

Abstract
Objective: Bumetanide has been reported to attenuate ischemia-evoked cerebral edema. However, whether bumetanide can protect cerebral ischemia-reperfusion injury (IRI) in vivo is unclear. In the present study, we aim to determine whether intravenously injection bumetanide can attenuate cerebral IRI and if its protection effect might be related to the modification of cerebral NKCC1 and KCC2 protein expression. Methods: Focal cerebral ischemia was induced by occluding the right middle cerebral artery (MCAO) for 2-h, followed by 3-h, 24-h or 48-h of reperfusion respectively. Brain edema, neurological deficits, and infarction volume were determined by (wet weights - dry weights)/dry weights ×100, 5-point neurological function score evaluation system, and TTC staining, respectively. The expression levels of NKCC1 and KCC2 were determined by immunohistochemical staining. Results: Reperfusion increased brain edema, neurological deficits, and infarction volume. Bumetanide decreased brain edema, attenuated the neurological defects and reduced post-ischemic cerebral infarction. Cerebral ischemia-reperfusion injury increased NKCC1 expression level and decreased KCC2 expression level. Interestingly, bumetanide down-regulated the NKCC1 protein expression level without changing the KCC2 protein expression level in rat brain cortex. Conclusion: These results suggest that bumetanide protects focal cerebral ischemia-reperfusion injury in rat, which might through the inhibition of NKCC1.
Keywords: Bumetanide, Na+-K+-Cl--cotransporter 1 (NKCC1), Na+-K+-cotransporter 2 (KCC2), cerebral ischemia-reperfusion injury, neuroprotection
Introduction
Imbalance of ion homeostasis is a crucial mechanism leading to ischemia-induced cell damage [1]. Cation chloride co-transporters play an essential role in the regulation of neuronal chloride homeostasis [1-3]. The Na+-K+-Cl--co-transporter 1 (NKCC1) and Na+-K+-co-transporter 2 (KCC2) belong to the cation-Cl- co-transporter family, which mediates the coupled movement of Na+ and/or K+ with Cl- across the plasma membrane of cells under physiologic conditions [1-3]. NKCC1 has a broad tissue distribution, whereas KCC2 is found mainly in the nervous system [1-3]. In detail, KCC2 is predominately expressed in mature neurons in the cortex, cerebellum, and the dorsal horn of the spinal cord, while NKCC1 is expressed at a high level in immature neurons [4,5]. NKCC1 is important for the maintenance of intracellular Cl- in neurons and contributes to GABA-mediated depolarization in immature neurons [6,7] while KCC2 is the main chloride extruder in the adult nervous system [8].
In ischemic conditions, particularly after oxygen glucose deprivation in hippocampal slices [9] and in in vivo models of global ischemia [10], KCC2 is generally down-regulated while NKCC1 is up-regulated. In vitro studies have shown that bumetanide, a member of sulfamoyl benzoic acid loop diuretic family which exerts its diuretic action by blockade of the Na+-K+-2Cl cotransporter, significantly reduces the oxygen-glucose-deprivation and glutamate-mediated neuronal excitotoxicity and apoptosis by inhibiting NKCC1. Moreover, bumetanide has also been reported to attenuate ischemia-evoked cerebral edema [11]. However, whether bumetanide can protect cerebral ischemia-reperfusion injury (IRI) in vivo is still unclear.
In the present study, we aim to determine whether intravenously injection bumetanide can attenuate cerebral IRI and if its protection effect might be related to the modification of cerebral NKCC1 and KCC2 protein expression.
Materials and methods
This study was conducted in accordance to the guidelines for the care and use of animals in research, and under the protocols approved by the Guangzhou University of Connecticut Animal Care and Use Committee.
Animal
Adult male rats, weighing 250~320 g (8~10 weeks), were purchased from Guangdong Province Animals Center (Guangzhou, China). Rats were anesthetized with intraperitoneal injection of 10% chloral hydrate (3.5 ml/kg body weight). Rectal temperatures were monitored and maintained at 37°C±0.5°C with a heating blanket and a heating lamp. Rats were randomly divided into 3 groups (n=45 per group) including sham operation group (S), saline plus ischemia reperfusion group (N+I/R group), and bumetanide plus ischemia reperfusion group (B+I/R group).
Rats in the B+I/R group and N+I/R group were respectively received injection of bumetanide (30 mg/kg) [12] or the same volume of saline through tail vein 10 min before cerebral ischemia inducing. All rats except those in the sham group underwent 2-h right middle cerebral artery occlusion (MCAO) as previously reported [13] followed by 3-h, 24-h or 48-h reperfusion respectively.
Edema measurement
Rats were decapitated under deep anesthesia with 10% chloral hydrate at 3-h, 24-h and 48-h of reperfusion. The ipsilateral and contralateral hemispheres were dissected and the wet weight of the tissue was determined. The tissues were dried at 120°C for 24 hours. The percent cerebral water was determined as (wet weights - dry weights)/dry weights ×100.
Neurological evaluation
Rats were examined for neurological deficits 3-h, 24-h or 48-h after MCAO by two investigators who were blinded to this study using a 5-point neurological function score [13]: 0, no deficit; 1, failure to extend right forepaw fully; 2, circling to the right; 3, failing to the right; 4, no spontaneous walking with a depressed level of consciousness. Only those rats showed no or incomplete forelimb placing with rotational asymmetry at 3-h, 24-h or 48-h after MCAO were included in the subsequent analysis [14].
TTC staining
After 3-h, 24-h or 48-h of reperfusion, rat brains were removed and frozen at -80°C for 5 min. Two millimeter coronal slices were made with a rodent brain matrix. The sections were stained for 20 min at 37°C with 2% 2,3,5-triphenyltetrazolium chloride monohydrate (TTC, Sigma, USA) and then were fixed with 4% formalin. Infarction volume was calculated as previous reported [15]. Briefly, the sections were scanned, and the infarction area in each section was calculated by subtracting the noninfarct area of the ipsilateral side from the area of the contralateral side with Image-J analysis software. Infarction areas on each section were summed and multiplied by section thickness to give the total infarction volume.
Immunohistochemical staining
Rat brains were removed, and postfixed in 4% paraformaldehyde overnight at 4°C, and cryoprotected with 30% sucrose in PBS. Brains were cut into coronal sections (30 um) on a freezing microtome (Leica SM 2000 R; Leica, Nussloch, Germany). Coronal sections (0.2 mm anterior to bregma) were selected and processed for immuno-histochemistry. The sections from each rat were used and the free-floating method was used for avidin-biotin-peroxidase labeling. Coronal sections were rinsed with PBS and treated with 0.3% H2O2 (v/v) in PBS for 10 min at room temperature to quench endogenous peroxidase. After washing with PBS, sections were incubated with a blocking solution (10% normal goat serum, 0.3% Triton X-100 in PBS) for 30 min at 37°C. The slices were probed respectively with primary anti-NKCC1 polyclonal antibody (1:200; Santa Cruz, U.S.A.) and anti-KCC2 polyclonal antibody (1:200; Santa Cruz, U.S.A.) for 1 hour at 37°C, followed by overnight incubation at 4°C. After rinsing, sections were incubated with goat anti-rabbit IgG (1:200; Sigma, U.S.A.) for 1 hour at 37°C. Color reaction was developed with 0.05% diaminobenzidime tetrachloride and 0.03% H2O2 in PBS (pH 7.4). Slides were visualized using a Leica SP5 confocal microscope at 40x magnification using standard procedure. All imaging was performed with group identity blinded where at least 10 random images were obtained from each slide or group.
Statistical analysis
All values are presented as means±SD. Data shown were analyzed for significance using analysis of either the nonparametric Mann-Whitney test or ANOVA by SPSS13.0 software. If a significant difference was observed, Bonferroni’s post-hoc test was conducted to identify groups with significant differences. P-values that were less than 0.05 were considered to be statistically significant.
Results
Bumetanide reduces brain edema after focal cerebral ischemia
To determine whether bumetanide reduces edema formation resulting from MCAO, bumetanide or vehicle was administered intravenously 10 minutes before initiation of MCAO (or sham MCAO). Bumetanide reduced the increase of water content in ischemic brains by 33.3%, 49.7% and 45.8% respectively at 3-h, 24-h, and 48-h post-reperfusion (P<0.05) (Figure 1).
Figure 1.
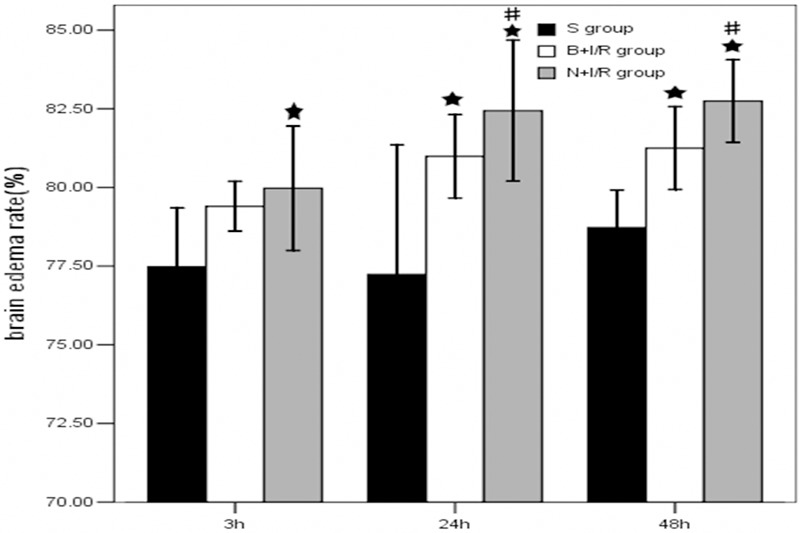
Bumetanide reduces brain edema after focal cerebral ischemia. Edema in bumetanide group (B+I/R) and saline group (N+I/R) was significantly higher than that in the sham group. Bumetanide significantly reduces brain edema after focal cerebral ischemia. Data are expressed as means±s.d. Bumetanide group and saline group rats, n=6; sham group rats, n=3; ★p<0.05 bumetanide and saline versus sham group. #p<0.05 versus bumetanide or saline group.
Bumetanide decreases the volume of cerebral infarction 3-h post-reperfusion
Figure 2 shows the representative results of TTC staining in rats subjected to N+I/R compared with rats subjected to B+I/R. The average total infarction volume in saline group was 231.5±40.6 mm3 while the average total infarction volume was reduced to 193.6±30.7 mm3 after 3-h reperfusion in the bumetanide group (p<0.05). However, the average total infarction volume was not significantly changed in the bumetanide group at 24-h, and 48-h post-reperfusion (p>0.05).
Figure 2.

Bumetanide decreases the volume of cerebral infarction 3-h post-reperfusion. A. Representative results of TTC staining. B. Bumetanide decreases the volume of cerebral infarction 3-h post-reperfusion but not 24-h, and 48-h post-reperfusion. Data are expressed as means±s.d. n=6. ★p<0.05 saline versus bumtanide group rats.
Bumetanide reduces neurological deficit scores 24-h and 48-h post-reperfusion
In the sham group, no rats have symptoms of neurological deficits while the neurological deficits were obvious in the saline and bumetanide group. Moreover, the neurological deficits was significantly reduced in the bumetanide group rats 24-h and 48-h post-reperfusion compared to that in the saline group (P<0.05) (Figure 3).
Figure 3.
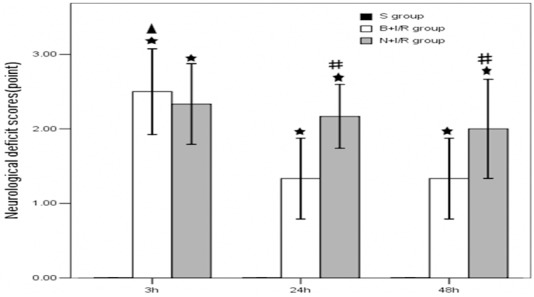
Bumetanide reduces neurological deficit scores 24-h and 48-h post-reperfusion. Data are expressed as means±s.d. n=6. ★p<0.01 versus sham group rats; #p<0.05 bumetanide versus saline group rats, ▲p<0.05 3-h versus 24-h and 48-h reperfusion in bumetanide group rats.
Bumetanide down-regulates the NKCC1 protein expression level in rat brain cortex
It has been reported that the expression level of NKCC1 was increased in cortex after 24-h reperfusion following 2-h ischemia [16]. As shown in Figure 4, an enhanced immunostaining of the NKCC1 in neurons scattered throughout ischemic cortex 3-h, 24-h, 48-h post-reperfusion. Compared with saline groups, pretreatment with bumeitanide significantly reduced the protein expression level of NKCC1.
Figure 4.
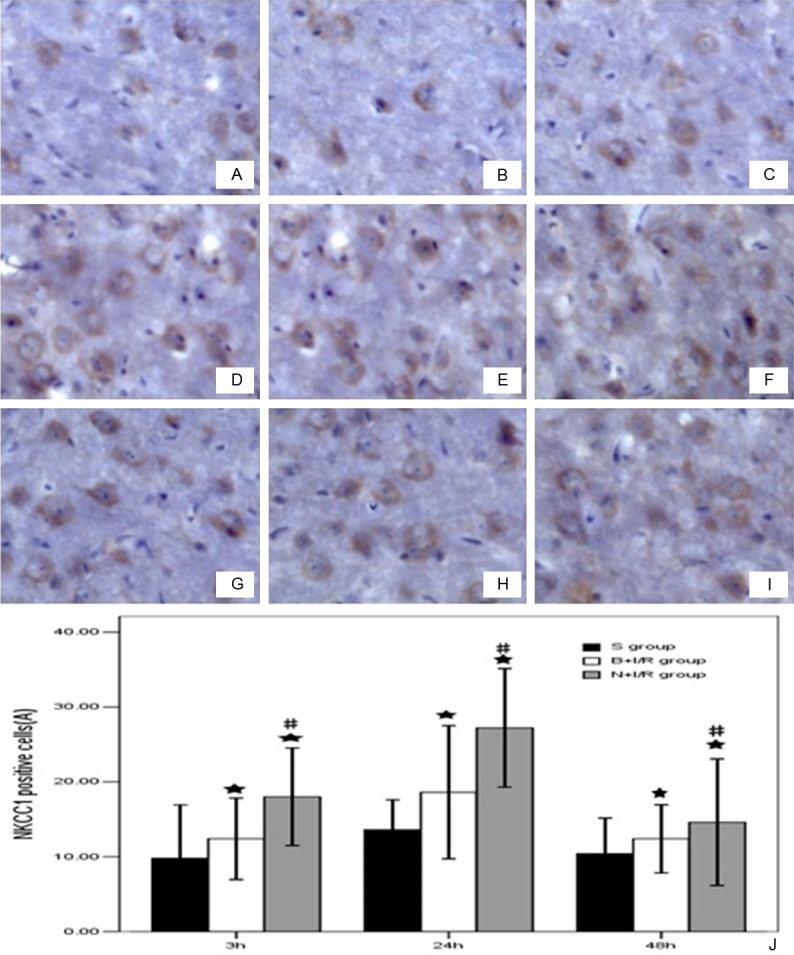
Bumetanide down-regulates the NKCC1 protein expression level in rat brain cortex. A-C. Respectively were sham group rats at 3h, 24h, 48h. D-F were saline group, G-I were bumetanide groups. J. Bumetanide down-regulates the NKCC1 protein expression level in rat brain cortex. Data are expressed as means±s.d. n=6. ★p<0.05 versus sham group rats; ▲p<0.05 bumetanide versus saline group rats.
Bumetanide does not change the KCC2 protein expression level in rat brain cortex
As shown in Figure 5, KCC2 protein was expressed in the plasma membrane and dendrites of neurons. The expression level of KCC2 is significantly down-regulated in the ischemic hemisphere 3-h, 24-h, and 48-h post-reperfusion. However, bumetanide does not change the KCC2 protein expression level in rat brain cortex (Figure 5).
Figure 5.
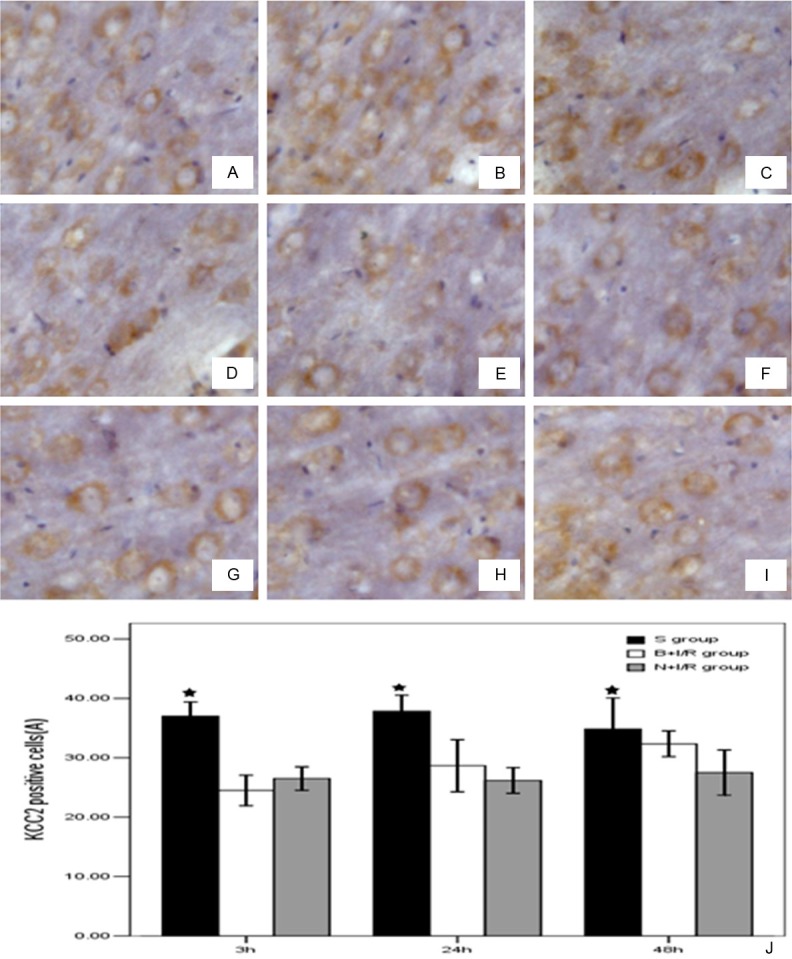
Bumetanide does not change the KCC2 protein expression level in rat brain cortex. A-C were the S group 3h, 24h, 48h post-reperfusion. D-F were N+I/R group 3h, 24h, 48h post-reperfusion and G-I were B+I/R group 3h, 24h, 48h post-reperfusion. J. Bumetanide does not change the KCC2 protein expression level in rat brain cortex. Data are expressed as means±s.d. n=6. ★p<0.05 versus sham group rats.
Discussion
The major findings of the present study are as follows. Firstly, bumetanide reduces brain edema after focal cerebral ischemia. Secondly, bumetanide decreases the volume of cerebral infarction 3-h post-reperfusion. Thirdly, bumetanide reduces neurological deficit scores 24-h and 48-h post-reperfusion. Finally, bumetanide down-regulates the NKCC1 protein expression level without changing the KCC2 protein expression level in rat brain cortex.
NKCC1 transports Na+, K+, and Cl- into cells under both physiological and pathophysiological conditions with a stoichiometry of 1Na+:1K+:2Cl- and it can be inhibited by either bumetanide or furosemide. Bumetanide can compete with Cl- for the second Cl- binding site and thus inhibits NKCC1 function [17-19]. After focal ischemia, extracellular K+ increases, glutamate is released, intracellular Ca2+ increases, excessive Na+ and Cl- accumulates, resulting in the enhancement of NKCC1 activity [20]. Pathologic activation of NKCC1 seems to be an important mechanism of cerebral edema following ischemia. However, in certain pathological conditions, such as ischemia, hypoxia and trauma, NKCC1 expression was increased which led to cell swelling. NKCC1 has been suggested to play a role in K+ uptake and swelling in astrocytes [21]. In this study, we found that bumetanide reduced brain edema, decreases the volume of cerebral infarction, and reduces neurological deficit scores after focal cerebral ischemia that might be related to its inhibition effects in NKCC1.
KCC2 abundantly expresses in the adult mammalian nervous system, and it plays an important role in maintaining the low Clenvironment of mature neurons. In addition, KCC2 participates in the regulation of ion balance, nerve growth and maturation, synaptic development and neuronal plasticity [22]. A decrease of KCC2 expression was previously described under ischemic conditions [9]. In the present study, we also demonstrated that KCC2 expression level was down-regulated after ischemia-reperfusion in rat cerebral cortex, which is consistent with the previous report [23]. However, we did not observe significant changes in KCC2 expression following a single intravenous bumetanide (30 mg/kg) injection, which might be due to the fact that low concentrations of bumetanide (2~10 μM) specifically inhibits NKCC1 but does not affect KCC2 [24,25]. Taken together, the present study suggests that bumetanide might confer its neuroprotection effect mainly by inhibiting NKCC1.
In summary, cerebral ischemia-reperfusion injury increases NKCC1 expression level and decreases KCC2 expression level. Bumetanide attenuates the neurological defects and reduces post-ischemic cerebral infarction, which might through the inhibition of NKCC1.
Acknowledgements
This work was supported by the grants from Guang Zhou Medical University (HS. Huang).
Disclosure of conflict of interest
None to declare.
References
- 1.Haas M, Forbush B 3rd. The Na-K-Cl cotransporter of secretory epithelia. Ann Rev Physiol. 2000;62:515–534. doi: 10.1146/annurev.physiol.62.1.515. [DOI] [PubMed] [Google Scholar]
- 2.Russell JM. Sodium-potassium-chloride co-transport. Physiol Rev. 2000;80:211–276. doi: 10.1152/physrev.2000.80.1.211. [DOI] [PubMed] [Google Scholar]
- 3.Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-Chloride Cotransporters and Neuronal Function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Hebert SC, Mount DB, Gamba G. Molecular physiology of cation-coupled Cl- cotransport: the SLC12 family. Pflugers Arch. 2004;447:580–593. doi: 10.1007/s00424-003-1066-3. [DOI] [PubMed] [Google Scholar]
- 5.Khirug S, Yamada J, Afzalov R, Voipio J, Khiroug L, Kaila K. GABAergic depolarization of the axon initial segment in cortical principal neurons is caused by the Na-K-2Cl cotransporter NKCC1. J Neurosci. 2008;28:4635–4639. doi: 10.1523/JNEUROSCI.0908-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamada J, Okabe A, Toyoda H, Kilb W, Luhmann HJ, Fukuda A. Cl- uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J Physiol. 2004;557:829–841. doi: 10.1113/jphysiol.2004.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kahle KT, Simard JM, Staley KJ, Nahed BV, Jones PS, Sun D. Molecular mechanisms of ischemic cerebral edema: role of electroneutral ion transport. Physiology (Bethesda) 2009;24:257–265. doi: 10.1152/physiol.00015.2009. [DOI] [PubMed] [Google Scholar]
- 8.Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K–C co-transporter kcc2 rendersgaba hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- 9.Galeffi F, Sah R, Pond BB, George A, Schwartz-Bloom RD. Changes in intracellular chloride after oxygen-glucose deprivation of the adult hippocampal slice: Effect of diazepam. J Neurosci. 2004;24:4478–4488. doi: 10.1523/JNEUROSCI.0755-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Papp E, Rivera C, Kaila K, Freund TF. Relationship between neuronal vulnerability and potassium-chloride cotransporter 2 immunoreactivity in hippocampus following transient forebrain ischemia. Neuroscience. 2008;154:677–689. doi: 10.1016/j.neuroscience.2008.03.072. [DOI] [PubMed] [Google Scholar]
- 11.Lu KT, Wu CY, Cheng NC, Wo YY, Yang JT, Yen HH, Yang YL. Inhibition of the Na+-K+-2Cl--cotransporter in choroid plexus attenuatestraumatic brain injury-induced brain edema and neuronal damage. Eur J Pharmacol. 2006;548:99–105. doi: 10.1016/j.ejphar.2006.07.048. [DOI] [PubMed] [Google Scholar]
- 12.Liu Y, Shangguan Y, Barks JD, Silverstein FS. Bumetanide augments the neuroprotective efficacy of phenobarbital plus hypothermia in a neonatal hypoxia-ischemia model. Pediatr Res. 2012;71:559–565. doi: 10.1038/pr.2012.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 14.Thored P, Wood J, Arvidsson A, Cammenga J, Kokaia Z, Lindvall O. Long-term neuroblast migration along blood vessels in an area with transient angiogenesis and increased vascularization after stroke. Stroke. 2007;11:3032–3039. doi: 10.1161/STROKEAHA.107.488445. [DOI] [PubMed] [Google Scholar]
- 15.Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- 16.Yan Y, Dempsey RJ, Sun D. Na+-K+-Cl- contransporter in rat focal cerebral ischemia. J Cereb Blood Flow Metab. 2001;21:711–721. doi: 10.1097/00004647-200106000-00009. [DOI] [PubMed] [Google Scholar]
- 17.Yan Y, Dempsey RJ, Flemmer A, Forbush B, Sun D. Inhibition of Na+-K+-Cl- cotransporter during focal cerebral ischemia decreases edema and neuronal damage. Brain Res. 2003;961:22–31. doi: 10.1016/s0006-8993(02)03832-5. [DOI] [PubMed] [Google Scholar]
- 18.Chen H, Luo J, Kintner DB, Shull GE, Sun D. Na+-dependent chloride transporter (NKCC1)-null mice exhibit less gray and white matter damage after focal cerebral ischemia. J Cereb Blood Flow Metab. 2005;25:54–66. doi: 10.1038/sj.jcbfm.9600006. [DOI] [PubMed] [Google Scholar]
- 19.Forbush B 3rd, Palfrey HC. [3H] bumetanide binding to membranes isolated from dog kidney outer medulla. Relationship to the Na,K, Cl co-transport system. J Biol Chem. 1983;258:11787–11792. [PubMed] [Google Scholar]
- 20.O’Donnell ME, Tran L, Lam TI, Liu XB, Anderson SE. Bumetanide inhibition of the blood-brain barrier Na-K-Cl cotransporter reduces edema formation in the rat middle cerebral artery occlusion model of stroke. J Cereb Blood Flow Metab. 2004;24:1046–1056. doi: 10.1097/01.WCB.0000130867.32663.90. [DOI] [PubMed] [Google Scholar]
- 21.Su G, Haworth RA, Dempsey RJ, Sun D. Regulation of Na(+)-K(+)-Cl(-) cotransporter in primary astrocytes by dibutyryl cAMP and high [K(+)] (o) Am J Physiol Cell Physiol. 2000;279:C1710–21. doi: 10.1152/ajpcell.2000.279.6.C1710. [DOI] [PubMed] [Google Scholar]
- 22.Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- 23.Jaenisch N, Witte OW, Frahm C. Downregulation of potassium chloride cotransporter KCC2 after transient focal cerebral ischemia. Stroke. 2010;41:e151–e159. doi: 10.1161/STROKEAHA.109.570424. [DOI] [PubMed] [Google Scholar]
- 24.Gillen CM, Brill S, Payne JA, Forbush B 3rd. Molecular cloning and funetional expression of the K-Cl cotransporter from rabbit, rat, and human. A new member of the cation-chloride cotransporter family. J Biol Chem. 1996;271:16237–1624. doi: 10.1074/jbc.271.27.16237. [DOI] [PubMed] [Google Scholar]
- 25.Tyzio R, Cossart R, Khalilov I, Minlebaev M, Hubner CA, Represa A, Ben-Ari Y, Khazipov R. Maternal oxytocin triggers a transient inhibitory switch in gaba signaling in the fetal brain during delivery. Science. 2006;314:1788–1792. doi: 10.1126/science.1133212. [DOI] [PubMed] [Google Scholar]