

Abstract
Cancer risk at low doses of ionizing radiation remains poorly defined because of ambiguity in the quantitative link to doses below 0.2 Sv in atomic bomb survivors in Hiroshima and Nagasaki arising from limitations in the statistical power and information available on overall radiation dose. To deal with these difficulties, a novel nonparametric statistics based on the ‘integrate-and-fire’ algorithm of artificial neural networks was developed and tested in cancer databases established by the Radiation Effects Research Foundation. The analysis revealed unique features at low doses that could not be accounted for by nominal exposure dose, including (i) the presence of a threshold that varied with organ, gender and age at exposure, and (ii) a small but significant bumping increase in cancer risk at low doses in Nagasaki that probably reflects internal exposure to 239Pu. The threshold was distinct from the canonical definition of zero effect in that it was manifested as negative excess relative risk, or suppression of background cancer rates. Such a unique tissue response at low doses of radiation exposure has been implicated in the context of the molecular basis of radiation–environment interplay in favor of recently emerging experimental evidence on DNA double-strand break repair pathway choice and its epigenetic memory by histone marking.
Keywords: cancer risk, low-dose radiation, A-bomb survivors, artificial neural networks, integrate-and-fire model, DSB repair pathway choice
INTRODUCTION
The Life-Span Study (LSS) of the Radiation Effects Research Foundation (RERF), which involves a long-term follow-up of atomic bomb (A-bomb) survivors in Hiroshima and Nagasaki, has provided fundamental information on cancer risk in humans following exposure to ionizing radiation. The dose dependency of cancer risk for survivors exposed to moderate-to-high radiation doses has been unequivocally established [1, 2]. However, statistical properties still limit the analysis at lower doses (<0.2 Sv) where the dose–response is imprecise, and its resolution is hampered by limited statistical power. In this circumstance, despite its unproven assumptions, a linear non-threshold (LNT) model has been pragmatically adopted in the setting of radiation protection standards [3, 4]. However, the projection of the dose–response to publicly or occupationally relevant low doses has long been a matter of intense debate [5, 6]. Limitations are also due to uncertainties with regard to radiation doses that are accountable for residual radiation, radionuclide fallout and internal exposure. Furthermore, some unexplained transient elevations of cancer risk have been noted in the 0.15–0.3 Sv dose range [7], which cannot simply be described by current radiobiological knowledge on issues such as genomic instability, the bystander effect, adaptive responses or heterogeneous genetic susceptibility [5, 8].
To deal with these complications, we developed here a novel non-parametric statistical procedure based on the noise cancellation process of the artificial neural networks (ANN) theorem, and evaluated cancer risk in A-bomb survivors exposed to low doses of radiation. ANN is a mathematical construct modeled on the ‘integrate-and-fire’ excitation response of neurons [9, 10], in which the firing of an action potential occurs when the membrane potential arising from the accumulation of small, depolarizing input signals reaches a threshold. Apart from its application to neurophysiology, the mathematical algorithm of the threshold process has been used extensively in a variety of information technology fields, including image processing, robotics, etc. [11, 12]. The rationale behind the use of the ANN theorem in cancer statistics is that, if we consider the cancer rate at given radiation dose as an input variable, the same algorithm would apply to the robust performance in dealing with noisy incomplete cancer incidence data at low doses to disclose the threshold or the dose-proportionate increase in cancer risk. When analyzing the LSS cancer databases of the RERF, the method unequivocally disclosed the threshold for some cancers where survivors were most likely exposed to an uppermost organ dose of 0.1–0.2 Sv. Surprisingly, however, instead of a zero effect or Gaussian white noise, the threshold appeared as a negative excess relative risk, indicating the suppression of spontaneously or environmentally arising background cancer.
APPLICATION OF ANN THEOREM TO CANCER RISK STATISTICS
In a single perceptron model of ANN [13], the ‘integrate-and-fire’ response, or a special case of noise cancellation process, is described by
 |
(1) |
where φ(..) is a gain function, in which f(xi) is the ith input variable with weight wi, and μ is the threshold. When the weighted sum of variables exceeds the threshold, the system is activated. In the application of the ANN theorem to cancer risk assessment, the relative risk (RR) of cancer at radiation dose x, i.e. f(x), is regarded as an input variable, and we tentatively dropped the threshold term because the threshold in cancer development is not known a priori. Consequently, we will hereafter consider a simplified ANN for a model-free random pattern describing a weighted sum of RR:
 |
(2) |
where n is the number of datapoints and w is a weighting defined by humming distance wi = xi − xi−1. RR is denoted as the number of cancer cases in the exposed population relative to that in the control population. If the study population is sufficiently large and cancer is refractory to radiation doses, the RR may be randomly distributed around the mean μ0 = 1 (Gaussian white noise). Since the excess relative risk (ERR) is given by ERR = RR − 1, the weighted sum of ERR is expressed by
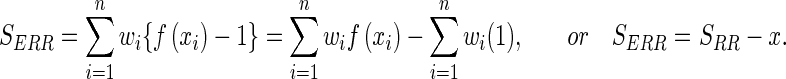 |
(3) |
SRR is optimized by fitting to a continuously differentiable polynomial function by the maximum likelihood method combined with bootstrap resampling with weighting by the inverse function of RR. The optimized SRR and its differential at dose x, RR(x) and ERR(x), are expressed by continuous probability density functions:
 |
(4) |
where θi is the coefficient of the ith term of the polynomial. The optimal degree of polynomial, m, was determined by Akaike information criterion (AIC) [14] that minimizes
 |
(5) |
where s2 is the maximum likelihood.
DATA SOURCES AND DATA ASSESSMENT FOR ANN ANALYSIS
The ANN statistics test was performed on two cancer databases, ‘lssinc07’ and ‘DS02can’, of the LSS cohort of A-bomb survivors, which are publicly available from RERF (http://www.rerf.or.jp). The database ‘lssinc07’ is the source term data used in Preston et al. [1] and contains cancer incidence (morbidity) data for 111 952 subjects followed up during the period 1958–1998, in which information on the occurrence of cancer (X) and its site is cross-tabulated in a total of 26 806 data cells (records) with city (c), gender (s), person year at risk (PY), age at the time of bombing (ATB, a), attained age (t), radiation dose (x) determined by the dosimetry system 2002 (DS02), and so on. The database ‘DS02can’ is the source term data used in Richardson et al. [2] and contains data on deaths (mortality) from all solid cancers combined or liquid cancers followed up during the 1950–2000 period on a total of 86 611 subjects compiled in 33 219 data cells, together with additional information as in ‘lssinc07’. Liquid cancers include leukemia, lymphoma and multiple myeloma, in which leukemia constitutes 46.5% of all cases. The analyses were carried out for solid cancer in the ‘lssinc07’ incidence database and for liquid cancer in the ‘DS02can’ mortality database. Subjects exposed to doses below 0.005 Gy, including persons who were not in the city at the time of bombing (NIC), were treated as controls. The RR was calculated according to the formula RR = [X/PY]csat/λcsat, where X is the observed number of cancer cases in the exposed population, and λcsat is the expected frequency in the control population, in which cancer rate is expressed as a logistic function of attained age (t), λcsat = u/[1 + k1·exp(−k2t)], for each city-, gender- and ATB-category. The calculation of age-dependency, t and a, was made with a 5-year interval. Neutron dose was weighted by the dose-dependent variable relative biological effectiveness (RBE) against γ-rays, Rγ,n, as previously described [15], except for dose to the skin in which only neutron dose weighted by constant RBE = 10 was available. Liver dose was applied to organs in the upper trunk, i.e. oral, brain, esophagus, liver, breast, lung, stomach, gallbladder, pancreatic and thyroid cancers, and the colon dose was applied to all solid cancers combined and organs in the lower trunk, i.e. colon, rectum, ovary, cervix, urinary organ and prostate cancers. Bone marrow dose was used for liquid cancer.
To avoid systematic distortion of the distribution of RR around the mean, a population size that was large enough to satisfy RR > 0 was determined by moving window averaging (MWA). For instance, an MWA = w1000s50 implies that a group of 1000 data cells is consecutively moved forward in increasing order of dose with a step size of 50 data cells. The mean dose of the window is expressed as the PY-weighted dose. Although the minimum window size that satisfied RR > 0 was w100 for most solid cancers and w500 for liquid and thyroid cancers, the analysis was carried out with an MWA = w1500s50 and a cut-off dose to survivors, Dmax, of 3 Sv unless otherwise stated.
THRESHOLD AT LOW DOSES IS MANIFESTED AS A NEGATIVE ERR
To begin, we performed a cross-validation of methods—the conventional piecewise dose-category method (e.g. [1]) and the present ANN method—used to calculate the ERR of solid cancers in a combined Hiroshima and Nagasaki cohort. To avoid any effects of the use of different neutron weighting systems, the analysis was performed using a constant RBE = 10 for neutrons. The results of the two methods matched reasonably well for the overall pattern of dose–response; including an abnormally elevated ERR at low doses (see Supplementary figure Fig. S1). However, unless otherwise stated, the ANN analysis was performed using the dose-dependent variable RBE of neutrons (RBE = Rγ,n), and extended to the characterization of city differences, gender specificity and ATB effects with particular attention paid to the response at low doses.
The procedures of the ANN analysis are shown in Fig. 1 using an example of lung cancer cases in Hiroshima male survivors exposed at ATB (20+), this being 20 years or older at the time of bombing. Unexpectedly, the integrated ERR, SERR = SRR − x, was less than zero at low doses, it progressively decreased with dose to reach a minimum, and then increased with further increases in dose (Fig. 1a). This implies that, instead of zero risk (canonical definition of the threshold or random Gaussian fluctuation), the threshold was identified as a negative ERR, or manifested as a reduction of the background cancer rate, in a dose-independent manner at low doses. Here we call this negative ERR at low doses the ‘threshold’ although it differs from the conventional definition. The dose limit of the threshold, t1, was determined that satisfies the derivative S′ERR = 0 by the Lagrange derivative interpolation method. The linearly decreasing part (x ≤ t1) of SERR was fitted to a linear regression SERR = κ+ μx, the derivative of which gives the ERR at threshold, ERR = μ. The dose, t2, at which SERR crosses the SERR = 0 axis, corresponds to the dose above which all survivors within a moving block were exposed to a supra-threshold dose of radiation (dose x > t1). Thus, the ERR is discontinuous with a breakpoint at t1, followed by a transient phase t1 < x < t2, and then a dose range without threshold response (x > t2; Fig. 1b). The suppression-independent, non-threshold, dose–response may be constructed only for ERR datapoints in the dose range x > t2 (dotted line in Fig. 1b). We call this the ‘reference dose–response’, which represents induction kinetics unrelated to the low-dose suppression, or the universally valid induction function; this can be fitted to a linear-quadratic function of dose described by the equation: y(x > t2) = αx + βx2. Parameters for the threshold and polynomial coefficients of the best fit to the probability density function of ERR calculated in this way for each cancer type are presented in Supplementary data Table S1.
Fig. 1.

Data processing by the ANN method (example). Lung cancer incidence in Hiroshima A-bomb male survivors exposed at ATB (20+). MWA = w1000s50, Dmax= 3 Sv. (a) SRR and SERR are the weighted sum of RR and ERR, respectively, fitted to a polynomial function of dose by maximum likelihood method. (b) The continuous probability density function of ERR (solid line) obtained by calculating S′RR − 1 according to Eq. 4 is compared with the observed ERR data with 80% confidence intervals (CIs) connected by spline interpolation. t1 = 0.32, t2 = 0.55, μ = −0.12. The reference dose–response (dotted line) is calculated according to: y(x > t2) = (0.359 ± 0.066)x + (0.219 ± 0.066)x2.
SOLID CANCERS: RESPONSE CHARACTERISTICS BY CITY, GENDER AND CANCER SITE
Figure 2a shows the ANN analysis of all solid cancers in a combined Hiroshima and Nagasaki cohort. Although the neutron dose was weighted by dose-dependent RBE, an overall dose–response was not much different from that obtained using an RBE = 10. A small-scale threshold was indicative in SERR response at low doses (Fig. 2a-1), which was followed by an atypical elevation of ERR at 0.2–0.4 Sv (Fig. 2a-2). However, when the data for Hiroshima and Nagasaki survivors were treated separately, it became evident that this elevation of ERR at low doses was a reflection of the situation in Nagasaki survivors (Fig. 2b), and mainly due to cancers of the lung, liver and gallbladder (Fig. 2c–e), but not to other solid cancers included in the screening protocol. When males and females were treated separately, the threshold was evident in males in Hiroshima (Fig. 2f). However, in Nagasaki, a transient elevation of ERR at low doses occurred both in males and females and tended to mask the threshold that was potentially present in survivors from that city (Fig. 2g).
Fig. 2.

Solid cancers by city and gender. (a) All solid cancers in a combined Hiroshima and Nagasaki cohort (T). Panel 1: SRR and SERR. Panel 2: ERR. (b) City-based difference of all solid cancers. (c) Difference by city for ERR for lung cancer. (d) Difference by city for ERR for liver cancer. (e) Difference by city for ERR for gallbladder cancer. (f) Gender difference for ERR for all solid cancers in Hiroshima. (g) Gender difference for ERR for all solid cancers in Nagasaki. (h) City- and gender-adjusted ERR for all solid cancers in general populations with anticipated 1:1 gender ratio. H = Hiroshima, N = Nagasaki, T = two cities combined, M = male. F = female.
A significant difference in dose–response pattern between the two cities of Hiroshima and Nagasaki at low doses may give rise to a problem in the use of combined two-city data for risk projection extrapolated to the general population. The male/female ratio differed considerably from unity, as was also the case for other areas during the second world war, and varied with ATB; for instance, it was 0.3–0.4 for ATB (20–40), whereas it was 0.8–0.9 for ATB (0–20) and ATB (40–60). Since the same trend was also the case for control populations, such age structures might have had little effect on the relative risk of cancer for each sex. Adjustments for the city in question may however be needed because, in terms of population size, the Hiroshima-to-Nagasaki ratio of the exposed populations is 0.76:0.24. Figure 2h shows the ERR of solid cancers in an anticipated 1:1 gender ratio for Hiroshima (H), Nagasaki (N) and combined-city (T) populations after adjustment for city ratio and gender ratio differences. As expected, adjustment according to the gender ratio distribution among ATB groups had no effect on outcome. In contrast, however, although the Nagasaki cohort comprised 24% of the total combined population, an abnormal elevation of ERR at low doses (≤0.4 Sv) was still apparent for the two cities combined, and hindered the construction of a joint dose–response relationship in a simple form. The reference dose–responses (threshold- and low-dose abnormality-unrelated) are described by yH(x>t2) = (0.501 ± 0.065)x + (0.088 ± 0.008)x2, yN(x>0.4) = (0.512 ± 0.017)x + (0.061 ± 0.020)x2 and yT(x > 0.4) = (0.511 ± 0.070)x + (0.057 ± 0.081)x2, for Hiroshima (H), Nagasaki (N) and the two cities combined (T), respectively. The reference dose–responses were very close to each other at high doses, and characterized by the presence of a small but significant dose-quadratic term.
The ANN statistics were then applied for to analyze organ specificity and gender specificity in Hiroshima survivors (Fig. 3). The threshold level differed by cancer site and gender, being prominent in males for cancers of the esophagus, stomach, pancreas, gallbladder, liver, lung, urinary organs and brain (central nervous system), and both in males and females for skin cancer. The threshold was also seen in breast, prostate and ovarian cancer, but not in colorectal or thyroid cancer for males or for females. A small threshold was evidenced for oral cancer in females but not in males. Breast cancer in females increased proportionally to dose following a small threshold at low doses (t1 = 0.107 Sv, μ = − 0.022). This contrasted to cervical cancer, which showed a complete lack of response to dosage (Fig. 3n). Such was also the case for breast and cervical cancer in Nagasaki (data not shown), indicating a prevailing role of human papillomavirus (HPV) in the causation of cervical cancer.
Fig. 3.

Difference in ERR by gender and organ (Hiroshima). (a) Oral cancer. (b) Esophageal cancer. (c) Stomach cancer. (d) Colorectal cancer. (e) Pancreatic cancer. (f) Gallbladder cancer. (g) Liver cancer. (h) Lung cancer. (i) Thyroid cancer. (j) Cancer of urinary system. (k) Skin cancer (non-melanoma). (l) Brain cancer (cancer of central nervous system). (m) Prostate cancer. (n) Breast and cervical cancer. (o) Ovarian cancer. M = male, F = female.
LIQUID CANCERS, INCLUDING LEUKEMIA
Liquid cancers (combined leukemia, lymphoma and multiple myeloma) were studied in the mortality data. The dose–response of ERR was quite different between cities (Fig. 4a). In Hiroshima, the dose–response of ERR was similar to that of incidence (morbidity) data for solid cancers described above; i.e. there was a distinct threshold that was followed by a dose-proportional increase in ERR both in males and females (Fig. 4b). The reference dose–response was y(x > t2) = (0.924 ± 0.066)x + (0.563 ± 0.053)x2 and y(x > t2) = (0.888 ± 0.147)x + (0.737 ± 0.104)x2, for males and females, respectively. The threshold parameters were t1 = 0.187 Sv, t2 = 0.271 and μ = − 0.105 for males and t1 = 0.239 Sv, t2 = 0.598 and μ = − 0.190 for females. For the Nagasaki data, the dose–response patterns differed not only from those of Hiroshima but also by gender (Fig. 4c). Surprisingly, however, when factory workers were excluded from the analysis, gender differences completely disappeared (Fig. 4d). However, the overall response pattern still differed from that of Hiroshima in that it showed a large-scale threshold expanding over a wide dose range up to 1 Sv, although it was superimposed by a transient increase of risk at low doses as seen for solid cancers in Nagasaki. Because of this unique feature of Nagasaki survivors, data for the two cities cannot be combined for generalization. Figure 4e shows the gender-adjusted dose–response for Hiroshima (H), Nagasaki (N, non-factory workers) and the two cities combined (T) cohorts. In Hiroshima, the ERR increases in a relatively monotonic fashion following the appearance of a distinct threshold at low doses. This contrasts to Nagasaki, where a small increase in ERR at low doses was followed by a long-range suppression of ERR up to 1 Sv, and then by a dose-proportional increase at higher doses. The increase at high doses was comparable to that in Hiroshima. For generality, the reference dose–response could be represented only by that of Hiroshima, which is yH(x > t2) = (0.433 ± 0.025)x + (0.731 ± 0.028)x2, where t1 = 0.178 Sv, t2 = 0.404 Sv and μ = − 0.125. The response kinetics was very similar to those of solid cancers.
Fig. 4.
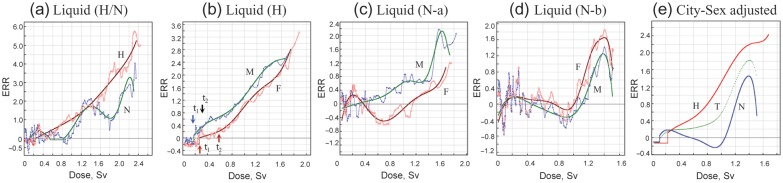
Liquid cancer. (a) City differences for ERR. (b) Gender difference for ERR in Hiroshima. (c) Gender difference for ERR in Nagasaki (including factory workers) (N − a). (d) Gender difference for ERR in Nagasaki (excluding factory workers) (N − b). (e) The dose–responses after city- and gender-adjustment. For Nagasaki, adjustments were made using non-factory workers. M = male, F = female, H = Hiroshima, N = Nagasaki, T = two cities combined.
EFFECTS OF AGE AT THE TIME OF BOMBING (ATB)
The effects of age at the time of bombing (ATB effects) were studied in Hiroshima survivors, with the results summarized in Fig. 5. The threshold was more pronounced in younger ATB groups for solid cancers, being particularly more evident for liquid cancers (Fig. 5a and b). This can be seen more clearly in the weighted sum of ERR (Fig. 5a-1 and b-1). The age-dependence of ERR was generally the same as that reported previously [16–18], in that the ERR was higher for younger ATB both for solid and liquid cancers (Fig. 5a and b). An exception to this rule was ATB (20–30), which showed the highest ERR at low doses. Since the male-to-female ratio of this group was low (0.29:1), the ERR patterns for males and females were treated in proportion to the gender ratio and compared with those of ATB (0–20), where the male-to-female ratio was nearly 1:1. As seen in Fig. 5c–f, the atypical aspect of the ATB (20–30) group was due to the elevated ERR at low doses in female survivors, both for solid and liquid cancers. The reason for this is not clear, but may be due to an artificial downwards shift of the dose rather than a consequence of biological factors. In the early years after the bombing, when knowledge of the heritable effects was lacking, it is likely that there were some biases in registering dose-related exposure conditions experienced by young unmarried women. Indeed, a decreased marriage rate was noted for Hiroshima and Nagasaki women of young ATB due probably to the anticipated discriminatory bias for marriage [19].
Fig. 5.

Effects of ATB (Hiroshima). (a) Solid cancers. (b) Liquid cancers. Panel 1 shows SERR and Panel 2 shows ERR at each ATB. (c) ERR for solid cancers for ATB (0–20). (d) ERR for solid cancers for ATB (20–30). (e) ERR for liquid cancers for ATB (0–20). (f) ERR for liquid cancers for ATB (20–30). M = male, F = female.
DISCUSSION AND CONCLUSION
A new approach to using ANN statistics has been shown here to provide a powerful means for defining cancer risk in response to exposure to low doses of radiation. The systematic distortion of the dose–response pattern of cancer risk at low doses of radiation exposure was evident in relation to (i) an abnormal elevation of risk for some cancers, specifically in Nagasaki, and (ii) the organ- and gender-specific appearance of a threshold, appearing as a negative excess relative risk at low doses. The origin of the transient heightening of cancer risk at low doses of radiation exposure in Nagasaki remains to be elucidated, but it nevertheless had an impact on the overall dose–response pattern of cancer risk. For any effects of radiation to be observed other than direct flash radiation, such as the spreading of radioactive fallout and/or secondary radionuclides, the distribution of standardized incidence (or mortality) ratio, SIR or SMR, of solid cancers against ground distance from the hypocenter must be studied. SIR and SMR were calculated using the ANN method, as described above for RR, i.e. SIR = RR = [X/PY]csat/λcsat, but in this case as a function of ground distance from the hypocenter rather than radiation dose (Fig. 6). Cancer incidence was standardized in the NIC cohort that constituted residents who were not in the city at the time of the bombing. In terms of ground distance from the hypocenter, the controls (<0.005 Gy) correspond to those at about 2.5 km or farther from the hypocenter. In Hiroshima, the SIR in distal survivors (<0.005 Gy) was significantly higher than unity (Fig. 6a). A similar result was noted by Watanabe et al. [20], who used the entire populations of the Hiroshima and Okayama prefectures as reference populations. A monotonic increase in SIR in distal survivors over a wide range of ground distance suggests that this elevated level of SIR could be a false positive result due to geographic variability in the reference populations, as suggested by Grant et al. [21]. The SIR was then normalized to that of the farthest distance, which was 3.84 km (range, 2.30–7.58 km) for Hiroshima and 3.79 km (2.49–6.97 km) for Nagasaki. As seen in Fig. 6b, there was no indication of an increase in SIR that could not be accounted for by primary flash radiations in Hiroshima. However, in Nagasaki, the elevation of SIR continued by passing across the 2.5 km point and beyond. The same trend was also observed for SMR (Fig. 6c), in which the mortality ratio was standardized in the control cohort because the ‘DS02can’ mortality database did not include NIC groups.
Fig. 6.
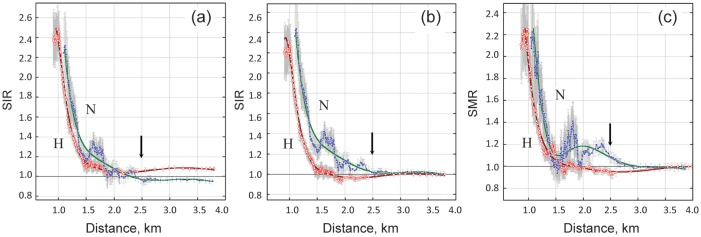
Distribution of SIR and SMR of solid cancers with ground distance from the hypocenter. (a) SIR standardized by NIC as the reference population (not normalized). (b) SIR standardized by NIC as the reference population (normalized by farthest value set at SIR = 1). (c) SMR standardized by nominal control population (<0.005 Gy) as the reference population (normalized by farthest value being set at SMR = 1). Arrows indicate the critical distance beyond which no survivors were exposed to doses of x ≥ 0.005 Gy.
The long tail of the SIR or SMR in Nagasaki is likely to be a reflection of continued exposure to alpha particles emitted from internally deposited 239Pu. Indeed, 239Pu fallout has been observed in the eastern area of Nagasaki at a distance ≥ 1 km from the hypocenter [22–25]. Furthermore, an unusually high level of alpha tracks has been observed in autopsy samples from Nagasaki victims [26]. The biological half-life of 239Pu (fuel material in the Nagasaki bomb) is about 200 years, which is extremely long compared with the 15–100 days for the 235U used in the Hiroshima bomb [27].
A most interesting finding made here was the threshold response at low doses, which was unique in that it manifested as a negative ERR, or in other words a suppression of the background cancer rate. The biological mechanism behind this threshold response is unclear. However, allowing for a considerable variability in the estimated dose within an organ, the critical dose below 0.1–0.2 Sv is reminiscent of that for the radio-adaptive response (ADR) [28–30]. ADR is a well-documented phenomenon by which irradiation with low linear energy transfer (LET) radiation, such as X- or γ-rays, at low doses (typically < 0.1 Gy) renders cells resistant to the induction of mutation and chromosome aberrations by subsequent exposures to radiation or chemical genotoxins. The low doses of low-LET radiation alone reduce spontaneously occurring chromosome aberrations and malignant transformation in cultured mammalian cells [31–34]. Moreover, irradiation of mice with low dose, or low dose-rate, γ-rays has been reported to suppress spontaneously occurring as well as chemical-induced mutations and tumors [35–42]. All available evidence points the possibility that low-doses of low-LET radiation activate a sustained-response, error-free DNA repair system.
Obviously, the challenge of understanding the underlying molecular basis of cancer epidemiology requires further research and discussion. It is tempting nevertheless to speculate on possibilities within the context of recently emerging experimental evidence concerning the DNA repair pathway choice of cells in response to DNA double-strand breaks (DSBs). DSBs are produced by ionizing radiation, DNA replication forks stalled by bulky adducts, or by other causes, and present a major threat to genetic integrity; they are, consequently, a leading cause of chromosome aberrations and cancer in cells exposed to ionizing radiation and chemicals [43, 44]. DSBs are repaired either by homologous recombination (HR) or by non-homologous end-joining (NHEJ) processes that are present in cells as distinct and competitively operating pathways [45, 46]. Recently, two subpathways have been identified for NHEJ, i.e. canonical NHEJ (C-NHEJ, also called D-NHEJ or the DNA-PK-dependent pathway) and alternative NHEJ (Alt-NHEJ, also known as B-NHEJ or the backup pathway) [refs. 47–49 for review]. The molecular nature and biological significance of these pathways are distinct; C-NHEJ is a rapid and precise high-fidelity process that mediates direct joining of two broken ends, whereas Alt-NHEJ is a slow and error-prone process thought to be a primary mediator of mutations since it often requires end processing and uses sequence homology of various lengths at a junction (microhomology-mediated end-joining, MMEJ) [50]. Briefly, the former employs Ku70/80 end-binding proteins associated with DNA-PKcs to form a holoenzyme that phosphorylates Artemis (Cernunnos) endonuclease, DNA polymerase μ/λ, and the XRCC4/ligase IV/XLF complex for ligation, whereas the latter uses PARP1 for DSB recognition followed by CtIP/MRN endonuclease, DNA polymerase β and XRCC1/ligase III. In mammalian cells, the two NHEJ pathways have been proposed to contribute equally [51]. HR is initiated by DSBs generated during replication, where the DSB end is resected to give rise to a 3′ overhang, which recruits Rad51 recombinase and invades an intact homologous duplex to use it as a template for repair by polymerase δ/ε and ligase I. HR is often described as ‘error-free’, but its recombination processes at replication arrest can result in large-scale genome rearrangements such as deletions and translocations that could contribute to cancer [52–54]. Low doses of low-LET radiation stimulate the DNA-PKcs/Ku-dependent NHEJ pathway, and then render cells resistant to subsequent high doses of radiation [55]. The choice of the precise rejoining pathway by low-dose irradiation has been confirmed in the in vitro DSB rejoining assay system [56, 57], where the high fidelity rejoining pathway is activated in low-dose irradiated cells, which in turn suppresses microhomology-mediated misrejoining that is otherwise observed in high-dose irradiated cells. Similar to the dose limit for ADR [29] the critical dose for pathway switching is ∼ 0.1 Gy [58].
A most significant observation of the repair process is the crosstalk among repair pathways; i.e. the activation of C-NHEJ suppresses Alt-NHEJ and HR [47–49]. The discriminating signaling processes, e.g. cell cycle phase dependence, end-stabilization (Ku70/80 vs PARP1), end-processing (inhibition by Rif1/53BP) and/or kinase specificity (DNA-PKcs vs ATM/ATR), have been reported for their co-regulation and pathway choice [59–77]. It is noteworthy that 53BP1 (p53 binding protein) is essential for the Ku70/DNA-PKcs/Artemis-dependent NHEJ pathway (C-NHEJ), but not for HR [74, 75]. Rif1 (Rap1 interaction factor 1) cooperates with 53BP1 to inhibit the 5' end resection needed for Alt-NHEJ and HR [76, 77]. Interaction between tumor suppressor protein p53 and 53BP1 is crucial for end resection, where 53BP1 binds to the core residue at the BRCT domain but not to mutant p53 [78]. These lines of evidence are consistent with cytogenetic observations that activation of the ADR response is strictly dependent on wild-type p53 but refractory to mutant p53 or abrogation of p53 by transcriptional silencing [29, 30, 79, 80]. The p53 is directly phosphorylated by p38MAPK [81, 82]. We previously found that p38MAPK is specifically activated by low-dose X-rays but downregulated by high doses [83]. p38MAPK has been also reported to mediate dose-dependent biphasic activation of signaling molecules by γ-rays and hydrogen peroxide [84–86]. It is thus likely that the p38MAPK/p53/53BP1 damage-sensing network plays a pivotal role in ensuring a switching DSB repair pathway to error-free mode. During S phase, BRCA1 antagonizes 53BP1, where there is also redundancy with p53 [77, 87–89]. The impairment of HR has been described as suppressing spontaneous and radiation- or chemically induced sister-chromatid exchange (SCE, a cytogenetic manifestation of HR) [90] and chromatid aberration, but at a cost of cell killing [91–94]. In support of this, low-dose irradiation renders cells hypersensitive to killing [95] and reduces chemically induced SCE [96].
Unlike the direct breakage of the DNA duplex by ionizing radiation, DSBs are also generated when replicating DNA encounters endogenous lesions (mostly single-strand breaks) or lesions associated with exogenous genotoxins including ultraviolet light (UV) and chemicals. The replicating DNA disrupted at the replication fork by DNA lesions may eventually be collapsed, producing DSBs that are subject to repair by HR and NHEJ. The suppression of error-prone HR and Alt-NHEJ by low-dose radiation may reduce chromosome aberrations and erroneous crossover (albeit enhancing cell killing) and hence mitigate cancer. Recently, DSBs of endogenous origin have been estimated to be 20–100 times lower in prevalence than previously estimated, and more likely to occur at a rate of ∼ 1 DSB/replication cycle in the human genome [97]. This raises the importance of DNA lesions associated with exogenous genotoxins, such as metabolites of cigarette smoke, alcohol, environmental sources, nutrition and dietary mutagens, which are major causes of human cancer [98–100]. The prevalence of threshold in cancer of the esophagus, lung, stomach and pancreas in male survivors suggests that smoking and alcohol consumption could be significant factors, since they are major cancers causally related to smoking and alcohol consumption [101]. Tobacco smoke contains at least 60 chemical carcinogens, of which many form bulky DNA adducts [102]. Acetaldehyde, a metabolite of ethanol, also forms DNA adducts and is causally related to the development of various cancers of the gastrointestinal tract, typically esophageal cancer [103]. The DNA adducts are removed by base excision repair (BER) or nucleotide excision repair (NER), or when they meet to DNA replication stall replication at fork and give rise to DSB, which is subject to repair by HR [104]. UV-induced lesions also cause replication arrest, leading to the development of DSBs and activation of HR; these processes have been correlated with the development of skin cancer [105]. Collectively, it is likely that the HR pathway is repressed by low-dose ionizing radiation, reducing the mutagenic rejoining at fork arrests and eventually suppressing cancer associated with smoking, alcohol and UV. Given the interplay between low-dose radiation and genotoxins, of serious concern is the fact that the acetaldehyde-detoxifying gene, aldehyde dehydrogenase-2 (ALDH2) is polymorphic, and its inactive variant ALDH2*Glu540Lys is prevalent in east Asian populations (about 40% in Japan) in contrast to populations of Caucasian ancestry where the prevalence of this variant is almost nil [106, 107].
Smoking and alcohol consumption are also risk factors for the development of oral cancer. While the dose–response pattern of ERR was similar to that of esophageal cancer (Fig. 3a and b), the low-dose threshold was not observed in males. The reason for this is not clear, but it could be related to the interaction of two risk factors that are supermultiplicative for oral cancer and submultiplicative for esophageal cancer [108]. Moreover, an involvement of human papilloma viruses has also been noted for oral cancer [109].
The reason for the unique dose–response pattern for liquid cancers in Nagasaki remains an open question. Difference associated with gender, and their disappearance when factory workers were excluded from the cohort analysis, strongly suggest an involvement of adult T-cell leukemia/lymphoma (ATLL), because the ATLL is endemic in southwestern Japan, including Nagasaki, and the factory workers were not necessarily Nagasaki native citizens, but many would have been conscripted or mobilized from other parts of Japan. ATLL is a pathological manifestation of HTLV-1 infection with long-term latency period. The infection itself is asymptomatic but the stable integration of viral DNA into transcriptionally active regions of the host genome is critical for the onset of disease [110]. The integration of retroviral DNA is a multistep process and anticipated to be associated with the DSB repair system [111]. The role played by DSB repair mechanisms in retroviral integration remains unclear and is not free of controversy; some cell-based studies have shown that deficiencies in early sensors of NHEJ, i.e. DNA-PK kinase and MRN nuclease, or overexpression of Rad51 or Rad52 recombination proteins, but not other Rad52 paralogs, downregulate the retroviral integration [112–117], while others have shown that damage sensors for NHEJ, including DNA-PK, are not critical [118]. The suppressive effects of Rad51/Rad52 overexpression could be due to a steric hindrance of integrase, a retroviral recombinase, by competition between functional domains shared by the two proteins [113, 119], since the overexpression of Rad51 is known to suppress DSB-induced HR [120, 121]. For instance, in cells infected with HIV-1 (human immunodeficiency virus 1), Rad51 proteins (a major component of HR) are recruited and promote viral DNA integration [122]. On the other hand, the activation of DNA-PK, a key component of C-NHEJ, inhibits retroviral DNA integration [123]. These findings are consistent with the DSB repair pathway choice in response to low-dose radiation; i.e. suppression of HR by activation of the C-NHEJ pathway. Since the impairment of HR does not alter the efficiency of NHEJ [124], HR may play a critical role in the retroviral integration. Therefore, the suppression of liquid cancers at low radiation doses (<1 Sv) in Nagasaki could be a consequence of the activation of the C-NHEJ pathway by low doses of radiation, leading to downregulation of HR-mediated HTLV-1 DNA integration in virus carriers. Incidentally, in a recent survey dealing with ATLL in Nagasaki, HTLV-1 seroprevalence and lifetime risk of ATLL in carriers were found to be 11.82 and 7.29, and 15.6 and 3.78%, for males and females, respectively [125]. The lifetime HTLV-1 prevalence rate in non-endemic areas of Japan is currently about 1.5% although a progressive yearly increase has been noted that is due to population movement [126]. The inclusion of migrant factory workers from non-endemic areas may have distorted the dose–response of liquid cancers, particularly at low radiation doses.
Here, the low-dose response of cancer induction has been discussed in favor of the DSB repair pathway choice. For the pathway choice to have an effect, the activation should be sustainable for a long period of time after exposure. ADR by low doses of low LET radiation has been associated with the enhanced repair of DSBs by C-NHEJ pathway, since it is dependent on Ku and DNA-PKcs [127, 128] and inhibited by high-LET radiation [129]. The experimental evidence for the sustainability of ADR is not consistent; it rarely exceeds 2 d in mammalian cells cultured in vitro. However, it is much longer in animals irradiated in vivo, e.g. lasting at least for weeks, months, and even life-long depending on the experimental protocol used [130–132]. Thus, the sustainability of the signaling process seems to be different between cultured cells and whole body, probably due to its incorporation in stem cells in the latter. ADR is also inducible in spermatogonial cells of mice, but it is not transmitted to offspring [131]. In A-bomb survivors, radiation exposure influences the risk of lung cancer associated with tobacco smoking, including post-bombing smoking history, where the joint effects are less than additive; in this way it has been reported that radiation exposure tends to decrease the risk of smoking-associated lung cancer more efficiently in heavier smokers [133–135]. A more pronounced effect of radiation exposure on heavier smokers is likely because the repair mechanism depends on the types and amount of damage in the genome. Frankenberg-Schwager et al. [136] showed that, in the repair of post-replication-derived DSBs, HR is more important than NHEJ for the repair of complex DSBs, while NHEJ plays a major role in the repair of simple DSBs. Unlike HR, status of C-NHEJ and p53 does not affect the efficiency of other pathways such as NER, mismatch repair (MMR) and SSB repair (TDP1) [80]. Tobacco smoking- and alcohol-related cancers are not simply proportional to the dose, but often show a J-shaped dose–response reflecting a different burden on the repair pathways depending on the amount of damage to the genome [137–139]. In this context, evidence is accumulating concerning the epigenetic memory of the DSB repair pathway choice, namely a crosstalk between the particular type of DSB repair and histone modification, called ‘histone code’ theory [140–142]. The tail domains of histones are modified by acetylation, methylation, phosphorylation or sumoylation in a manner that is specific to the repair pathway, i.e. NHEJ or HR [143, 144]. It is noteworthy that the lifetime epigenetic memory of the stress response in Drosophila is associated with the phosphorylation of activating transcription factor 2 (ATF2) by stress-activated protein kinase p38MAPK [145]. Indeed, ATF2 is activated in response to low doses of X-rays by p38MAPK via PKCα-p38MAPK-PLCδ autoregulatory circuitry signaling loops and downregulated by high doses [38, 146].
PERSPECTIVES
Due largely to a limited statistical power at low doses in A-bomb survivors, cancer risk is often expediently correlated linearly with dose down to zero dose without threshold and expressed on a ‘per-Sv’ basis [147]. The application of the ANN method developed here circumvents this difficulty and unequivocally demonstrates for the first time the presence of a threshold of excess relative risk in humans exposed to ionizing radiation. However, the threshold was fundamentally different from that of the canonical definition of zero effect until the dose reached a critical point, but instead it was manifested as a reduction of background cancer rate. Therefore, cancer risk at low doses is not a simple extension of that seen with moderate-to-high radiation doses. Obviously, much remains to be determined when considering the reasons underlying such a unique response. Here we hypothesized that the response should be considered in the context of radiation–environment interplay, favoring recently emerging experimental evidence involving DSB repair pathway choice and epigenetic memory by histone code theory. In this way, activation of the high fidelity C-NHEJ (or ADR) pathway, although not excluding joining of illegitimate ends, by low doses of low-LET radiation suppresses the microhomology-mediated error-prone Alt-NHEJ or HR pathways, and hence mitigates the genomic insult caused by DSBs arising from radiation exposure or fork arrest by genotoxins. Such a choice is not the case for high doses (>0.1 Gy) or high-LET radiation, which not only are unable to elicit ADR but also invalidate the ADR response to low-LET radiation [148, 149]. The low dose abnormality in Nagasaki could be a case in question.
Considering the appearance of the threshold as a consequence of radiation–environment interplay or lifetime genotoxin experience that is not intrinsic to radiation per se, the setting of radiation protection standards cannot be made on the premise of lifestyle due to vast individual variability. Otherwise, cancer risk will be underestimated for persons with a healthy lifestyle, children and for persons exposed to high-LET radiation. As a precautionary principle, we propose the use of a ‘reference dose–response’ function as a risk factor for a universally valid protection standard, which provides a threshold-unrelated measure of cancer risk (x > t2). In this way, a gender-averaged ERR is best expressed in the following ways:
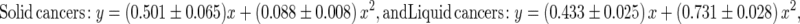 |
where x is the radiation dose in Sv. With the exception of high-LET radiation, the response function itself does not necessarily provide a measure of risk assessment at low doses, because the extrapolation to zero dose, or the presence or absence of the threshold, is related to the unforeseen factors such as genotoxin experience, and probably of post-exposure history as well. The issue also evokes questions concerning the so-called ‘radiation hormesis’ (beneficial effect) and ‘healthy worker effect’ in some cancers in populations exposed to low doses of low-LET radiation [150–153]. These must be reconsidered in view of the negative interaction between radiation and environment. To date, confounding factors have often been considered in terms of additive or synergistic interactions. The present observations may lead to a new paradigm for explaining the molecular basis of cancer epidemiology in human populations exposed to low doses of ionizing radiation. Furthermore, granted the interplay between low doses of ionizing radiation and environmental carcinogens, caution must be taken in risk transferring across populations in the context of genetic polymorphisms of redox genes such as ALDH2 and the prevalence of HTLV-1, in which disease mechanisms are closely related with the DSB repair pathway.
SUPPLEMENTARY DATA
Supplementary data is available at the Journal of Radiation Research online.
CONFLICT OF INTEREST
The authors have no conflicts of interest to report.
Supplementary Material
ACKNOWLEDGEMENTS
We thank T. Sobue (Department of Epidemiology, Graduate School of Medicine, Osaka University) and D. A. Pierce (Department of Public Health and Preventive Medicine, Oregon Health and Science University) for valuable comments. This report makes use of data obtained from the Radiation Effects Research Foundation (RERF) in Hiroshima, Japan. RERF is a non-profit private foundation funded equally by the Ministry of Health, Welfare and Labor of Japan and the U.S. Department of Energy through the U.S. National Academy of Sciences. The conclusions in this report are those of the authors and do not necessarily reflect the scientific judgment of the RERF or its funding agencies. The authors alone are responsible for the content and writing of this report.
REFERENCES
- 1.Preston DL, Ron E, Tokuoka S, et al. Solid cancer incidence in atomic bomb survivors: 1958–1998. Radiat Res. 2007;168:1–64. doi: 10.1667/RR0763.1. [DOI] [PubMed] [Google Scholar]
- 2.Richardson D, Sugiyama H, Nishi N, et al. Ionizing radiation and leukemia mortality among Japanese atomic bomb survivors, 1950–2000. Radiat Res. 2009;172:368–82. doi: 10.1667/RR1801.1. [DOI] [PubMed] [Google Scholar]
- 3.US National Academy of Sciences, Committee of the Biological Effects of Ionizing Radiation. Washington DC: Academic Press; 2006. BEIR VII Phase 2. Health Effects from Exposure to Low Level of Ionizing Radiation. [Google Scholar]
- 4.ICRP (International Commission on Radiological Protection) The 2007 recommendations of the ICRP. Publication 103. Ann ICRP. 2007;37:1–332. doi: 10.1016/j.icrp.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 5.Brenner DJ, Doll R, Goodhead DT, et al. Cancer risks attributable to low doses of ionizing radiation: assessing what we really know. Proc Natl Acad Sci U S A. 2003;100:13761–6. doi: 10.1073/pnas.2235592100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mullenders L, Atkinson M, Paretzke H, et al. Assessing cancer risks of low-dose radiation. Nat Rev Cancer. 2009;9:596–603. doi: 10.1038/nrc2677. [DOI] [PubMed] [Google Scholar]
- 7.Pierce DA, Preston DL. Radiation-related cancer risks at low doses among atomic bomb survivors. Radiat Res. 2000;154:178–86. doi: 10.1667/0033-7587(2000)154[0178:rrcral]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 8.Dropkin G. Low dose radiation and cancer in A-bomb survivors: latency and non-linear dose–response in the 1950–90 mortality cohort. Environ Health. 2007;6:1–25. doi: 10.1186/1476-069X-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCulloch S, Pitts W. A logical calculus of ideas immanent in neuron activity. Bull Math Biophys. 1943;5:115–33. [PubMed] [Google Scholar]
- 10.Stein RB. A theoretical analysis of neuronal variability. Biophys J. 1965;5:173–94. doi: 10.1016/s0006-3495(65)86709-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hertz J, Krogh A, Palmer RG. New York: Addison-Wesley; 1991. Introduction to the Theory of Neural Computation. [Google Scholar]
- 12.Golden RM. Cambridge, Massachusetts, London: MIT Press; 1996. Mathematical Methods for Neural Network Analysis and Design. [Google Scholar]
- 13.Rosenblatt F. The perceptron: a probabilistic model for information storage and organization in the brain. Psychol Rev. 1958;65:386–408. doi: 10.1037/h0042519. [DOI] [PubMed] [Google Scholar]
- 14.Akaike H. Likelihood of a model and information criteria. J Econometrics. 1981;16:3–14. [Google Scholar]
- 15.Sasaki MS, Nomura T, Ejima Y, et al. Experimental derivation of relative biological effectiveness of A-bomb neutrons in Hiroshima and Nagasaki and implication for risk assessment. Radiat Res. 2008;170:101–17. doi: 10.1667/RR1249.1. [DOI] [PubMed] [Google Scholar]
- 16.Preston DL, Kato H, Kopecky KJ, et al. Studies on the mortality of A-bomb survivors. 8. Cancer mortality, 1950–1982. Radiat Res. 1987;111:151–78. [PubMed] [Google Scholar]
- 17.Shimizu Y, Kato H, Shull WL, et al. Studies of the mortality of A-bomb survivors. 9. Mortality. 1950–1985: Part 1. Comparison of risk coefficients for site-specific cancer mortality based on the DS86 and T65DR shielded kerma and organ doses. Radiat Res. 1989;118:502–24. [PubMed] [Google Scholar]
- 18.Shimizu Y, Kato H, Schull WJ. Studies of the mortality of A-bomb survivors. 9. Mortality, 1950–1985: Part 2. Cancer mortality based on the recently revised doses (DS86) Radiat Res. 1990;121:120–41. [PubMed] [Google Scholar]
- 19.The Committee for the Compilation of Materials on the Damage by the Atomic Bombs in Hiroshima and Nagasaki (ed) Tokyo: Iwanami-Shoten; 1979. The Damage by the Atomic Bombings in Hiroshima and Nagasaki and Their After-Effects. (in Japanese) [Google Scholar]
- 20.Watanabe T, Miyao M, Honda R, et al. Hiroshima survivors exposed to very low doses of A-bomb primary radiation showed a high risk for cancer. Environ Health Prev Med. 2008;13:264–70. doi: 10.1007/s12199-008-0039-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grant EJ, Shimizu Y, Kasagi, et al. Commentary: radiation unlikely to be responsible for high cancer rates among distal Hiroshima A-bomb survivors. Environ Health Prev Med. 2009;14:247–9. doi: 10.1007/s12199-009-0087-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakanoue M, Tsuji T. Plutonium content of soil at Nagasaki. Nature. 1971;234:92–3. [Google Scholar]
- 23.Kudo A, Mahara Y, Santry DC, et al. Geographical distribution of fractionated local fallout from the Nagasaki A-bomb. J Environ Radioact. 1991;14:305–16. [Google Scholar]
- 24.Shimasaki T, Okumura Y, Yoshida M, et al. Distribution of plutonium and cesium of fallout from atomic bomb in Nagasaki. Gencho Hiroshima Igaku. 1994;47:418–22. (in Japanese) [Google Scholar]
- 25.Saito-Kokubu Y, Yasuda K, Magura M, et al. Distribution of plutonium isotopes and 137Cs found in the surface soils of Nagasaki, Japan. J Geosci (Osaka City Univ) 2007;50:7–13. [Google Scholar]
- 26.Shichijo K, Takatsuji T, Fukumoto M, et al. Methodology of detecting internal radiation on materials of Nagasaki atomic bomb casualties – No. 2. Gencho Hiroshima Igaku. 2010;63:265–6. (in Japanese) [Google Scholar]
- 27.Eisenbud M. New York: Academic Press; 1973. Environmental Radioactivity. [Google Scholar]
- 28.Wolff S, Afzal V, Wiencke JK, et al. Human lymphocytes exposed to low doses of ionizing radiations become refractory to high doses of radiation as well as to chemical mutagens that induce double-strand breaks in DNA. Int J Radiat Biol Relat Stud Phys Chem Med. 1988;53:39–47. doi: 10.1080/09553008814550401. [DOI] [PubMed] [Google Scholar]
- 29.Sasaki MS. On the reaction kinetics of the radioadaptive response in cultured mouse cells. Int J Radiat Biol. 1995;68:281–91. doi: 10.1080/09553009514551211. [DOI] [PubMed] [Google Scholar]
- 30.Sasaki MS, Ejima Y, Tachibana A, et al. DNA damage response pathway in radioadaptive response. Mutat Res. 2002;504:101–18. doi: 10.1016/s0027-5107(02)00084-2. [DOI] [PubMed] [Google Scholar]
- 31.George KA, Hada M, Jackson LJ, et al. Dose response of γ rays and iron nuclei for induction of chromosomal aberrations in normal and repair-deficient cell lines. Radiat Res. 2009;171:752–63. doi: 10.1667/RR1680.1. [DOI] [PubMed] [Google Scholar]
- 32.Zyugikov NA, Coates PJ, Pally JM, et al. Lack of nontargeted effects in murine bone marrow after low-dose in vivo X-irradiation. Radiat Res. 2011;175:322–7. doi: 10.1667/RR2386.1. [DOI] [PubMed] [Google Scholar]
- 33.Azzam EI, de Toledo SM, Raaphorst GP, et al. Low-dose ionizing radiation decreases the frequency of neoplastic transformation to a level below the spontaneous rate in C3H 10T1/2 cells. Radiat Res. 1996;146:369–73. [PubMed] [Google Scholar]
- 34.Elmore E, Lao X-Y, Kapadia R, et al. Low doses of very low-dose-rate low-LET radiation suppress radiation-induced neoplastic transformation in vitro and induce an adaptive response. Radiat Res. 2008;169:311–8. doi: 10.1667/RR1199.1. [DOI] [PubMed] [Google Scholar]
- 35.Sakai K, Hoshi Y, Nomura T, et al. Suppression of carcinogenic processes in mice by chronic low dose rate gamma-irradiation. Int J Low Radiat. 2003;1:142–6. [Google Scholar]
- 36.Ina Y, Tanooka H, Yamada T, et al. Suppression of thymic lymphoma induction by life-long low-dose-rate irradiation accompanied by immune activation in C57BL/6 mice. Radiat Res. 2005;163:153–8. doi: 10.1667/rr3289. [DOI] [PubMed] [Google Scholar]
- 37.Sakai K, Nomura T, Ina Y. Enhancement of bio-protective function by low dose/dose-rate radiation. Dose–response. 2006;4:327–32. doi: 10.2203/dose-response.06-115.Sakai. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ishii-Ohba H, Kobayashi S, Nishimura M, et al. Existence of a threshold-like dose for γ-ray induction of thymic lymphomas and no susceptibility to radiation-induced solid tumors in SCID mice. Mutat Res. 2007;619:124–33. doi: 10.1016/j.mrfmmm.2007.02.028. [DOI] [PubMed] [Google Scholar]
- 39.Yamaguch K, Kakinuma S, Sudo S, et al. Differential effects of low- and high-dose X-rays on N-ethyl-N-nitrosourea-induced mutagenesis in thymocytes of B6C3F1 gpt-delta mice. Mutat Res. 2008;640:27–37. doi: 10.1016/j.mrfmmm.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 40.Shin SC, Kang YM, Kim HS. Life span and thymic lymphoma incidence in high- and low-dose-rate irradiated AKR/J mice and commonly expressed genes. Radiat Res. 2010;174:341–6. doi: 10.1667/RR1946.1. [DOI] [PubMed] [Google Scholar]
- 41.Shimada Y, Nishimura M, Amasaki Y, et al. Interaction of low dose radiation and other factors. Health Phys. 2010;100:278–9. doi: 10.1097/hp.0b013e3182080e07. [DOI] [PubMed] [Google Scholar]
- 42.Kakinuma S, Nishimura M, Amasaki Y, et al. Combined exposure to X-irradiation followed by N-ethyl-nitrosourea treatment alters the frequency and spectrum of Ikaros point mutations in murine T-cell lymphoma. Mutat Res. 2012;737:43–50. doi: 10.1016/j.mrfmmm.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 43.Hoeijmarker JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–74. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 44.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27:247–54. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 45.Takata M, Sasaki MS, Sonoda E, et al. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping role in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998;17:5497–508. doi: 10.1093/emboj/17.18.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sonoda E, Hochegger H, Saberi A, et al. Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair (Amst) 2006;5:1021–9. doi: 10.1016/j.dnarep.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 47.Lieber MR, Ma Y, Rannicke U, et al. Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol. 2003;4:712–20. doi: 10.1038/nrm1202. [DOI] [PubMed] [Google Scholar]
- 48.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2011;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mladenov E, Iliakis G. Induction and repair of DNA double strand breaks: the increasing spectrum of non-homologous end joining pathways. Mutat Res. 2010;711:61–72. doi: 10.1016/j.mrfmmm.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 50.McVey M, Lee SE. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–38. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin Y, Lukacsovich T, Waldman AS. Multiple pathways for repair of double-strand breaks in mammalian chromosomes. Mol Cell Biol. 1999;19:8353–60. doi: 10.1128/mcb.19.12.8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clikeman JA, Khalsa GJ, Barton SL, et al. Homologous recombinational repair of double-strand breaks in yeast is enhanced by MAT heterozygosity through yKu-dependent and -independent mechanisms. Genetics. 2001;157:579–89. doi: 10.1093/genetics/157.2.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reliene R, Bishop AJR, Schiestl RH. Involvement of homologous recombination in carcinogenesis. Adv Genet. 2007;58:67–87. doi: 10.1016/S0065-2660(06)58003-4. [DOI] [PubMed] [Google Scholar]
- 54.Delacôte F, Lopez BS. Importance of the cell cycle phase for the choice of the appropriate DSB repair pathway, for genome stability maintenance: the trans-S double-strand break repair model. Cell Cycle. 2008;7:33–8. doi: 10.4161/cc.7.1.5149. [DOI] [PubMed] [Google Scholar]
- 55.Mizuno K, Miyabe I, Schalbetter SA, et al. Recombination-restarted replication makes inverted chromosome fusions at inverted repeats. Nature. 2013;493:246–9. doi: 10.1038/nature11676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu X, Wang H, Wang P, et al. The Ku-dependent non-homologous end-joining pathway contributes to low-dose radiation-stimulated cell survival. J Cell Physiol. 2010;226:369–74. doi: 10.1002/jcp.22342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tachibana A. Genetic and physiological regulation of non-homologous end-joining in mammalian cells. Adv Biophys. 2004;38:21–44. [PubMed] [Google Scholar]
- 58.Klammer H, Zhang L-H, Kadhim M, et al. Dependence of adaptive response and its bystander transmission on the genetic background of tested cells. Int J Radiat Biol. 2012;88:720–6. doi: 10.3109/09553002.2012.691613. [DOI] [PubMed] [Google Scholar]
- 59.Klammer H, Kadhim M, Illiakis G. Evidence of an adaptive response targeting DNA nonhomologous end joining and its transmission to bystander cells. Cancer Res. 2010;70:8498–506. doi: 10.1158/0008-5472.CAN-10-1181. [DOI] [PubMed] [Google Scholar]
- 60.Fukushima T, Takata M, Morrison C, et al. Genetic analysis of the DNA-dependent protein kinase reveals an inhibitory role of Ku in late S-G2 phase DNA double-strand break repair. J Biol Chem. 2001;276:4413–8. doi: 10.1074/jbc.M106295200. [DOI] [PubMed] [Google Scholar]
- 61.Nea JA, Dang V, Douglas P, et al. Inhibition of homologous recombination by DNA-dependent protein kinase requires kinase activity, is titratable, and is modulated by autophosphorylation. Mol Cell Biol. 2011;31:1719–33. doi: 10.1128/MCB.01298-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pierce AJ, Hu P, Han M, et al. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001;15:3237–42. doi: 10.1101/gad.946401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stark JM, Pierce AJ, Oh J, et al. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol. 2004;24:9305–16. doi: 10.1128/MCB.24.21.9305-9316.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Audebert M, Salles B, Calsau P. Involvement of poly(ADP-ribose)polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J Biol Chem. 2004;279:5517–26. doi: 10.1074/jbc.M404524200. [DOI] [PubMed] [Google Scholar]
- 65.Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008;18:134–47. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- 66.Shrivastav M, Miller CA, De Haro LP, et al. DNA-PKcs and ATM co-regulate DNA double-strand break repair. DNA Repair (Amst) 2009;8:920–9. doi: 10.1016/j.dnarep.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang M, Wu W, Wu W, et al. PARP1 and Ku compete for repair of DNA double-strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–82. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Simsek D, Jasin M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat Struct Mol Biol. 2010;17:410–5. doi: 10.1038/nsmb.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fattah F, Lee EH, Weisensel N, et al. Ku regulates the non-homologous end joining pathway choice of DNA double strand break repair in human somatic cells. PLoS Genet. 2010;6:1–14. doi: 10.1371/journal.pgen.1000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Ann Rev Genet. 2011;45:247–71. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 71.Paddock MN, Bauman AT, Higdon R, et al. Competition between PARP-1 and Ku70 control the decision between high-fidelity and mutagenic DNA repair. DNA Repair (Amst) 2011;10:338–43. doi: 10.1016/j.dnarep.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grabarz A, Barascu A, Guirouilh-Barbat J, et al. Initiation of DNA double strand break repair: signaling and single-stranded resection dictate the choice between homologous recombination, non-homologous end-joining and alternative end-joining. Am J Cancer Res. 2012;2:249–68. [PMC free article] [PubMed] [Google Scholar]
- 73.Kocher S, Rieckmann T, Rohaly G, et al. Radiation-induced double-strand breaks require ATM but not Artemis for homologous recombination during S-phase. Nucleic Acids Res. 2012;40:8336–47. doi: 10.1093/nar/gks604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nakamura K, Sakai W, Kawamoto T, et al. Genetic dissection of vertebrate 53BP1: a major role in non-homologous end joining of DNA double strand breaks. DNA Repair (Amst) 2006;5:741–9. doi: 10.1016/j.dnarep.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 75.Orsburn B, Escudero B, Prakas M, et al. Differential requirement for H2AX and 53BP1 in organismal development and genome maintenance in the absence of poly(ADP)ribosyl polymerase 1. Mol Cell Biol. 2010;30:2341–52. doi: 10.1128/MCB.00091-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zimmermann M, Lottersberger F, Buonomo SB, et al. 53BP 1 regulates DSB repair using Rif 1 to control 5' end resection. Science. 2013;339:700–4. doi: 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chapman JR, Barral P, Vannier J-B, et al. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell. 2013;49:858–71. doi: 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Iwabuchi K, Bartel PL, Li B, et al. Two cellular proteins that bind to wild-type but not mutant p53. Proc Natl Acad Sci U S A. 1994;91:6098–102. doi: 10.1073/pnas.91.13.6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang L, Sasaki MS. Trans-regulated silencing and reactivation of TP53 tumor suppressor gene in malignant transformation and reversion. Jpn J Cancer Res. 2000;91:1111–8. doi: 10.1111/j.1349-7006.2000.tb00893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lin WY, Wilson JH, Lin Y. Chromosomal double-strand breaks by precise ligation in human cells. DNA Repair (Amst) 2013;12:480–7. doi: 10.1016/j.dnarep.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bulavin DV, Saito S, Hollander MC, et al. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 1999;18:6845–54. doi: 10.1093/emboj/18.23.6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yu E, Ahn YS, Jang SJ, et al. Overexpression of the wip1 gene abrogates the p38MAPK/p53/wip1 pathway and silences p16 expression in human breast cancers. Breast Cancer Res Treat. 2007;101:269–78. doi: 10.1007/s10549-006-9304-y. [DOI] [PubMed] [Google Scholar]
- 83.Shimizu T, Kato T, Tachibana A, et al. Coordinated regulation of radioadaptive response by protein kinase C and p38 mitogen-activated protein kinase. Exptl Cell Res. 1999;251:424–32. doi: 10.1006/excr.1999.4582. [DOI] [PubMed] [Google Scholar]
- 84.Daum G, Kalmes A, Levkau B, et al. Pervanadete inhibits mitogen-activated protein kinase kinase-1 in p38MAPK-dependent manner. FEBS Lett. 1998;427:271–4. doi: 10.1016/s0014-5793(98)00448-7. [DOI] [PubMed] [Google Scholar]
- 85.Sen P, Chakraborty PK, Raha S. 38 mitogen-activated protein kinase (p38MAPK) upregulates catalase levels in response to low dose H2O2 treatment through enhancement of mRNA stability. FEBS Lett. 2005;579:4402–6. doi: 10.1016/j.febslet.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 86.Rizvi A, Pecaut MJ, Slater JM, et al. Low-dose γ-rays modify CD4+ T cell signaling response to simulated solar particle event protons in a mouse model. Int J Radiat Biol. 2011;87:24–35. doi: 10.3109/09553002.2010.518206. [DOI] [PubMed] [Google Scholar]
- 87.Bunting SF, Callén E, Wong N, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–54. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bouwman P, Aly A, Escandel JM, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17:688–95. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lin WY, Wilson JH, Lin Y. Repair of chromosomal double-strand breaks by precise ligation in human cells. DNA Repair (Amst) 2013;12:480–7. doi: 10.1016/j.dnarep.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sonoda E, Sasaki MS, Morrison C, et al. Sister chromatid exchanges are mediated by homologous recombination in vertebrate cells. Mol Cell Biol. 1999;19:5166–9. doi: 10.1128/mcb.19.7.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Takata M, Sasaki MS, Tachiiri S, et al. Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol Cell Biol. 2001;21:2858–66. doi: 10.1128/MCB.21.8.2858-2866.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sasaki MS, Takata M, Sonoda E, et al. Recombination repair pathway in the maintenance of chromosomal integrity against DNA interstrand crosslinks. Cytogent Genome Res. 2004;104:28–34. doi: 10.1159/000077463. [DOI] [PubMed] [Google Scholar]
- 93.Nagasawa H, Wilson PF, Chen DJ, et al. Low dose of alpha particles do not induce sister chromatid exchanges in bystander Chinese hamster cells defective in homologous recombination. DNA Repair (Amst) 2008;7:515–22. doi: 10.1016/j.dnarep.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 94.Beucher A, Birraux J, Tchouandrong L, et al. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009;28:3413–27. doi: 10.1038/emboj.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Joiner MC, Marples B, Lambin P, et al. Low-dose hypersensitivity: current status and possible mechanisms. Int J Radiat Oncol Biol Phys. 2001;49:379–89. doi: 10.1016/s0360-3016(00)01471-1. [DOI] [PubMed] [Google Scholar]
- 96.Moquet JE, Prosser JS, Edwards AA, et al. Sister-chromatid exchanges induced by mitomycin C after acute or chronic exposure of human lymphocytes to a low dose of X-rays. Mutat Res. 1989;227:207–13. doi: 10.1016/0165-7992(89)90098-5. [DOI] [PubMed] [Google Scholar]
- 97.Pennington JM, Rosenberg SM. Spontaneous DNA breakage in single living Escherichia coli cells. Nature Gene. 2007;39:797–802. doi: 10.1038/ng2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Doll R, Peto R. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J Natl Cancer Inst. 1981;66:1191–308. [PubMed] [Google Scholar]
- 99.Sugimura T. Nutrition and dietary carcinogens. Carcinogenesis. 2008;21:387–95. doi: 10.1093/carcin/21.3.387. [DOI] [PubMed] [Google Scholar]
- 100.Anand P, Kunnumakara AB, Sundaram C, et al. Cancer is a preventable disease that requires major lifestyle changes. Pharmaceut Res. 2008;25:2097–116. doi: 10.1007/s11095-008-9661-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.IARC. Lyon: International Agency for Research on Cancer; 2012. IARC monograph on the evaluation of carcinogenic risks to humans. A review of human carcinogens: personal habits and indoor combustions. Vol. 100E. [PMC free article] [PubMed] [Google Scholar]
- 102.Phillip DH, Venitt S. DNA and protein adducts in human tissues resulting from exposure to tobacco smoke. Int J Cancer. 2012;131:2733–53. doi: 10.1002/ijc.27827. [DOI] [PubMed] [Google Scholar]
- 103.Brooks PJ, Enoch M-A, Goldman D, et al. The alcohol flushing response: an unrecognized risk factor for esophageal cancer from alcohol consumption. PLoS Med. 2009;6 doi: 10.1371/journal.pmed.1000050. e1000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kotova N, Vare D, Schultz N, et al. Genotoxicity of alcohol is linked to DNA replication-associated damage and homologous recombination repair. Carcinogenesis. 2013;34:325–30. doi: 10.1093/carcin/bgs340. [DOI] [PubMed] [Google Scholar]
- 105.Limoli CL, Giedzinski E, Bonner WM, et al. UV-induced replication arrest in the xeroderma pigmentosum variant leads to DNA double-strand breaks, γ-H1AX formation, and Mre11 relocation. Proc Natl Acad Sci U S A. 2002;99:233–8. doi: 10.1073/pnas.231611798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yokoyama A, Muramatsu T, Ohmori T, et al. Esophageal cancer and aldehyde dehydrogenase-2 genotypes in Japanese males. Cancer Epidemiol Biomarkers Prev. 1996;5:99–102. [PubMed] [Google Scholar]
- 107.Li H, Borinskaya S, Yoshimura K, et al. Refined geographic distribution of the oriental ALDH2*504Lys (nee 487Lys) variant. Ann Hum Genet. 2009;73:335–45. doi: 10.1111/j.1469-1809.2009.00517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Johnson N. Tobacco use and oral cancer. J Dental Educ. 2001;65:328–39. [PubMed] [Google Scholar]
- 109.Nair S, Pillai MR. Human papillomavirus and disease mechanisms; relation to oral and cervical cancers. Oral Dis. 2005;11:350–9. doi: 10.1111/j.1601-0825.2005.01127.x. [DOI] [PubMed] [Google Scholar]
- 110.Meekings KN, Leipzig J, Bushman FD, et al. HTLV-1 integration into transcriptionally active genomic regions is associated with proviral expression and with HAM/TSP. PLOS Pathog. 2008;4 doi: 10.1371/journal.ppat.1000027. e1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Studamire B, Goff SP. Interaction of host proteins with murine leukemia virus integrase. Viruses. 2010;2:1110–45. doi: 10.3390/v2051110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Daniel R, Katz RA, Skalka AM. A role for DNA-PKI in retroviral DNA integration. Science. 1999;284:644–8. doi: 10.1126/science.284.5414.644. [DOI] [PubMed] [Google Scholar]
- 113.Lau A, Kanaar R, Jackson SP, et al. Suppression of retroviral infection by the Rad52 DNA repair protein. EMBO J. 2004;23:3421–9. doi: 10.1038/sj.emboj.7600348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Desfarges S, Filippo JS, Fournier M, et al. Chromosomal integration of LTR-flanked DNA in yeast expressing HIV-1 integrase: down regulation by Rad 51. Nucleic Acids Res. 2006;34:6215–24. doi: 10.1093/nar/gkl843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Smith JA, Wang F-X, Zhang H, et al. Evidence that the Nijmegen breakage syndrome protein, an early sensor of double-strand DNA breaks (DSB), is involved in HIV-1 post-integration repair by recruiting the ataxia telangiectasia-mutated kinase in a process similar to, but distinct from, cellular DSB repair. Virol J. 2008 doi: 10.1186/1743-422X-5-11. doi:10.1186/1743-422X-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sakurai Y, Komatsu K, Agematsu K, et al. DNA double strand break repair enzymes function at multiple steps in retroviral infection. Retrovirol. 2009;6:114–26. doi: 10.1186/1742-4690-6-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cosnefroy O, Tocco A, Lesbats P, et al. Stimulation of the human Rad51 nucleofilament restricts HIV-1 integration in vitro and in infected cells. J Virol. 2010;86:513–26. doi: 10.1128/JVI.05425-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ariumi Y, Turelli P, Masutani M, et al. DNA damage sensors ATM, ATR, DNA-PKcs, and PARP-1 are dispensable for human immunodeficiency virus type I integration. J Virol. 2005;79:2973–8. doi: 10.1128/JVI.79.5.2973-2978.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Goulaovic H, Chow SA. Directed integration of viral DNA mediated by fusion proteins consisting of human immunodeficiency virus type 1 integrase and Escherichia coli LexA protein. J Virol. 1996;70:37–46. doi: 10.1128/jvi.70.1.37-46.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kim PM, Allen C, Wagener BM, et al. Overexpression of human RAD51 and RAD52 reduces double-strand break-induced homologous recombination in mammalian cells. Nucleic Acids Res. 2001;29:4352–60. doi: 10.1093/nar/29.21.4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Paffett KS, Clikeman JA, Palmer S, et al. Overexpression of Rad51 inhibits double-strand break-induced homologous recombination but does not affect gene conversion tract length. DNA Repair (Amst) 2005;4:687–98. doi: 10.1016/j.dnarep.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 122.Rom I, Darbinyan A, White MK, et al. Activation of HIV-1 LTR by Rad51 in microglial cells. Cell Cycle. 2010;9:3715–22. doi: 10.4161/cc.9.18.12930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Song S, Lu Y, Chol Y-K, et al. DNA-dependent PK inhibits adeno-associated virus DNA integration. Proc Natl Acad Sci U S A. 2004;101:2112–6. doi: 10.1073/pnas.0307833100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang H, Zen Z-C, Bui T-A, et al. Efficient rejoining of radiation-induced DNA double-strand breaks in vertebrate cells deficient in genes of the RAD52 epistasis group. Oncogene. 2001;20:2212–24. doi: 10.1038/sj.onc.1204350. [DOI] [PubMed] [Google Scholar]
- 125.Koga Y, Iwanaga M, Soda M, et al. Trends in HTLV-1 prevalence and incidence of adult T-cell leukemia/lymphoma in Nagasaki, Japan. J Med Virol. 2010;82:668–74. doi: 10.1002/jmv.21738. [DOI] [PubMed] [Google Scholar]
- 126.Satake M, Yamaguchi K, Tadokura K. Current prevalence rate of HTLV-1 in Japan as determined by screening of blood donors. J Med Virol. 2012;84:327–35. doi: 10.1002/jmv.23181. [DOI] [PubMed] [Google Scholar]
- 127.Yu X, Wang H, Wang P, et al. The Ku-dependent non-homologous end-joining pathway contributes to low-dose radiation-stimulated cell survival. J Cell Physiol. 2004;226:369–74. doi: 10.1002/jcp.22342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Klammer H, Zhang L-H, Kadhim M, et al. Dependence of adaptive response and its bystander transmission on the genetic background of tested cells. Int J Radiat Biol. 2012;88:720–6. doi: 10.3109/09553002.2012.691613. [DOI] [PubMed] [Google Scholar]
- 129.Wang H, Wang X, Zhang P, et al. The Ku-dependent non-homologous end-joining but not other repair pathway is inhibited by high linear energy transfer ionizing radiation. DNA Repair (Amst) 2008;7:725–33. doi: 10.1016/j.dnarep.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 130.Saenko AS, Semenets TN, Semina OV. The enhanced radioresistance (adaptive response) in vivo of splenic colony-forming units (CFU-S) following the exposure of mice to low-level radiation from radioisotopes or X-rays. Mutat Res. 1989;211:7–12. [Google Scholar]
- 131.Cai L, Wang P. Induction of cytogenetic adaptive response in germ cells of irradiated mice with very low-dose rate of chronic gamma-irradiation and its biological influence on radiation-induced DNA or chromosomal damage and cell killing in their male offspring. Mutagenesis. 1995;10:95–100. doi: 10.1093/mutage/10.2.95. [DOI] [PubMed] [Google Scholar]
- 132.Klokov DY, Zaichkina SI, Rozanova OM, et al. The duration of radioadaptive response in mouse bone narrow cells in vitro. In: Yamada T, Mothersill C, Michael BD, et al., editors. Biological Effects of Low Dose Radiation. Tokyo: Elsevier; 2000. pp. 87–91. [Google Scholar]
- 133.Pierce DA, Sharp GB, Mabuchi K. Joint effects of radiation and smoking on lung cancer risk among atomic bomb survivors. Radiat Res. 2003;159:511–20. doi: 10.1667/0033-7587(2003)159[0511:jeoras]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 134.Furukawa K, Preston DL, Lönn S, et al. Radiation and smoking effects on lung cancer incidence among atomic bomb survivors. Radiat Res. 2010;174:72–82. doi: 10.1667/RR2083.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Egawa H, Furukawa K, Preston D, et al. Radiation and smoking effects on lung cancer incidence by histological types among atomic bomb survivors. Radiat Res. 2012;178:191–201. doi: 10.1667/rr2819.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Frankenberg-Schwager M, Gebauer A, Koppe C, et al. Single-strand annealing, conservative homologous recombination, nonhomologous DNA end joining, and the cell cycle-dependent repair of DNA double-strand breaks induced by sparsely and densely ionizing radiation. Radiat Res. 2009;171:265–73. doi: 10.1667/RR0784.1. [DOI] [PubMed] [Google Scholar]
- 137.Tsugane S, Fahey MT, Sasaki S, et al. Alcohol consumption and all-cause and cancer mortality among middle-aged Japanese men: seven-year follow-up at the JPHC study cohort 1. Amer J Epidemiol. 1999;150:1201–7. doi: 10.1093/oxfordjournals.aje.a009946. [DOI] [PubMed] [Google Scholar]
- 138.Yun YH, Lim MK, Jung KW, et al. Relative and absolute risk of cigarette smoking on major histologic types of lung cancer in Korean men. Cancer Epidemiol Biomarker Predict. 2005;14:2125–30. doi: 10.1158/1055-9965.EPI-05-0236. [DOI] [PubMed] [Google Scholar]
- 139.Yang L, Zhou M, Sherliker P, et al. Alcohol drinking and overall and cause-specific mortality in China: nationally representative prospective study of 220,000 men with 15 years of follow-up. Int J Epidemiol. 2012;41:1101–13. doi: 10.1093/ije/dys075. [DOI] [PubMed] [Google Scholar]
- 140.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 141.Turner BM. Histone acetylation and an epigenetic code. BioEssays. 2000;22:836–45. doi: 10.1002/1521-1878(200009)22:9<836::AID-BIES9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 142.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 143.Moore JD, Krebs JE. Histone modifications and DNA double-strand break repair. Biochem Cell Biol. 2002;82:446–52. doi: 10.1139/o04-034. [DOI] [PubMed] [Google Scholar]
- 144.Escargueil AE, Soares DG, Salvador M, et al. What histone code for DNA repair? Mutat Res. 2008;658:259–70. doi: 10.1016/j.mrrev.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 145.Seong K-H, Li D, Shimizu H, et al. Inheritance of stress-induced, ATF-2-dependent epigenetic change. Cell. 2011;145:1049–61. doi: 10.1016/j.cell.2011.05.029. [DOI] [PubMed] [Google Scholar]
- 146.Chappell LJ, Whalen MK, Gurai S, et al. Analysis of flow cytometry DNA damage response protein activation kinetics after exposure to x rays and high-energy ion nuclei. Radiat Res. 2010;174:691–702. doi: 10.1667/RR2204.1. [DOI] [PubMed] [Google Scholar]
- 147.Ozasa K, Shimizu Y, Suyama A, et al. Studies of the mortality of atomic bomb survivors, report 14, 1950–2003: an overview of cancer and noncancer diseases. Radiat Res. 2012;177:229–43. doi: 10.1667/rr2629.1. [DOI] [PubMed] [Google Scholar]
- 148.Swant SG, Randers-Pehrson G, Metting NF, et al. Adaptive response and the bystander effect induced by radiation in C3H-10T(1/2) cells in culture. Radiat Res. 2001;156:177–80. doi: 10.1667/0033-7587(2001)156[0177:aratbe]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 149.Zaichkina SI, Rozanova OM, Aptikaeva GF, et al. Peculiarities of the effect of low-dose-rate radiation simulating high-altitude flight conditions on mice in vivo. Radiat Environ Biophys. 2007;46:131–5. doi: 10.1007/s00411-007-0107-2. [DOI] [PubMed] [Google Scholar]
- 150.Kondo S. Evidence that there are threshold effects in risk of radiation. J Nucl Sci Technol. 1999;36:1–9. [Google Scholar]
- 151.Feinendegen LE. Evidence for beneficial low level radiation effects and radiation hormesis. Brit J Radiol. 2005;78:3–7. doi: 10.1259/bjr/63353075. [DOI] [PubMed] [Google Scholar]
- 152.Wagner LK. The “Healthy worker effect”: science or prejudice? Radiology. 2003;229:16–7. doi: 10.1148/radiol.2291030810. [DOI] [PubMed] [Google Scholar]
- 153.Wakefort R. Radiation in the workplace – a review of studies of the risks of occupational exposure to ionizing radiation. J Radiol Prot. 2009;29:A61–79. doi: 10.1088/0952-4746/29/2A/S05. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.