementation was put into place, CNS has been largely replaced in the industrial field. Further, in this previous approach, solvent molecules were excluded from the model, and the bulk-solvent correction and temperature factors of protein atoms were not refined. The method also did not allow multiple QM regions, alternative atom positions and so on. In the present work, we present a new, streamlined QM X-ray refinement implementation based on the popular, modern crystallographic package PHENIX (Adams et al., 2010 ▶; Afonine et al., 2012 ▶). QuantumBio’s linear scaling, semiempirical QM C++/Python toolkit libQB, upon which DivCon (Dixon & Merz, 1996 ▶, 1997 ▶) is built, has been integrated with PHENIX, resulting in a user-friendly tool referred to as PHENIX/DivCon. Unlike conventional refinement, this method does not require CIF generation using a priori knowledge of the expected outcome, and since the QM functional is by definition more rigorous then the conventional functional, PHENIX/DivCon can be applied to complex protein–ligand structures, including metal-containing systems and structures with covalently bound ligands, without specifying coordination-sphere parameters, link restraints and the like.
2. Methods
2.1. DivCon discovery suite
DivCon employs divide-and-conquer (D&C), linear scaling, semiempirical quantum-mechanics (SE-QM) methods as described previously (van der Vaart, Gogonea et al., 2000 ▶; van der Vaart, Suarez et al., 2000 ▶; Wang et al., 2007 ▶; Dixon & Merz, 1996 ▶, 1997 ▶). The D&C methodology makes routine QM characterization of protein–ligand systems with thousands or even tens of thousands of atoms feasible, and it has been applied to a number of key SBDD applications including a patented QMScore (Merz & Raha, 2011 ▶; Diller et al., 2010 ▶; Raha & Merz, 2005 ▶; Zhang et al., 2010 ▶) along with an NMRScore (Williams et al., 2009 ▶; Wang et al., 2004 ▶, 2007 ▶), QM-based QSAR (Peters & Merz, 2006 ▶; Dixon et al., 2005 ▶; Zhang et al., 2010 ▶) and X-ray refinement (Li et al., 2010 ▶; Yu et al., 2005 ▶). While DivCon includes support for modern Hamiltonians such as PM6 (Řezáč et al., 2009 ▶; Stewart, 2009 ▶) and soon PM7 (Stewart, 2013 ▶; Hostaš et al., 2013 ▶), in order to better compare with the state of the art in CIF generation such as the program eLBOW (Moriarty et al., 2009 ▶), for the present project the traditional AM1 (Dewar et al., 1985 ▶) Hamiltonian was chosen for all SE-QM calculations (except when otherwise indicated).
2.2. QM X-ray refinement
The typical refinement protocol in PHENIX involves fitting bulk-solvent parameters and anisotropic scaling, atomic coordinate refinement, temperature-factor refinement and occupancy refinement. These stages are executed in sequential order as depicted in the flow chart in Fig. 1 ▶, and are repeated for each refinement macro-cycle. The overall refinement target E total in PHENIX is usually presented as follows:
where Ωxray and Ωgeom are weights assigned to X-ray data and geometry (stereochemical) restraints, respectively, and wcxscale is the additional scale factor implemented in PHENIX (Afonine et al., 2012 ▶).
Figure 1.
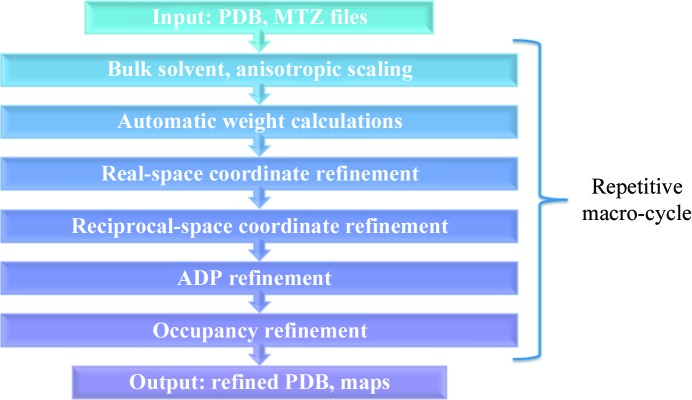
Refinement flow-chart in PHENIX.
The macro-cycle is broken into a number of minimization-driven micro-cycles, each of which, in PHENIX, is built on top of a minimization engine from the quasi-Newton family of minimizers called limited memory–Broyden–Fletcher–Goldfarb–Shanno (L-BFGS; Liu & Nocedal, 1989 ▶). This engine takes the function to be minimized (e.g. the coordinates) and the corresponding gradients (e.g. the atomic forces) as input and determines the approximate inverse Hessian matrix. The Hessian matrix is a matrix of second derivatives of energy with respect to nuclear motion, and it determines the direction of the minimum search. The matrix is iteratively updated at each step based upon updated gradients. This reliance on gradients underscores the importance of high-quality gradients in the minimization. During the coordinate-refinement stage in PHENIX, gradients on each atom in the structure are evaluated both from X-ray data (e.g. X-ray gradients) and from the stereochemical restraint function (e.g. geometry gradients). X-ray and geometry gradients are then added together, taking into account the corresponding weights, and the sum of the gradients is provided to L-BFGS. For each macro-cycle there are generally 15–30 L-BFGS iterations, and there are three (default) macro-cycles in the entire refinement.
In SBDD, where an accurate representation of ligand(s), cofactor(s) and surrounding active-site residues is of paramount importance, the stereochemical restraint function is often not rigorous enough to represent the chemistry within the complex. Therefore, in order to improve the description of the atomic forces experienced by each atom within the complex, the SE-QM-based function available in DivCon is employed in the present method at each step and the updated coordinates and gradients are generated in ‘real time’ during the entire course of the refinement. This DivCon (libQB) toolkit is a C++-based library on which input/output processing functions are layered in order to facilitate communication between itself and the Python-based PHENIX package. During the coordinate-refinement stage of each macro-cycle, the atomic coordinates of the structure are passed to DivCon through a lightweight Python class each time geometry gradients are required to complete an L-BFGS minimization step. The QM engine within DivCon then generates the single-point SE-QM energy and atomic gradients, and the same lightweight Python class processes the results and replaces the native PHENIX stereochemistry gradients with the QM gradients. The sequential organization of the refinement flow in PHENIX allows us to fully employ the other steps of the procedure such as bulk-solvent correction and B-factor refinement without any modifications or corrections owing to inclusion of the SE-QM data without an external run script and with all communication performed underneath the phenix.refine executable.
2.3. Region-specific QM X-ray refinement
DivCon can be used to treat the entire protein–ligand complex; however, owing to the repeated iterations required for each PHENIX macro-cycle, this treatment becomes computationally expensive for large protein structures even when the linear scaling methodology is employed. For example, L-BFGS in PHENIX requires a new set of computed gradients 20–30 times (micro-cycles) for each refinement macro-cycle. Instead of treating the entire protein, one can reduce the size of the problem by only treating the region of interest (for example, a ligand/cofactor along with its surrounding active site). In the previous CNS-based implementation (Li et al., 2010 ▶), the third-party package AMBER (Case et al., 2010 ▶) and its QM/MM implementation was used. The active site was treated at an ab initio or semiempirical level of theory, and the rest of the protein utilized the AMBER force field for the MM portion of the computations.
In this work, in order to limit dependence on third-party software and to improve the general usability of the software, we implemented an automatic region-specific methodology to carry out QM-based refinement on the area(s) of interest within the protein–ligand complex (Fig. 2 ▶). As depicted in Fig. 2 ▶, firstly we define all residues within a certain distance of any atom of the ligand as a part of the main or core region. This core region is the region for which the atomic SE-QM gradients are determined and returned for use in the refinement. Instead of layering an MM region on top of this core region, we employ a second SE-QM region referred to as a buffer region, which includes any residues surrounding the core region. By using a buffer region to chemically insulate the core region, we gain a significant speed-up versus a full QM treatment and, at the same time, we limit any errors that may occur in the gradients owing to capping or to some artificial chemical environment surrounding the core region. Granted, we lose longer-range interactions of a true QM/MM Hamiltonian; however, we gain greater usability, improved QM convergence characteristics and software independence by not being dependent upon the AMBER package in our implementation. Finally, while not explored in the present paper, this regional QM method can also be applied concurrently to more than one region within the complex. Since the atoms within the buffer region are only provided in order to chemically separate the QM core from the rest of the biomolecular structure, the QM gradients generated from the atoms within the buffer region are not used in the refinement and are thrown out, and the standard stereochemical restraints are used instead.
Figure 2.

Schematic view of the region-refinement concept.
To explore the performance of regional QM refinement, we set the residue-selection radius of the core region to 5 Å from the ligand and the residue-selection radius for the buffer region to 3 Å from the core region. Both regions within PHENIX/DivCon are extended in a residue-inclusive manner, meaning that if a single residue atom falls within a particular distance of the ligand/core then the entire residue is included in the calculation. The complete core + buffer region is treated with SE-QM, and where the buffer region is separated or cut from the protein, any ‘dangling bonds’ are addressed through the addition of protons. For the remainder of the protein, standard PHENIX stereochemistry restraints are used, as schematically shown in Fig. 2 ▶. While the stereochemistry restraints are not as sophisticated as an MM force field, they are still well parameterized for standard residues such as amino acids and hence produce reasonable results for the protein as assessed by several studies (Kleywegt & Jones, 1996 ▶; Jaskolski et al., 2007 ▶; Dodson et al., 1996 ▶). During QM regional refinement, QM and stereochemical gradients on each atom with coordinates x are combined according to
where the weight ϖ is set to 1 for the core QM region(s) and 0 for the rest of the atoms, including the buffer region. In order to gauge the overall performance of this regional QM refinement, full QM refinement was also performed, in which the weight ϖ is 1 for all atoms in the whole system.
2.4. Local ligand strain energy calculations
In addition to crystallographic metrics to measure the agreement between model and experiment, local ligand strain has been used and provided throughout this project. The local ligand strain energy, defined as the difference between the energy of the isolated ligand conformation and the protein-bound ligand conformation, serves as an important quality indicator of protein–ligand structures as it shows how much strain the ligand must take on in order to bind to the protein. Some strain is expected as it captures ligand deformation upon binding; however, large ligand strains can imply problems in the ligand. As suggested by Fu et al. (2011 ▶), ligand strain energy E strain is computed as
where E ligand xray is the single-point energy computed for the ligand X-ray geometry and E ligand optimized is the energy of the optimized ligand that corresponds to the local minimum.
Upon the completion of each refinement, the starting and ending ligand strain using the Hamiltonian chosen in PHENIX/DivCon (e.g. AM1 in this case) is provided as output, and these strains are reported. In order to compare these AM1 strains with a higher level of theory, the strains calculated using the ab initio HF/6-311+G** method as implemented within GAMESS v. October 1, 2010 (Schmidt et al., 1993 ▶) are also reported. Protons were added as per the ReadySet! workflow. Since the added protons are not optimized using the AM1 Hamiltonian in ReadySet!, prior to calculating the strain energy for deposited structures the positions of any added H atoms were optimized. AM1 as implemented in GAMESS v. October 1, 2010 was chosen for this step because it provides a mechanism to refine protons separately from heavy atoms. Therefore, the error associated with proton addition has been canceled when reported. Solvation (implicit or explicit) was not included in any strain calculations owing to methodological and implementation limitations. Finally, both the raw strain and the normalized strain values are reported, where the normalized strain is the raw strain divided by the number of atoms.
2.5. Structure preparation
Validation of the QM-augmented refinement method was conducted in two stages. The first stage involved a detailed description of the QM-based refinement of the high-quality protein structure PDB entry 1lri (Lascombe et al., 2002 ▶; Table 1 ▶) with a well resolved ligand. When this structure was originally deposited in the PDB, it was determined at a resolution of 1.45 Å with an R free below 20% and with sound Ramachandran plot statistics (99% of residues in the most favorable region). It therefore serves as a positive test to reveal flaws (if any) in our QM protocol.
Table 1. Crystallographic data, refinement statistics and ligand strain energy.
| Refinement type | Non-regional QM | Regional QM | Conventional | PDB |
|---|---|---|---|---|
| Structure | 1lri | |||
| Space group | C2221 | |||
| Unit-cell parameters (Å) | a = 30.96, b = 94.80, c = 65.30 | |||
| Molecules per asymmetric unit | 1 | |||
| Resolution (Å) | 16.3–1.45 | |||
| No. of reflections (work/R free) | 17471/1397 | |||
| No. of atoms | 1795 | |||
| R work (%) | 16.6 | 16.7 | 15.8 | 17.8† |
| R free (%) | 17.4 | 17.8 | 17.8 | 18.4 |
| Average B factor (Å2) | 25.0 | 24.5 | 24.3 | 23.1 |
| Ramachandran plot† | ||||
| Most favored (%) | 99 | 99 | 99 | 99 |
| Allowed (%) | 1 | 1 | 1 | 1 |
| R.m.s. deviations from ideality† | ||||
| Bonds (Å) | 0.02 | 0.018 | 0.017 | 0.012 |
| Angles (°) | 1.99 | 1.868 | 1.515 | 2.258 |
| Ligand strain energy (kcal mol−1) | 15.73 | 15.25 | 19.34 | 28.89 |
Evaluated using MolProbity (Chen, Arendall et al., 2010 ▶)
The second stage of the validation involved high-throughput PHENIX/DivCon refinement of 50 protein structures containing various ligands (Table 2 ▶). These structures were chosen through an internal strain energy survey for all ligand poses found in the PDB. Using these ligand strains, the structures were divided into three bins: 0–50 kcal mol−1 strain, 50–100 kcal mol−1 strain and >100 kcal mol−1 strain. The bins were further divided into those structures deposited in the years prior to 2007 and those deposited in the years since. A total of 30 structures for high-throughput validation were then randomly selected from the latter set of bins at a rate of ten examples from each bin. The remaining 20 structures were made up of two additional equal sized bins: ten metal-containing structures and ten models containing covalently bound ligands. This selection led to a total population of 50 structures with a large variety of chemistry and ligand strain that were deposited in approximately the last five years.
Table 2. Crystallographic data, refinement statistics and ligand strain energy (kcal mol−1) calculated at the AM1 and HF/6-311+G** levels.
The structures discussed in detail in the paper are highlighted in bold.
| R work/R free | Strain energy† | Strain energy‡ | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PDB code | Ligand ID | Deposition year | Resolution (Å) | Reported | QM refined | Original refinement program | R.m.s.d.§ (Å) | QM refined | Original xyz | Fold | QM refined | Original xyz |
| Low strain | ||||||||||||
| 3ix1 | NFM | 2009 | 2.4 | 0.203/0.264 | 0.186/0.258 | CNS 1.2 | 0.124 | 2.75 | 33.75 | 12.3 | 19.74 | 50.27 |
| 0.13 ¶ | 1.53 | 0.90 | 2.29 | |||||||||
| 2vts | LZC | 2008 | 1.9 | 0.210/0.261 | 0.217/0.263 | BUSTER-TNT 2.1.1 | 0.182 | 2.58 | 35.93 | 13.9 | 31.80 | 36.45 |
| 0.06 | 0.78 | 0.69 | 0.79 | |||||||||
| 2yhd | TES | 2011 | 2.2 | 0.209/0.259 | 0.202/0.260 | REFMAC 5.5.0102 | 0.065 | 5.88 | 26.98 | 4.6 | 28.66 | 29.01 |
| 0.12 | 0.55 | 0.58 | 0.59 | |||||||||
| 3qyi | PG4 | 2011 | 2.18 | 0.214/0.281 | 0.222/0.281 | REFMAC 5.5.0109 | 0.183 | 11.44 | 24.20 | 2.1 | 29.87 | 36.71 |
| 0.37 | 0.78 | 0.96 | 1.18 | |||||||||
| 3nc9 | TR3 | 2010 | 2.4 | 0.237/0.285 | 0.245/0.298 | phenix.refine 1.6.1_357 | 0.094 | 5.47 | 36.20 | 6.6 | 28.36 | 32.08 |
| 0.11 | 0.75 | 0.59 | 0.67 | |||||||||
| 3bea | IXH | 2007 | 2.02 | 0.193/0.230 | 0.184/0.229 | CNX 2005 | 0.138 | 5.86 | 48.11 | 8.2 | 39.39 | 53.79 |
| 0.08 | 0.70 | 0.57 | 0.78 | |||||||||
| 3rdp | NMF | 2011 | 2.8 | 0.200/0.244 | 0.191/0.248 | phenix.refine 1.6.2_432 | 0.169 | 6.07 | 30.35 | 5.0 | 31.33 | 43.79 |
| 0.20 | 0.98 | 1.01 | 1.41 | |||||||||
| 3suv | NOK | 2011 | 1.6 | 0.144/0.171 | 0.140/0.164 | REFMAC 5.2.0019 | 0.269 | 4.58 | 28.00 | 6.1 | 20.52 | 30.35 |
| 0.15 | 0.93 | 0.68 | 1.01 | |||||||||
| 2vww | 7X2 | 2008 | 1.9 | 0.184/0.229 | 0.182/0.239 | REFMAC 5.3.0034 | 0.351 | 9.12 | 35.28 | 3.9 | 39.22 | 49.67 |
| 0.19 | 0.72 | 0.80 | 1.01 | |||||||||
| 2q3y | 1CA | 2007 | 2.4 | 0.196/0.240 | 0.171/0.222 | CNS 1.1 | 0.156 | 8.65 | 25.89 | 3.0 | 32.44 | 35.30 |
| 0.16 | 0.48 | 0.60 | 0.65 | |||||||||
| Average | 0.199/0.246 | 0.194/0.246 | 6.24 | 32.70 | 5.2 | 30.13 | 39.75 | |||||
| 0.16 | 0.82 | 0.74 | 1.04 | |||||||||
| Medium strain | ||||||||||||
| 3nyx | TZ1 | 2010 | 2.5 | 0.215/0.275 | 0.213/0.264 | REFMAC 5.2.0019 | 0.084 | 2.86 | 65.68 | 23.0 | 38.74 | 55.95 |
| 0.08 | 1.93 | 1.14 | 1.65 | |||||||||
| 3vkj | FNR | 2011 | 1.7 | 0.185/0.217 | 0.175/0.210 | REFMAC 5.5.0109 | 0.255 | 25.98 | 85.30 | 3.3 | 49.79 | 75.89 |
| 0.50 | 1.64 | 0.96 | 1.46 | |||||||||
| 3r22 | D37 | 2011 | 2.9 | 0.269/0.313 | 0.255/0.310 | REFMAC 5.5.0109 | 0.163 | 5.49 | 53.52 | 9.7 | 53.73 | 61.91 |
| 0.11 | 1.05 | 1.05 | 1.21 | |||||||||
| 3o50 | LJE | 2010 | 2.0 | 0.285/0.351 | 0.253/0.334 | CNS | 0.559 | 10.71 | 71.36 | 6.7 | 44.67 | 91.52 |
| 0.21 | 1.43 | 0.89 | 1.83 | |||||||||
| 4dqc | 017 | 2012 | 1.94 | 0.182/0.230 | 0.198/0.266 | REFMAC 5.6.0117 | 0.115 | 15.96 | 45.30 | 2.8 | 55.30 | 58.57 |
| 0.21 | 0.60 | 0.74 | 0.78 | |||||||||
| 3l5e | BDW | 2009 | 1.53 | 0.186/0.200 | 0.175/0.193 | CNS | 0.102 | 16.34 | 67.18 | 4.1 | 71.29 | 82.25 |
| 0.19 | 0.76 | 0.81 | 0.93 | |||||||||
| 2xk4 | OL2 | 2010 | 2.1 | 0.168/0.211 | 0.181/0.227 | phenix.refine | 0.103 | 12.10 | 70.37 | 5.8 | 38.86 | 69.08 |
| 0.23 | 1.33 | 0.73 | 1.30 | |||||||||
| 3otu | BI4 | 2010 | 2.1 | 0.194/0.240 | 0.189/0.237 | phenix.refine 2010_01_09_2330 | 0.060 | 29.99 | 86.56 | 2.9 | 51.89 | 90.79 |
| 0.51 | 1.47 | 0.88 | 1.54 | |||||||||
| 3gus | N11 | 2009 | 1.53 | 0.169/0.195 | 0.146/0.178 | REFMAC 5.2.0005 | 0.145 | 3.93 | 91.48 | 23.3 | 33.86 | 105.59 |
| 0.11 | 2.61 | 0.97 | 3.02 | |||||||||
| 3owj | 1EL | 2010 | 1.85 | 0.196/0.242 | 0.177/0.211 | REFMAC 5.5.0044 | 0.140 | 5.71 | 51.72 | 9.1 | 24.75 | 62.10 |
| 0.16 | 1.48 | 0.71 | 1.77 | |||||||||
| Average | 0.205/0.247 | 0.196/0.243 | 12.90 | 68.80 | 5.3 | 46.29 | 75.37 | |||||
| 0.23 | 1.43 | 0.89 | 1.55 | |||||||||
| High strain | ||||||||||||
| 2zl9 | VDA | 2008 | 1.9 | 0.216/0.276 | 0.223/0.274 | CNS 1.1 | 0.270 | 23.76 | 283.35 | 11.9 | 61.14 | 285.44 |
| 0.30 | 3.54 | 0.76 | 3.57 | |||||||||
| 2y68 | T04 | 2011 | 1.49 | 0.179/0.204 | 0.191/0.218 | REFMAC 5.5.0109 | 0.167 | 48.43 | 139.43 | 2.9 | 86.29 | 151.15 |
| 0.93 | 2.68 | 1.66 | 2.91 | |||||||||
| 3ii4 | 3II | 2009 | 2.42 | 0.185/0.242 | 0.156/0.210 | CNS 1.2 | 0.280 | 14.63 | 156.64 | 10.7 | 57.78 | 161.10 |
| 0.19 | 2.03 | 0.75 | 2.09 | |||||||||
| 3h2o | B63 | 2009 | 2.7 | 0.271/0.311 | 0.299/0.357 | REFMAC 5.2.0019 | 0.283 | 14.94 | 41.76 | 2.8 | 53.25 | 60.14 |
| 0.38 | 1.07 | 1.37 | 1.54 | |||||||||
| 3v7y | A3N | 2011 | 1.97 | 0.193/0.229 | 0.212/0.255 | phenix.refine 1.6.4_486 | 0.283 | 25.67 | 268.15 | 10.4 | 56.64 | 243.66 |
| 0.68 | 7.06 | 1.49 | 6.41 | |||||||||
| 3drr | R8E | 2008 | 2.89 | 0.183/0.269 | 0.251/0.323 | BUSTER-TNT 2.1.1 | 0.187 | 31.45 | 117.16 | 3.7 | 55.04 | 89.23 |
| 0.75 | 2.79 | 1.31 | 2.12 | |||||||||
| 2wue | KEK | 2009 | 1.8 | 0.194/0.210 | 0.163/0.202 | phenix.refine | 0.408 | 18.63 | 176.56 | 9.5 | 36.93 | 179.27 |
| 0.55 | 5.19 | 1.09 | 5.27 | |||||||||
| 2xnp | WCX | 2010 | 1.98 | 0.157/0.201 | 0.169/0.211 | phenix.refine | 0.117 | 8.63 | 134.02 | 15.5 | 43.37 | 154.46 |
| 0.13 | 1.97 | 0.64 | 2.27 | |||||||||
| 3fe9 | CMJ | 2008 | 1.75 | 0.161/0.187 | 0.189/0.222 | REFMAC 5.4.0077 | 0.168 | 37.06 | 70.61 | 1.9 | 61.22 | 127.65 |
| 0.28 | 0.54 | 0.47 | 0.97 | |||||||||
| 2oga | PGU | 2007 | 2.05 | 0.165/0.247 | 0.159/0.206 | TNT | 0.375 | 96.24 | 185.56 | 1.9 | 138.67 | 206.24 |
| 2.41 | 4.64 | 3.47 | 5.16 | |||||||||
| Average | 0.190/0.238 | 0.200/0.247 | 31.90 | 157.30 | 4.9 | 65.03 | 165.83 | |||||
| 0.66 | 3.15 | 1.30 | 3.23 | |||||||||
| Metal-containing structures | ||||||||||||
| 2of1 | THP: Ca | 2007 | 1.92 | 0.218/0.244 | 0.200/0.229 | CNS 1.1 | 0.092 | 35.45 | 81.82 | 2.3 | 59.94 | 70.82 |
| 0.96 | 2.21 | 1.62 | 1.91 | |||||||||
| 3ovs | CTP: Ca | 2010 | 2.8 | 0.211/0.268 | 0.213/0.264 | REFMAC 5.2.0019 | 0.214 | 68.94 | 132.05 | 1.9 | 75.45 | 85.23 |
| 1.68 | 3.22 | 1.84 | 2.08 | |||||||||
| 3mtw | M3R: Zn | 2010 | 1.7 | 0.180/0.203 | 0.178/0.201 | phenix.refine 1.5_2 | 0.235 | 111.58 | 112.19 | 1.0 | 165.05 | 207.04 |
| 3.72 | 3.74 | 5.50 | 6.90 | |||||||||
| 3spz | UPC: Ca | 2011 | 2.43 | 0.200/0.257 | 0.208/0.263 | REFMAC 5.5.0109 | 0.293 | 92.95 | 237.02 | 2.5 | 107.71 | 245.00 |
| 2.27 | 5.78 | 2.63 | 5.98 | |||||||||
| 2wo9 | 68: Zn | 2009 | 1.7 | 0.172/0.218 | 0.169/0.216 | REFMAC 5.4.0073 | 0.195 | 13.19 | 25.90 | 2.0 | 36.54 | 44.16 |
| 0.31 | 0.62 | 0.87 | 1.05 | |||||||||
| 3oim | VZ5: Zn | 2010 | 1.45 | 0.144/0.170 | 0.164/0.186 | phenix.refine 1.6.3_467 | 0.106 | 49.51 | 88.56 | 1.8 | 58.76 | 81.00 |
| 0.93 | 1.67 | 1.11 | 1.53 | |||||||||
| 2pjc | 343: Zn | 2007 | 1.74 | 0.179/0.209 | 0.168/0.206 | CNX 2005 | 0.143 | 49.77 | 101.63 | 2.0 | 112.97 | 128.37 |
| 0.62 | 1.27 | 1.41 | 1.60 | |||||||||
| 3zyf | 147: Ca | 2011 | 1.94 | 0.186/0.210 | 0.199/0.228 | phenix.refine | 0.228 | 27.23 | 112.88 | 4.1 | 37.22 | 99.47 |
| 0.80 | 3.32 | 1.09 | 2.93 | |||||||||
| 2x7t | WZB: Zn | 2010 | 1.89 | 0.211/0.248 | 0.192/0.232 | CNS 1.2 | 0.359 | 14.50 | 96.71 | 6.7 | 38.77 | 101.81 |
| 0.25 | 1.64 | 0.66 | 1.73 | |||||||||
| 3qiy | QI1: Zn | 2011 | 2.3 | 0.226/0.263 | 0.253/0.304 | BUSTER 2.8.0 | 0.166 | 15.32 | 32.06 | 2.1 | 39.63 | 51.33 |
| 0.36 | 0.75 | 0.92 | 1.19 | |||||||||
| Average | 0.193/0.229 | 0.194/0/233 | 47.80 | 102.10 | 2.6 | 73.20 | 111.42 | |||||
| 1.19 | 2.42 | 1.77 | 2.69 | |||||||||
| Models containing covalently bound ligands | ||||||||||||
| 3nck | NFF | 2010 | 2.8 | 0.185/0.265 | 0.172/0.259 | REFMAC 5.5.0102 | 0.155 | 16.68 | 96.91 | 5.8 | 45.42 | 121.67 |
| 0.32 | 1.86 | 0.87 | 2.34 | |||||||||
| 3ume | HC4 | 2011 | 1.8 | 0.120/0.146 | 0.105/0.147 | CNS 1.1 | 0.230 | 7.25 | 24.90 | 3.4 | 14.54 | 21.80 |
| 0.38 | 1.31 | 0.77 | 1.15 | |||||||||
| 3lxs | 4MC | 2010 | 1.5 | 0.122/0.158 | 0.133/0.156 | REFMAC 5.5.0109 | 0.128 | 50.45 | 149.03 | 3.0 | 104.18 | 150.36 |
| 0.61 | 1.80 | 1.26 | 1.81 | |||||||||
| 2wzz | ZX1 | 2009 | 1.57 | 0.181/0.197 | 0.165/0.185 | REFMAC 5.5.0072 | 0.145 | 16.61 | 35.72 | 2.2 | 43.19 | 51.98 |
| 0.39 | 0.83 | 1.00 | 1.21 | |||||||||
| 2wj1 | S99 | 2009 | 1.84 | 0.186/0.214 | 0.192/0.221 | phenix.refine | 0.155 | 4.77 | 23.11 | 4.8 | 27.86 | 31.01 |
| 0.10 | 0.47 | 0.57 | 0.63 | |||||||||
| 3fa6 | LSR | 2008 | 1.54 | 0.171/0.221 | 0.192/0.237 | REFMAC 5.2.0005 | 0.280 | 3.48 | 21.95 | 6.3 | 21.69 | 41.92 |
| 0.09 | 0.56 | 0.56 | 1.07 | |||||||||
| 2uzj | E64 | 2008 | 1.55 | 0.181/0.227 | 0.176/0.212 | REFMAC 5.2.0019 | 0.162 | 72.52 | 108.84 | 1.5 | 124.57 | 146.08 |
| 1.34 | 2.02 | 2.31 | 2.71 | |||||||||
| 2vk7 | FSI | 2007 | 1.2 | 0.131/0.159 | 0.154/0.176 | REFMAC 5.0 | 0.270 | 25.93 | 23.18 | 0.9 | 52.17 | 62.76 |
| 0.68 | 0.61 | 1.37 | 1.65 | |||||||||
| 2v6n | XP1 | 2007 | 1.98 | 0.164/0.211 | 0.173/0.220 | REFMAC 5.2.0019 | 0.135 | 1.06 | 6.88 | 6.5 | 10.25 | 12.46 |
| 0.05 | 0.31 | 0.47 | 0.57 | |||||||||
| 3ixh | PCZ | 2009 | 2.3 | 0.194/0.248 | 0.177/0.248 | REFMAC 5.5.0072 | 0.265 | 41.66 | 77.53 | 1.9 | 97.78 | 105.86 |
| 1.04 | 1.94 | 2.44 | 2.65 | |||||||||
| Average | 0.164/0.205 | 0.164/0.206 | 23.70 | 56.80 | 2.4 | 54.17 | 74.59 | |||||
| 0.50 | 1.17 | 1.16 | 1.58 | |||||||||
| Overall | 0.190/0.233 | 0.190/0.235 | 24.60 ± 3.67 | 83.50 ± 9.03 | 53.76 ± 4.61 | 93.39 ± 9.00 | ||||||
| 0.55 ± 0.09 | 1.80 ± 0.20 | 1.17 ± 0.12 | 2.02 ± 0.21 | |||||||||
Ligand strain calculated at AM1. The first number is the raw strain, while the second, italicized number is the strain normalized by the number of atoms.
Ligand strain calculated at HF/6-311+G**. The first number is the raw strain, while the second, italicized number is the strain normalized by the number of atoms.
Structural (three-dimensional) r.m.s.d. calculated between initial and final ligand structures for all atoms.
Normalized strain energy values (strain energy/N atoms).
Coordinates and structure factors for all 50 structures were downloaded from the Protein Data Bank (PDB). Ligands(s), solvent molecules, metals or anions (e.g. Cl−) were included in our refinement, and H atoms were added to protein residues, ligands and water molecules using the PHENIX utility ReadySet! as implemented in the PHENIX package v.1.7.3-928. No optimization of hydrogen positions was carried out prior to the re-refinement. The correctness of the ligand protonation was verified using the molecular-perception algorithm (Labute, 2005 ▶) implemented in DivCon. While not required for QM refinement, the CIF is required for PHENIX ‘error traps’. Therefore, the geometry restraints library or CIF for each ligand in the structures under investigation was generated using eLBOW (Moriarty et al., 2009 ▶) from PHENIX utilizing the AM1 optimization (the -opt) option. To limit investigator bias, we used the CIF files from eLBOW and ReadySet! as provided, and we did not hand-edit the files to correct, change or otherwise ‘improve’ the ligand CIFs to address any missed atom type or other problems associated with automated methods.
2.6. The refinement protocol
For the first validation, the structure 1lri was chosen for detailed validation in this study and was refined in two ways: (i) full (non-regional) SE-QM PHENIX/DivCon refinement and (ii) regional SE-QM PHENIX/DivCon refinement.
In each case, the refinement began with the identical PDB-deposited coordinates and structure factors, and the QM energy and gradient calculations were performed using the AM1 Hamiltonian (Dewar et al., 1985 ▶) as implemented in DivCon. For the regional refinement, the core of the QM region was defined as the ligand/cofactor along with residues that had any atoms within 5.0 Å of any of the ligand/cofactor atoms at the start of the refinement. The buffer region was configured to include any residues with atoms 2.0 Å from any core QM atoms. In both refinements, in order to further limit investigator bias and to demonstrate the high-throughput nature of the workflow, the default phenix.refine v.1.7.3-928 (Afonine et al., 2012 ▶) refinement options were chosen. Finally, the quality of the refined structures was analyzed using PROCHECK (Laskowski et al., 1993 ▶) and MolProbity (Chen, Arendall et al., 2010 ▶; Davis et al., 2007 ▶).
The high-throughput validation focuses on the treatment of 50 additional recently deposited PDB structures. For this validation, only the 5.0 Å core, 3.0 Å buffer regional SE-QM PHENIX/DivCon refinement described above was performed. In each refinement, the center of the region was defined as the ligand listed in Table 2 ▶. Also as with the 1lri validation, the AM1 Hamiltonian was chosen, and all default PHENIX/DivCon settings were used in order to validate the automated high-throughput nature of the new package. The real-space correlation coefficients (CCs) for each refined ligand are reported (Supplementary Table S11) as calculated using the program phenix.get_cc_mtz_pdb from the PHENIX package (Adams et al., 2010 ▶).
3. Results
3.1. Detailed QM method validation: refinement of 1lri
The structure of β-cryptogein, a sterol carrier protein, includes 98 residues in the complex with cholesterol (Lascombe et al., 2002 ▶), and has been determined to the relatively high resolution of 1.45 Å. Originally, this structure was refined using the anisotropic approximation of atomic displacements for non-H atoms (Lascombe et al., 2002 ▶). However, a recent comprehensive study (Merritt, 2012 ▶) indicates that the anisotropic refinement at this resolution may produce questionable temperature factors. Therefore, we adopted the isotropic refinement protocol for this structure in this study. Conventional PHENIX refinement produced good statistics overall, with R free and R work values of 17.8 and 15.8%, respectively (Table 1 ▶). Non-regional SE-QM PHENIX/DivCon refinement yielded a slightly better R free of 17.4%, and with an R work of 16.5% the structure resulting from SE-QM refinement was noticeably less overfitted relative to conventional PHENIX refinement. As noted in Table 3 ▶, the average protein backbone C—C and C—O bond lengths resulting from the SE-QM refinement are within 1σ of the ideal values accepted in macromolecular crystallography literature. The protein backbone C—N bonds do deviate by 1.5–2σ from the target length of 1.33 (2) Å reported by Jaskolski et al. (2007 ▶). This deviation is associated with the use of the AM1 Hamiltonian, which is known to slightly overestimate the C—N bond length, as noted previously (Yu et al., 2005 ▶).
Table 3. Main-chain average bond lengths in the structure 1lri after QM and conventional refinements.
Regional SE-QM PHENIX/DivCon refinement was centered on the cholesterol ligand. The resulting 5.0 Å core QM region comprises 564 atoms and the 2.0 Å buffer region adds another 828 atoms to complete the (core + buffer) QM region. This protocol yields overall refinement statistics (R work of 16.7% and R free of 17.8%) and model stereochemistry similar to those produced by the non-regional QM method (Table 1 ▶). As expected, since the protein outside the core QM region was treated with the same stereochemical restraints as the conventional PHENIX refinement, the overall quality of the protein, including the Ramachadran plot statistics, is very similar in both QM and PHENIX refinement. Importantly, MolProbity and PROCHECK analyses revealed no geometry distortion at the border between the QM core and the part of the molecule treated with PHENIX stereochemical restraints.
Since refinement involves the minimization of the geometry of the protein structure, it is expected that the QM energy (e.g. the heat of formation) of the system will decrease over the course of QM refinement. As depicted in Fig. 3 ▶, there is indeed a dramatic drop in the energy that occurs during the first L-BFGS macro-cycle, followed by much smaller decreases in subsequent cycles. This observation suggests that the most important macro-cycle is the first one. However, we cannot exclude the possibility that protein structures with bad starting geometry might experience more significant energy changes during subsequent refinement macro-cycles than we have seen here.
Figure 3.

Heat of formation (kcal mol−1) computed in DivCon for the structure 1lri after each macro-cycle of the full QM refinement.
Detailed analysis of the ligand (cholesterol) molecule structure in 1lri allows us to evaluate the overall robustness of our QM procedure. Regardless of the quality of the molecular description provided in the CIF (Borbulevych et al., 2011 ▶; Borbulevych & Westerhoff, 2011 ▶), upon the completion of each QM refinement the geometry of the cholesterol is found to be chemically correct, with all bond lengths and bond angles close to their standard values (Allen et al., 1987 ▶; Table 4 ▶). This CIF-independent result is consistent with the fact that the PHENIX/DivCon calculations do not rely on the ligand library for the ligand structure, and this observation is of pivotal importance to the application of this method in high-throughput X-ray refinement. Furthermore, the ligand also exhibits an excellent fit to the electron-density map (Fig. 4 ▶). Finally, when comparing the regional refinement with the non-regional or ‘full’ QM refinement, we have shown that the ligand geometry is virtually identical between the two methods (Table 4 ▶, Fig. 4 ▶), which demonstrates that the lower computational cost regional refinement method will suffice.
Table 4. Selected bond lengths (Å) in cholesterol in the structure 1lri .
| Parameter | Non-regional QM | Regional QM | Conventional | Original PDB |
|---|---|---|---|---|
| C1—C10 | 1.54 | 1.54 | 1.57 | 1.55 |
| C1—C2 | 1.51 | 1.51 | 1.52 | 1.53 |
| C2—C3 | 1.53 | 1.53 | 1.55 | 1.54 |
| C3—C4 | 1.53 | 1.53 | 1.50 | 1.54 |
| C4—C5 | 1.49 | 1.49 | 1.48 | 1.52 |
Figure 4.
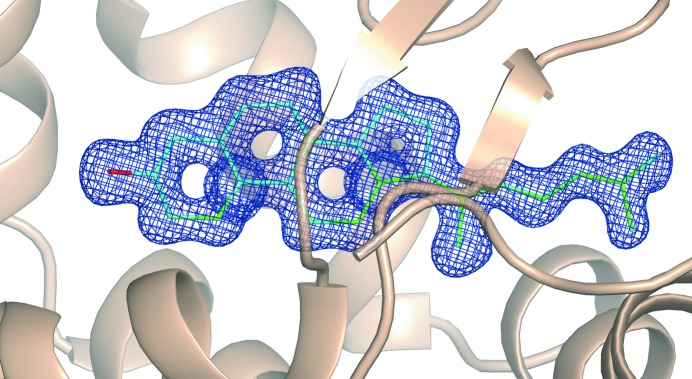
The σA-weighted 2mF o − DF c electron-density map contoured at 1σ around the ligand in the structure 1lri after full (green) and regional (cyan) QM refinement. The density map for other residues in the structure is omitted for clarity.
3.2. QM refinement validation on a set of 50 PDB structures
Once the method had been successfully validated against a single example, regional QM refinement for a set of 50 PDB structures was also performed. In each case, the center of the selection was the ligand found in the original PDB structure as specified in Table 2 ▶. To measure the impact that the QM method brings to the structure, we adopted strain energy (Fu et al., 2012 ▶) as a metric since traditional crystallographic metrics such as R-factor values or the stereochemical quality of the polypeptide chains (e.g. MolProbity) are typically not effective in assessing the quality of the ligand (Cooper et al., 2011 ▶). For example, we found that the average R work and R free factor values over 50 structures remains virtually unchanged after regional QM refinement, while the ligand strain energy dramatically improved. The insensitivity of the R-factor metrics can be explained by the fact that we apply the QM functional only to a relatively small region around the chosen ligand, while the majority of the structure is still treated using the conventional stereochemistry restraints within PHENIX. Furthermore, the method presented is focused on the actual refinement of the deposited structure, and does not involve steps that are beyond the scope of the present work such as novel ligand placement, docking or other tools for model building and ensemble generation.
The average ligand strain energy for the set of 50 structures calculated from the deposited coordinates is found to be 83.50 ± 9.03 kcal mol−1, where the minimum and maximum values are 6.88 and 283.35 kcal mol−1, respectively, or a range of 276.47 kcal mol−1. After regional PHENIX/DivCon refinement is performed, there is a dramatic improvement of the strain throughout the set (Table 2 ▶). The average strain energy of the refined set of structures is 24.60 ± 3.67 kcal mol−1, or 3.4 times smaller than that of the originally deposited structures. The normalized strain energy, which is the strain energy divided by the number of ligand atoms, shows a similar improvement of 3.3-fold, where the average of 50 structures decreased from 1.80 ± 0.20 kcal mol−1 (initial set) to 0.55 ± 0.09 kcal mol−1 (refined set).
Moreover, the energy distribution range also becomes significantly tighter (110.52 kcal mol−1) after the QM refinement. In recent work (Borbulevych et al., 2012 ▶), we studied the strain energy distributions over all structures deposited in the PDB using the semiempirical method PM6. In particular, we demonstrated that ∼55% of all ligand poses fall into the 0–40 kcal mol−1 strain bin, while ∼25% of all ligand poses exhibit a strain energy of over 100 kcal mol−1. Generally speaking, higher values of the strain increase the likelihood of an incorrect geometry (Perola & Charifson, 2004 ▶). Hence, the overall improvement of the ligand strain energy from an average of 83.5 kcal mol−1 to an average of 24.6 kcal mol−1 brings the average value into the most populated bin, likely owing to the elimination of gross errors in the ligand geometry.
Furthermore, the QM refinement protocol exhibits superior performance in cases that usually require tedious work to create library files in conventional refinement, such as ligands covalently bound to protein residues or ligands coordinated to metals. In both categories of structures studied in this work (Table 2 ▶), the overall decrease in the strain energy after the regional QM refinement was over twofold and was accompanied by significant corrections in the geometry of the linkage bonds or the metal coordination sphere, as discussed below in more detail.
We found that the average ligand strain improvement is similar in all three initial strain bins (e.g. low, 0–50 kcal mol−1; medium, 50–100 kcal mol−1; high, >100 kcal mol−1), ranging from 4.9-fold to 5.3-fold. Despite this similarity, the results for particular ligands vary significantly. For example, five ligands from the high-strain bin exhibit a significant decrease in strain of between 9.5-fold and 15.5-fold. The other five structures in this bin show a more modest improvement measured by twofold to 3.7-fold decreases. This observation underscores the fact that the strain energy consists of several components (both artificial and natural). Artificial strain is itself made up of both initial positional or docking-induced strain (e.g. DIS), which is strain associated with poor or questionable ligand starting geometry, and method-induced strain (MIS), which is from errors in the underlying force-field method, parameters or CIF file(s) used in the refinement. Natural ligand strain or target-induced strain (TIS), on the other hand, includes legitimate changes in the ligand conformation during the process of binding to the macromolecular receptor. In the present project, the initial binding conformation of the ligand has not been manipulated prior to QM refinement; therefore, it can be concluded that the sharp decrease in strain after QM refinement represents cases in which the main cause of strain in the deposited structure can be attributed to MIS (e.g. PDB entry 2wue, as detailed below). Interestingly, even a modest improvement in the strain energy corresponds to cases in which a ligand undergoes noticeable structure/conformation changes upon binding (e.g. PDB entry 3fe9). In cases where high strain is reported after QM refinement, it is probable that this strain is owing to poor initial conditions or DIS. Subsequent research will be performed in order to explore this hypothesis.
Table 5 ▶ shows the average strain energy for structures sorted by the original refinement software used. While this table should not be interpreted as a robust, statistical comparison of the various conventional refinement tools with one another, these results do illustrate that QM refinement gives rise to a significant improvement in ligand strain (e.g. threefold to fourfold) regardless of the software used by the authors of the published PDB entries. This observation underscores the fact that problematic ligand geometry is an inherent weakness of conventional refinement. For the set of 50 structures studied, REFMAC seemed to have performed marginally better than other refinement programs. However, even in the case of REFMAC, PHENIX/DivCon is still significantly superior.
Table 5. Average strain-energy improvement grouped by the type of original refinement program.
| Refinement program | No. of examples | Average strain original xyz | Energy QM refined | Improvement (fold) |
|---|---|---|---|---|
| REFMAC | 24 | 69.76 | 24.79 | 3 |
| PHENIX | 11 | 103.54 | 27.24 | 4 |
| CNS/CNX/TNT | 12 | 98.08 | 23.83 | 4 |
| BUSTER | 3 | 61.72 | 16.45 | 4 |
In order to validate these SE-QM energies, they have been compared with those calculated at the HF/6-311+G** ab initio level of theory, and these values are also provided in Table 2 ▶. Ab initio calculations can be quite expensive depending upon the size of the ligand and the level of theory. As one would expect, since the ab initio functional was not used in the refinement, the change in strain is not as pronounced with the ab initio method as with the SE-QM method. However, in all cases, especially in those with higher initial ligand strain, the SE-QM refinement yielded significantly improved ab initio-calculated ligand strains, suggesting that the SE-QM energies are quite robust. As PM6 replaces the much older AM1 Hamiltonian as the standard SE-QM method for refinement, we expect that the improvement in ligand strain will be even more robust.
A complete treatment of all individual examples would be beyond the space allowed for this paper. Instead, several specific examples from Table 2 ▶ are discussed below. For the individual analysis, we have chosen examples from different bins that also exhibit significant changes in strain energy and geometry of the ligand molecules after the QM refinement. All 50 structures are available for download as Supporting Information.
3.2.1. Refinement of 2wue
The crystal structure of the hydrolase inhibited with the synthetic analog of the cholesterol cleavage product (the ligand KEK) originally refined at 1.8 Å resolution was published in 2010 (Lack et al., 2010 ▶). The strain energy of this ligand computed using the deposited coordinates at the AM1 level was 176.56 kcal mol−1. Closer examination of the ligand molecule revealed that key geometry parameters are severely distorted (Fig. 5 ▶). In particular, all of the bond lengths in the phenyl ring were 1.53–1.54 Å, compared with the typical CAr—CAr bond length of 1.38 Å (Allen et al., 1987 ▶). Furthermore, the C—O bonds of the COO− group (1.41 and 1.18 Å) deviate significantly from the length of the delocalized double C=O bond of 1.254 Å (Allen et al., 1987 ▶) in carboxylate anions. After QM region-specific refinement using PHENIX/DivCon, it is found that the geometry parameters of the abovementioned moieties of the ligand approach the standard values for these bond lengths. This improvement in bond lengths is accompanied by a dramatic tenfold decrease of the ligand strain energy to 18.63 kcal mol−1.
Figure 5.
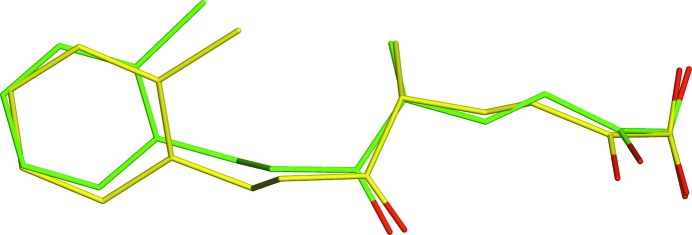
Superimposition of the QM re-refined PDB structure 2wue (green) with the original PDB structure (yellow).
3.2.2. Refinement of 2zl9
The structure 2zl9 was originally refined using CNS 1.1 against 1.9 Å resolution data (Shimizu et al., 2008 ▶). The vitamin D analog (ligand VDA) in the structure is found to have a strain energy of 283.35 kcal mol−1 that is likely to indicate significant errors in the ligand. Upon further inspection, almost all of the bond lengths in the ligand VDA significantly deviate from the corresponding standard values as listed in Table 6 ▶, and this deviation contributes to the high strain energy observed. For example, two C—S bonds (1.52 and 1.68 Å) are considerably shorter than the typical 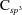 —S bond length of 1.819 Å (Allen et al., 1987 ▶). QM refinement within PHENIX gives rise to a significant improvement of the bond lengths throughout the ligand structure (Table 6 ▶, Fig. 6 ▶), and overall the strain energy after the QM refinement decreases by almost 12-fold to a value of 23.76 kcal mol−1.
—S bond length of 1.819 Å (Allen et al., 1987 ▶). QM refinement within PHENIX gives rise to a significant improvement of the bond lengths throughout the ligand structure (Table 6 ▶, Fig. 6 ▶), and overall the strain energy after the QM refinement decreases by almost 12-fold to a value of 23.76 kcal mol−1.
Table 6. Selected bond lengths (Å) in the ligand VDA in the original PDB structure 2zl9 and after QM refinement.
| Parameters | Original PDB | QM refined | PDB-REDO | Standard length† |
|---|---|---|---|---|
| C16—C17 | 1.57 | 1.35 | 1.34 | 1.323 |
| C1—C2 | 1.62 | 1.53 | 1.52 | 1.535 |
| C2—C3 | 1.68 | 1.53 | 1.52 | 1.535 |
| C3—C4 | 1.64 | 1.53 | 1.53 | 1.535 |
| C4—C5 | 1.56 | 1.48 | 1.51 | 1.535 |
| C5—C10 | 1.60 | 1.49 | 1.51 | 1.535 |
| C10—C1 | 1.58 | 1.53 | 1.52 | 1.535 |
| S22—C20 | 1.68 | 1.80 | 1.78 | 1.819 |
| S22—C23 | 1.52 | 1.77 | 1.76 | 1.819 |
Allen et al. (1987 ▶).
Figure 6.
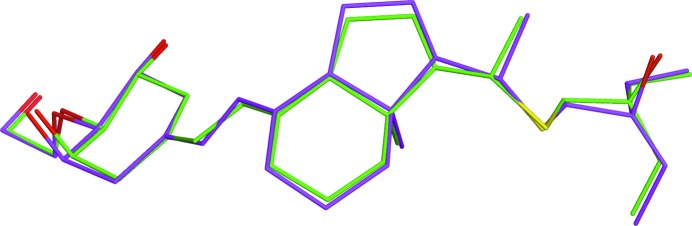
Superimposition of the QM re-refined PDB structure 2zl9 (green) with the original PDB structure (magenta).
3.2.3. Refinement of 2x7t
The structure 2x7t determined at 2.8 Å resolution (Cozier et al., 2010 ▶) features the enzyme carbonic anhydrase that has a tetrahedral zinc in the active site coordinated by three histidine residues (Fig. 7 ▶). The inhibitor (ligand ID WZB) is bound to zinc via an amino group to complete the coordination sphere of the metal. In the deposited structure, all Zn—N and Zn—O distances are in the range 2.14–2.25 Å, which are longer than the average length of 2.00 (2) Å for this bond type (Harding, 1999 ▶). This discrepancy leads to a distortion of the coordination sphere of the metal (Fig. 7 ▶, Table 7 ▶). Concerning the ligand geometry, the phenyl ring is distorted. In particular, one of the bond angles in the ring is 147°, and two bonds are shorter than the normal Car—Car bond length of 1.398 Å (Allen et al., 1987 ▶), while the other bonds are longer than the standard value. It is likely that these abnormities contribute to the high strain energy of 96.71 kcal mol−1 computed for this ligand using the deposited coordinates.
Figure 7.
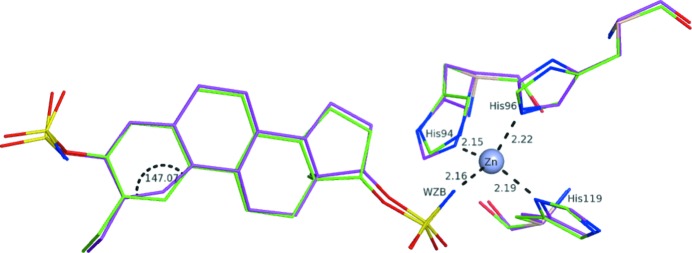
Superimposition of the residues in the coordination sphere of zinc in the structure 2x7t from the regional QM (green) refinements as well as the original PDB structure (magenta).
Table 7. Zn—N distances and selected geometry parameters (Å, °) in the ligand WZB in the deposited structure 2x7t and after QM refinement.
| Parameters | Original PDB | Regional QM | PDB-REDO |
|---|---|---|---|
| Zn—NE2His94 † | 2.16 | 2.02 | 2.13 |
| Zn—ND1His119 | 2.18 | 1.99 | 2.13 |
| Zn—NE2His96 | 2.25 | 1.99 | 2.15 |
| Zn—NADWZB1261 | 2.14 | 1.98 | 2.09 |
| CAW—CAJ | 1.31 | 1.40 | 1.41 |
| CAJ—CAT | 1.31 | 1.40 | 1.40 |
| CAV—CAT | 1.46 | 1.41 | 1.50 |
| CAW—CAU | 1.46 | 1.41 | 1.50 |
| CAW—CAJ—CAT | 147 | 122 | 121 |
Residue numbers are the same as in the deposited structure.
We carried out the QM refinement with no assumptions about the geometry of the coordination sphere expressed in the form of restraints. Upon completion of the PHENIX/DivCon refinement, the bond lengths in the coordination sphere of zinc are in the range 1.98–2.02 Å (Table 7 ▶), which are all remarkably close to the average values reported in the literature (Harding, 1999 ▶). Furthermore, the decrease in the ligand strain energy to 14.5 kcal mol−1 indicates significant improvements in the geometry of the bound molecule WZB, as is also seen from Table 7 ▶.
3.2.4. Refinement of 3nck
Suicide inhibitors represent a traditional challenge for conventional refinement (Kleywegt, 2007 ▶). Such ligands are covalently bound to certain amino acids (e.g. Ser or Lys), thus becoming part of the polypeptide chain. This bond makes it difficult to choose the correct sets of restraints for the chemically modified amino acid and the ligand. The structure 3nck determined at 2.8 Å resolution revealed the signal protein BlaC with a covalent bond to nafcilin, which belongs to the family of β-lactam antibiotics. The residue Ser84 of BlaC interacts with the four-membered β-lactam ring of the antibiotic and results in ring opening and in the formation of a covalent ester bond between the OG atom of the amino acid and the carbonyl C atom of the ligand. The deposited BlaC–nafcilin (Nff) structure exhibits severe geometry distortions in the region of the ester bond between the ligand and Ser84. In particular, the bond angle ONff1—CNff1—OGSer84 is 102°, CANff1—CNff1—OGSer84 is 123° and CBSer84—OGSer84—CNff1 is 144° (Table 8 ▶, Fig. 8 ▶), two of which differ significantly from the ideal sp 2 configuration arrangement of 120°.
Table 8. Selected bond lengths (Å) and bond angles (°) in the structure 3nck .
| Regional QM | Original PDB | PDB-REDO | |
|---|---|---|---|
| Bond lengths | |||
| CANff1—CNff1 | 1.53 | 1.63 | 1.54 |
| CANff1—NNff1 | 1.41 | 1.51 | 1.46 |
| CANff1—CBNff1 | 1.53 | 1.50 | 1.55 |
| CNff1—ONff1 | 1.24 | 1.48 | 1.43 |
| CNff1—OGSer84 | 1.39 | 1.45 | 1.35 |
| CBSer84—OGSer84 | 1.42 | 1.42 | 1.42 |
| Bond angles | |||
| CBSer84—OGSer84—C | 119 | 144 | 156 |
| CANff1—CNff1—ONff1 | 123 | 101 | 113 |
| CANff1—CNff1—OGSer84 | 122 | 123 | 127 |
| ONff1—CNff1—OGSer84 | 115 | 102 | 90 |
Figure 8.
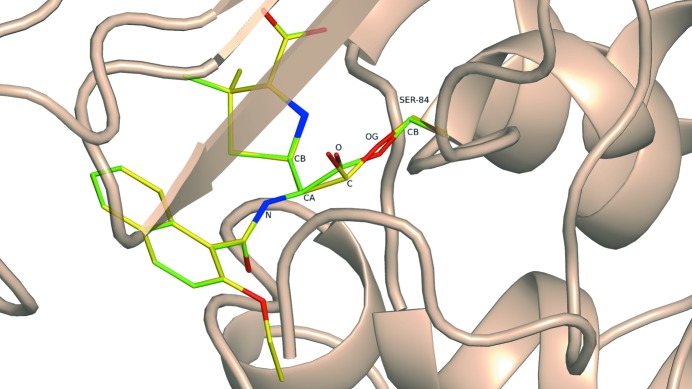
Superimposition of the QM re-refined PDB structure 3nck (green) with the original PDB structure (yellow).
As in all cases considered, no geometry restraints for the ligand and the surrounding active site, including the linkage bond CNff1—OGSer84, were utilized. Despite this, QM refinement leads to correct geometry of the ligand and the ester bond and the structure is completely fixed relative to the original PDB model. Notably, the sum of the bond angles around the CNff1 atom (360°) indicates the planarity of the C(O)—C moiety, with all bonds being close to standard values (Allen et al., 1987 ▶). In particular, the length of the linkage bond CNff1—OGSer84 (1.39 Å) corresponds to the ideal single C—O bond in esters of 1.40 Å (Allen et al., 1987 ▶). Overall, the strain energy of nafcilin after the SE-QM refinement is significantly improved in comparison to the PDB structure (Table 2 ▶). It should be noted that while in several of the cases presented the PDB-REDO project was able to partially recover or correct some of the erroneous structural details, in this case automatic re-refinement of the 3nck structure within the PDB-REDO project (Table 8 ▶) failed in the region of the linkage bond. PDB-REDO reports the bond angle CANff1—CNff1—OGSer84 as 127° and the bond angle CBSer84—OGSer84—CNff1 as 156°, which are worse than in the originally deposited structure.
3.2.5. Refinement of 3lxs
The structure 3lxs with the cysteine protease inhibitor 4MC was reported at 1.5 Å resolution (Chen, Brinen et al., 2010 ▶). In the deposited structure, one of the amide bonds in the inhibitor 4MC (Fig. 9 ▶) is notably distorted such that the length C17—O23 is 1.46 Å and the bond angles around the sp 2 C atom C17 (Table 9 ▶) are significantly different from the ideal bond angle of 120°. Furthermore, in the difference electron-density map drawn using deposited structure factors and phases (Fig. 9 ▶) we found both positive and negative electron-density difference peaks in this region associated with the fragment in question, underscoring its poor geometry. Superimposition of the original and of the QM re-refined structures indicates that the improvement in the geometry is also associated with the movement of the amide moiety towards the positive peak of the difference density and away from the negative peak. As a result, no difference density peaks at or above the 3σ level around this amide bond are observed in the re-refined structure. Overall, the strain energy for this ligand 4MC is improved threefold after the QM refinement (Table 2 ▶).
Figure 9.
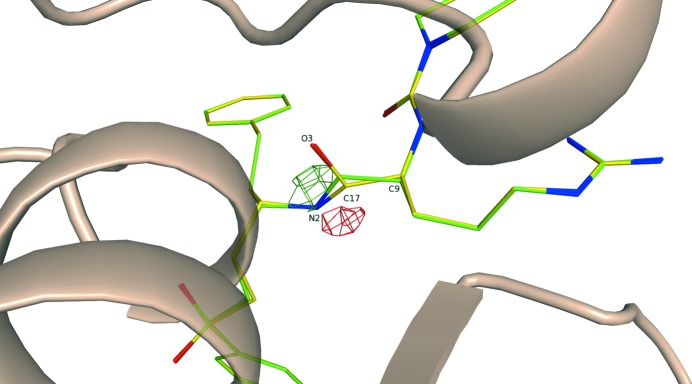
Superimposition of the QM re-refined PDB structure 3lxs (green) with the original PDB structure (yellow). The difference density is drawn at the 3σ level using the structure factors and phases for the original structure as obtained from the Electron Density Server at Uppsala.
Table 9. Selected bond lengths (Å) and bond angles (°) in the ligand 4MC of the structure 3lxs .
| Parameters | Regional QM | Original PDB | PDB-REDO |
|---|---|---|---|
| Bond lengths | |||
| C17—O3 | 1.22 | 1.46 | 1.26 |
| C17—N2 | 1.36 | 1.34 | 1.32 |
| C17—C9 | 1.54 | 1.43 | 1.49 |
| Bond angles | |||
| N2—C17—O3 | 121 | 112 | 121 |
| N2—C17—C9 | 118 | 137 | 120 |
| C9—C17—O3 | 120 | 108 | 119 |
4. Discussion
X-ray macromolecular structures deposited in the PDB have almost exclusively been refined using conventional geometry restraints-based protocols and very rudimentary energy functionals. However, the high percentage of ligands in the PDB with unusual or questionable geometry (Gore et al., 2011 ▶) raises concerns about the extent that this database can be considered to be a reliable source of information on the structures of small-molecule compounds (Cooper et al., 2011 ▶) and therefore the protein–ligand complexes so important to structure-based drug discovery. It follows that protein refinement using pre-calculated small-molecule restraint libraries is often not sufficient to determine the final ligand structure, and this observation is at least partially attributed to the fact that ligand dictionaries are very error-prone and frequently require manual editing. Theoretically, manual inspection should reduce a number of errors in the description; however, making advantageous corrections in the CIF is time-consuming and requires significant a priori knowledge of the chemistry and of the ligand(s) of interest in their bound state. This task might be especially challenging for unseasoned crystallographers, who are responsible for about 50% of structure depositions in the PDB (Cooper et al., 2011 ▶), some biochemically oriented crystallographers who may be more comfortable with biochemical structures than with small-molecule organic structures, and for industrial crystallographers pressed for time. The bottom line is that an incorrect ligand description inevitably gives rise to problematic geometry and the principle ‘garbage in/garbage out’ (Kleywegt, 2007 ▶) is well illustrated in these sorts of studies.
Efforts to improve the ligand representation for X-ray refinement have focused either on using semiempirical or ab initio QM methods (eLBOW; Moriarty et al., 2009 ▶) to create the CIF or deriving geometric values from small-molecule crystallographic data (Smart et al., 2011 ▶). The target ligand geometry is only part of the equation, and the shape of the harmonic function described by the weight Wk in (1) is also important. Those parameters determine how permissive restraints are and how well they allow a ligand to accommodate the bound geometry in the protein molecule. Unfortunately, there is not an easy recipe to tailor those parameters to a specific ligand–protein complex in situ. In essence, geometry restraints are biased towards isolated ligand geometry, while the X-ray diffraction method reports on the bound receptor–ligand structure. This contradiction might be a reason for the high ligand strain energy observed in conventional refinement (Fu et al., 2011 ▶) and some of the strain reported in the present project. Since the macromolecule to which the ligand is bound also influences the structure of the ligand, it is hypothesized that in cases where bond lengths or angles diverge from expected values that these divergences may in fact be owing to the influences of the active site on the ligand as captured by the improved functional. A future study will focus on this very important question.
SE-QM refinement demonstrated exceptional robustness and produced high-quality ligand stereochemistry independent of the ligand library. This result is owing to the fact that the SE-QM procedure is rooted in solving the Schrödinger equation for the system and hence does not require the ligand dictionary. This robustness also makes the QM X-ray refinement methodology ideally suited for high-throughput crystallography, especially in the industrial setting. Specifically, the geometric quality of the ligand structure can be quantitatively evaluated using the strain energy metrics (Fu et al., 2011 ▶), and in the present study we re-refined 50 protein structures as denoted in Table 2 ▶ and found that PHENIX/DivCon refinement consistently produces ligands of significantly lower strain energy values compared with those derived from the original PDB structures. Therefore, even though large differences in pure crystallographic metrics (e.g. R free, R work etc.) are rarely observed between QM and conventional refinements, differences in ligand strain are often quite large.
The intrinsic weaknesses of the geometry restraints can be observed in the cases of covalently bound ligand and/or metal-containing examples. From the QM perspective, since the concept of bound versus unbound is immaterial, the ligand and the active site are treated as a single unit and the QM Hamiltonian captures effects owing to electrostatics, polarization, dispersion, charge transfer and so on that are omitted in conventional refinement, leading to greater methodological robustness. In order to approximate the qualities that QM provides, the investigator would need a complete set of very accurate in situ restraints, and as observed with the PDB-REDO results, more accurate restraints can certainly aid in the refinement process. However, as demonstrated by decades of molecular-mechanics research, the goal of developing a robust, truly generalizable force field is exceedingly difficult to reach. For instance, inclusion of the PDB-REDO results also shows that a few missing or incorrect restraints, such as those involving the protein–ligand covalent bond, can lead to very deleterious results when the conventional functional is unable to compensate.
Overall, QM refinement addresses method-induced strain or MIS, and therefore the quality of the final model is limited by the parameterization of the semiempirical QM Hamiltonian. In recent years there has been significant Hamiltonian development including PM6 (Stewart, 2009 ▶; Řezáč et al., 2009 ▶), PM6-DH2 (Korth et al., 2010 ▶) and PM7 (Hostaš et al., 2013 ▶; Stewart, 2013 ▶). These advances in the development of semiempirical methods make the QM refinement methodology even more attractive and promising. In addition to the underlying Hamiltonian, there are a number of key determinants of success that are beyond the scope of the present work, including the protonation state(s) and chemical connectivity chosen by the investigator as well as the starting position(s) of the ligand(s), which is a key contributor to DIS.
5. Conclusions
In this work, we present a new methodology to perform X-ray refinement using the semiempirical quantum-mechanics functional implemented as a plug-in to the crystallographic package PHENIX. The PHENIX/DivCon refinement protocol has been validated against over 50 protein–ligand structures and the method clearly results in chemically reasonable ligand geometry even where conventional refinement exhibited difficulties. The problematic ligand geometries found in X-ray structures often originate with the use of small-molecule libraries that require a priori knowledge of the refined protein–ligand complex structure. The QM method derives the system characteristics such as energy and gradients by solving the Schrödinger equation in each refinement cycle, and therefore does not depend upon the ligand dictionary file, fixed atom types, link restraints, coordination-sphere parameters or other ‘user-supplied’ characteristics. It is therefore more robust then conventional methods when addressing ligand strain and artifacts induced by the rudimentary energy function (e.g. MIS) found in conventional refinement tools. A case in point is the comparison of several QM re-refined structures with those treated within the PDB-REDO project (Joosten et al., 2012 ▶; Tables 6 ▶, 7 ▶, 8 ▶ and 9 ▶). While the geometry has improved for some ligands in the PDB-REDO structures (e.g. 2zl9), it also failed to correct more challenging cases such as covalently bound ligands (e.g. 3nck) or a zinc-bound ligand (e.g. 2x7t). These results suggest that PDB-REDO structures also suffer from the same problems associated with reliance on explicit ligand descriptions and likely the rudimentary functional used.
When using the method as presented, the successful investigator must still take responsibility for providing a reasonable initial atom placement along with a correct ligand chemistry including connectivity and protonation. For example, while some bond breaking/making can occur with QM methods, a functional alone will not automatically or reliably transmute benzene (i.e. C6H6) into cyclohexane (i.e. C6H12) during refinement. Likewise, since the underlying method is gradient-based, it has the same limited radius of convergence as conventional methods, which often cannot overcome large potential energy barriers (Brünger & Rice, 1997 ▶). Therefore, simulated annealing followed by the QM refinement could be the right protocol when grossly wrong conformations are given as the input.
With this in mind, a logical next stage of the project is to pair PHENIX/DivCon with tools such as LigandFit (Terwilliger et al., 2006 ▶), AFIT (Wlodek et al., 2006 ▶) and/or simulated annealing in order to account for the major components of artificial strain (e.g. MIS and DIS) and to gain a much better understanding of the strain induced naturally through protein–ligand binding (e.g. TIS). With such a study, one could more easily decompose and measure the influence of the surrounding active site on structure and strain. Further, since strain is really only a measure of the chemical structure of the ligand versus the in vacuo structure, an additional metric to be considered in subsequent work is the change in predicted binding affinity and interaction energies upon QM refinement. One could imagine a final workflow in which the investigator uses a density-aware docking function to generate various starting geometries (perhaps with different connectivity or protonation), refines each structure using PHENIX/DivCon, and uses ligand strain, standard crystallographic metrics and QM interaction descriptors (Diller et al., 2010 ▶; Zhang et al., 2010 ▶; Raha & Merz, 2004 ▶, 2005 ▶) to inform his or her decision about which model is most correct.
Supplementary Material
50 QM refined PDB structures. DOI: 10.1107/S1399004714002260/rr5046sup1.zip
Supporting Information.. DOI: 10.1107/S1399004714002260/rr5046sup2.pdf
Acknowledgments
We thank the entire PHENIX development team, especially Drs Paul Adams, Nigel Moriarty, Ralf Grosse-Kunstleve and Nat Echols, for technical support and for instructional discussions early in the integration effort; Drs Nestor Concha and Patricia Elkins (GlaxoSmithKline) for discussion and perspective during the validation process; Dr Greg Warren (OpenEye Inc.) for suggested structures on which to focus our validation efforts; and NIH SBIR 1R42GM079899 for funding the research and development effort.
Footnotes
Supporting information has been deposited in the IUCr electronic archive (Reference: RR5046).
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Afonine, P. V., Grosse-Kunstleve, R. W., Echols, N., Headd, J. J., Moriarty, N. W., Mustyakimov, M., Terwilliger, T. C., Urzhumtsev, A., Zwart, P. H. & Adams, P. D. (2012). Acta Cryst. D68, 352–367. [DOI] [PMC free article] [PubMed]
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987). J. Chem. Soc. Perkin Trans. 2, pp. S1–S19.
- Borbulevych, O., Merz Jr, K. M. & Westerhoff, L. M. (2011). Acta Cryst. A67, C593.
- Borbulevych, O. Y., Plumley, J. A. & Westerhoff, L. M. (2012). Abstr. Pap. Am. Chem. Soc., abstract 478.
- Borbulevych, O. & Westerhoff, L. M. (2011). Abstr. Pap. Am. Chem. Soc., abstract 242.
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Brünger, A. T. & Rice, L. M. (1997). Methods Enzymol. 277, 243–269. [DOI] [PubMed]
- Case, D. A. et al. (2010). AMBER 11. University of California, San Francisco, USA.
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Chen, Y. T., Brinen, L. S., Kerr, I. D., Hansell, E., Doyle, P. S., McKerrow, J. H. & Roush, W. R. (2010). PLoS Negl. Trop. Dis. 4, e825. [DOI] [PMC free article] [PubMed]
- Cooper, D. R., Porebski, P. J., Chruszcz, M. & Minor, W. (2011). Expert Opin. Drug Discov. 6, 771–782. [DOI] [PMC free article] [PubMed]
- Cozier, G. E., Leese, M. P., Lloyd, M. D., Baker, M. D., Thiyagarajan, N., Acharya, K. R. & Potter, B. V. L. (2010). Biochemistry, 49, 3464–3476. [DOI] [PubMed]
- Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N., Kapral, G. J., Wang, X., Murray, L. W., Arendall, W. B., Snoeyink, J., Richardson, J. S. & Richardson, D. C. (2007). Nucleic Acids Res. 35, W375–W383. [DOI] [PMC free article] [PubMed]
- Dewar, M. J. S., Zoebisch, E. G., Healy, E. F. & Stewart, J. J. P. (1985). J. Am. Chem. Soc. 107, 3902–3909.
- Diller, D. J., Humblet, C., Zhang, X. & Westerhoff, L. M. (2010). Proteins, 78, 2329–2337. [DOI] [PMC free article] [PubMed]
- Dixon, S. L. & Merz, K. M. Jr (1996). J. Chem. Phys. 104, 6643.
- Dixon, S. L. & Merz, K. M. Jr (1997). J. Chem. Phys. 107, 879.
- Dixon, S., Merz, K. M. Jr, Lauri, G. & Ianni, J. C. (2005). J. Comput. Chem. 26, 23–34. [DOI] [PubMed]
- Dodson, E., Kleywegt, G. J. & Wilson, K. (1996). Acta Cryst. D52, 228–234. [DOI] [PubMed]
- Engh, R. A. & Huber, R. (1991). Acta Cryst. A47, 392–400.
- Evans, P. R. (2007). Acta Cryst. D63, 58–61. [DOI] [PMC free article] [PubMed]
- Fu, Z., Li, X. & Merz, K. M. Jr (2011). J. Comput. Chem. 32, 2587–2597. [DOI] [PubMed]
- Fu, Z., Li, X. & Merz, K. M. Jr (2012). J. Chem. Theory Comput. 8, 1436–1448. [DOI] [PMC free article] [PubMed]
- Gore, S., Olsson, T. S. G. & Zhuravleva, M. (2011). Acta Cryst. A67, C104.
- Harding, M. M. (1999). Acta Cryst. D55, 1432–1443. [DOI] [PubMed]
- Hostaš, J., Řezáč, J. & Hobza, P. (2013). Chem. Phys. Lett. 568–569, 161–166.
- Jaskolski, M., Gilski, M., Dauter, Z. & Wlodawer, A. (2007). Acta Cryst. D63, 611–620. [DOI] [PubMed]
- Joosten, R. P., Joosten, K., Murshudov, G. N. & Perrakis, A. (2012). Acta Cryst. D68, 484–496. [DOI] [PMC free article] [PubMed]
- Kleywegt, G. J. (2007). Acta Cryst. D63, 94–100. [DOI] [PMC free article] [PubMed]
- Kleywegt, G. J. & Jones, T. A. (1996). Structure, 4, 1395–1400. [DOI] [PubMed]
- Korth, M., Pitoňák, M., Řezáč, J. & Hobza, P. (2010). J. Chem. Theory Comput. 6, 344–352. [DOI] [PubMed]
- Labute, P. (2005). J. Chem. Inf. Model. 45, 215–221. [DOI] [PubMed]
- Lack, N. A., Yam, K. C., Lowe, E. D., Horsman, G. P., Owen, R. L., Sim, E. & Eltis, L. D. (2010). J. Biol. Chem. 285, 434–443. [DOI] [PMC free article] [PubMed]
- Lascombe, M.-B., Ponchet, M., Venard, P., Milat, M.-L., Blein, J.-P. & Prangé, T. (2002). Acta Cryst. D58, 1442–1447. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Li, X., Hayik, S. A. & Merz, K. M. Jr (2010). J. Inorg. Biochem. 104, 512–522. [DOI] [PMC free article] [PubMed]
- Liebeschuetz, J., Hennemann, J., Olsson, T. & Groom, C. R. (2012). J. Comput. Aided Mol. Des. 26, 169–183. [DOI] [PMC free article] [PubMed]
- Liu, D. C. & Nocedal, J. (1989). Math. Program. 45, 503–528.
- Merritt, E. A. (2012). Acta Cryst. D68, 468–477. [DOI] [PMC free article] [PubMed]
- Merz, K. M. Jr & Raha, K. (2011). US Patent 7904283.
- Moriarty, N. W., Grosse-Kunstleve, R. W. & Adams, P. D. (2009). Acta Cryst. D65, 1074–1080. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Perola, E. & Charifson, P. S. (2004). J. Med. Chem. 47, 2499–2510. [DOI] [PubMed]
- Peters, M. B. & Merz, K. M. Jr (2006). J. Chem. Theory Comput. 2, 383–399. [DOI] [PubMed]
- Pozharski, E., Weichenberger, C. X. & Rupp, B. (2013). Acta Cryst. D69, 150–167. [DOI] [PubMed]
- Raha, K. & Merz, K. M. Jr (2004). J. Am. Chem. Soc. 126, 1020–1021. [DOI] [PubMed]
- Raha, K. & Merz, K. M. Jr (2005). J. Med. Chem. 48, 4558–4575. [DOI] [PubMed]
- Řezáč, J., Fanfrlík, J., Salahub, D. & Hobza, P. (2009). J. Chem. Theory Comput. 5, 1749–1760. [DOI] [PubMed]
- Schmidt, M. W., Baldridge, K. K., Boatz, J. A., Elbert, S. T., Gordon, M. S., Jensen, J. H., Koseki, S., Matsunaga, N., Nguyen, K. A., Su, S., Windus, T. L., Dupuis, M. & Montgomery, J. A. (1993). J. Comput. Chem. 14, 1347–1363.
- Shimizu, M., Miyamoto, Y., Takaku, H., Matsuo, M., Nakabayashi, M., Masuno, H., Udagawa, N., DeLuca, H. F., Ikura, T. & Ito, N. (2008). Bioorg. Med. Chem. 16, 6949–6964. [DOI] [PubMed]
- Smart, O. S., Womack, T. O., Flensburg, C., Keller, P., Paciorek, W., Sharff, A., Vonrhein, C. & Bricogne, G. (2011). Acta Cryst. A67, C134. [DOI] [PMC free article] [PubMed]
- Stewart, J. J. P. (2009). J. Mol. Model. 15, 765–805. [DOI] [PubMed]
- Stewart, J. J. P. (2013). J. Mol. Model. 19, 1–32. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C., Klei, H., Adams, P. D., Moriarty, N. W. & Cohn, J. D. (2006). Acta Cryst. D62, 915–922. [DOI] [PMC free article] [PubMed]
- Tronrud, D. E. (2004). Acta Cryst. D60, 2156–2168. [DOI] [PubMed]
- Vaart, A. van der, Gogonea, V., Dixon, S. L. & Merz, K. M. Jr (2000). J. Comput. Chem. 21, 1494–1504.
- Vaart, A. van der., Suarez, D. & Merz, K. M. Jr (2000). J. Chem. Phys. 113, 10512–10523.
- Wang, B., Raha, K. & Merz, K. M. Jr (2004). J. Am. Chem. Soc. 126, 11430–11431. [DOI] [PubMed]
- Wang, B., Westerhoff, L. M. & Merz, K. M. Jr (2007). J. Med. Chem. 50, 5128–5134. [DOI] [PMC free article] [PubMed]
- Williams, D. E., Peters, M. B., Wang, B., Roitberg, A. E. & Merz, K. M. Jr (2009). J. Phys. Chem. A, 113, 11550–11559. [DOI] [PubMed]
- Wlodek, S., Skillman, A. G. & Nicholls, A. (2006). Acta Cryst. D62, 741–749. [DOI] [PubMed]
- Yu, N., Yennawar, H. P. & Merz, K. M. Jr (2005). Acta Cryst. D61, 322–332. [DOI] [PubMed]
- Zhang, X., Gibbs, A. C., Reynolds, C. H., Peters, M. B. & Westerhoff, L. M. (2010). J. Chem. Inf. Model. 50, 651–661. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
50 QM refined PDB structures. DOI: 10.1107/S1399004714002260/rr5046sup1.zip
Supporting Information.. DOI: 10.1107/S1399004714002260/rr5046sup2.pdf