

Abstract
Decalcified formalin-fixed and paraffin-embedded (dFFPE) bone marrow trephines remain the primary source of gDNA in hematopathological diagnostics. Here, we investigated the applicability of next-generation sequencing (NGS) to dFFPE samples. Chronic myelomonocytic leukaemia (CMML) is a haematopoietic stem cell malignancy delineated by genetic heterogeneity. Recently characteristic mutations have been identified for this entity in a distinct group of genes (TET2, CBL, KRAS). We comparatively investigated DNA extracted from fresh mononuclear cells as well as dFFPE samples from four CMML patients employing a commercially available primer set covering the above mentioned and well characterized mutational hotspots in CMML followed by an amplicon based next-generation deep-sequencing (NGS) approach. As we observed high quality run data as well as complete concordance between both sample types in all cases, we further validated the potential of NGS in hematopathology on a larger cohort of CMML patients (n=39), detecting sequence variations in 84.6% of patients. Sequence analysis revealed 92 variants, including five known polymorphisms, ten silent mutations, 36 missense mutations, 14 nonsense mutations, 24 frame shift mutations and three potential splice site mutations. Our findings ultimately demonstrate the applicability of NGS to dFFPE biopsy specimen in CMML and thus allowing the pathologist to evaluate prognostically relevant mutations at a high resolution and further contribute to risk stratification for the individual patient.
Keywords: Next-generation sequencing, decalcified, formalin-fixed, paraffin-embedded samples, chronic myelomonocytic leukaemia
Introduction
Although, modern high throughput sequencing approaches have greatly advanced our understanding of various hematological malignancies, they were primarily applied to fresh mononuclear cell samples [1,2]. However, diagnostics in hematopathology mainly rely on the use of decalcified, formalin-fixed, paraffin-embedded (dFFPE) bone marrow trephine biopsy specimen in order to elucidate immunoreactivity and morphological aspects in the topographical context of the hematopoietic system.
Chronic myelomonocytic leukaemia (CMML), for instance, is a clonal haematopoietic stem cell malignancy characterized by the presence of both myeloproliferative and myelodysplastic features as defined by the WHO Classification of myeloid neoplasms [3]. Peripheral and bone marrow blast counts are employed to divide CMML into two subtypes (CMML I <10% bone marrow and 5% peripheral blasts, CMML II up to 19% peripheral and/or bone marrow blasts) [4-6].
Clinical and haematological features regularly include monocytosis, cytopenia and/or hypercatabolic state. The clinical course however appears to be highly variable and no standardized treatment protocol could be demonstrated to positively affect overall or event-free survival [7,8]. Moreover blastic transformation into secondary AML with dismal prognosis is frequently observed [7-9].
No single pathognomonic morphological, immunohistochemical or cytogenetical hallmark has been established and few factors indicating prognosis and clinical course of disease have so far been reported, necessitating the search and validation of new markers for risk stratification [9,10].
Recently a group of genes (TET2, CBL, KRAS), carrying mutations of prognostic relevance, in CMML was characterized by Kohlmann et al. by means of next-generation deep-sequencing [11]. Their initial findings were subsequently validated on a large cohort in a multi-center study and a commercial primer set (Roche, Mannheim, Germany) covering the above mentioned well characterized mutational hotspots in CMML was developed [12].
TET2 has been shown to operate as a tumor suppressor maintaining haematopoietic cell homeostasis with mutations, particularly affecting its two highly conserved regions, thus compromising protein function, inducing myeloproliferative and/or myelodysplastic malignancies [13]. CBL shows ubiquitin ligase activity and inactivating mutations have recurrently been associated with pathogenetic abnormality in clinically aggressive MPNs (Myeloproliferative neoplasms) [14]. Mutations in RAS protein isoforms resulting in a constitutively activated RAS/RAF/MAP pathway are known to play an important role in a variety of malignancies, including colorectal, lung, bladder and thyroid cancer [15]. Further, KRAS mutations were recently depicted to play an important role in myeloid leukaemia [16].
In previous studies, investigating TET2, CBL and KRAS mutation status in myeloid neoplasia, native peripheral blood and bone marrow mononuclear cells were analyzed based on massively parallel pyrosequencing in picotiter-sized wells on the 454 platform (Roche) [11,12].
In order to assess the potential and applicability of massively parallel pyrosequencing to dFFPE samples on the 454 platform (454 Life Sciences, Branford, CT, USA) we comparatively investigated DNA extracted from fresh mononuclear cells as well as bone marrow trephine biopsy specimen from four CMML I patients for mutations in TET2, CBL and KRAS and further validated our findings on an extended cohort (n=39).
Materials and methods
Patient samples
For comparative evaluation of mutation status four paired CMML I samples of fresh mononuclear cells and dFFPE bone marrow trephine biopsies were retrieved from the registry of the Reference Center for Lymph Node Pathology and Hematopathology, University Hospital of Schleswig-Holstein, Campus Luebeck.
In order to subsequently validate our initial findings on a larger cohort additional dFFPE bone marrow trephine biopsies from 26 patients with CMML I and 13 patients with CMML II were recruited.
All samples were collected as part of standard clinical care and all studies were approved by the Ethics Committee at the University of Luebeck and are in accordance with the Declaration of Helsinki. All cases were reassessed for independent pathology review by two experienced Hematopathologists (HM & ACF) without knowledge of mutation status. Diagnosis was confirmed according to the World Health Organization classification criteria, integrating clinical, morphological and immunohistochemical findings. Immunohistochemical studies were performed on formalin-fixed paraffinembedded (FFPE) sections according to a standard, three-step immunoperoxidase technique using the automated TechMate system (DAKO, Glostrup, Denmark) and the BrightVision Kit (ImmunoLogic, Duiven, Netherlands). Clinical and haematological features of the study group are briefly summarized in Table 1.
Table 1.
Clinical characteristics of the patient cohort (n=39)
| Gender | male | 25 (64.1%) |
| female | 14 (35.9%) | |
| Age [y] | Median | 76.0 |
| Range | 54.7-91.1 | |
| Diagnosis | CMML I (WHO) | 25 (64.1%) |
| CMML II (WHO) | 14 (35.9%) | |
| CMML MDS (FAB) | 12 (32.4%) | |
| CMML MPD (FAB) | 25 (67.6%) | |
| no data available | 2 | |
| Leukocyte count [Gpt/l] | Median | 18.730 |
| Range | 3.700-100.320 | |
| no data available | 2 | |
| Erythocyte count [Tpt/l] | Median | 3.600 |
| Range | 2.400-7.720 | |
| no data available | 4 | |
| Haemoglobin level [g/dl] | Median | 10.15 |
| Range | 7.1-16.6 | |
| no data available | 3 | |
| PB monocyte count [%] | Median | 26.0 |
| Range | 6.0-67.0 | |
| no data available | 4 | |
| PB monocyte count [Gpt/l] | Median | 3.600 |
| Range | 1.080-26.300 | |
| no data available | 4 |
Based on peripheral leukocyte counts, the FAB group proposed to distinguish between two subtypes of CMML: CMML MDS (WBC ≤13x109/l) and CMML MPD (WBC >13x109/l) [31].
Next-generation sequencing
Genomic DNA was obtained from fresh mononuclear cells and dFFPE specimen using QiaAmp mini kit 250 according to the manufacturer’s instructions as described [17]. Quality and quantity of extracted DNA samples was assessed using a Nano Drop 1000 system (Thermo Scientific, Wilmington, DE, USA). DNA fragmentation was comparatively evaluated between fresh and dFFPE samples employing a multiplex PCR approach.
Next we applied amplicon-based next-generation deep-sequencing using the GS GType TET2/CBL/KRAS primer sets (Roche, Mannheim, Germany), designed for the investigation of three samples per 96 well plate, on a GS Junior platform (454 Life Sciences, Branford, CT, USA) with slight modifications as described [12]. Briefly, 31 PCR products covering the coding regions of TET2, exons 8 and 9 of CBL as well as exons 2 and 3 of KRAS were amplified. PCR reactions were performed using the FastStart High Fidelity PCR System Kit (Roche) and PCR products were pooled and purified using the Agencourt AMPure XP beads (Beckmann Coulter, Krefeld, Germany). The concentration of the amplicon pool was determined with the Quant-iT PicoGreen dsDNA Assay Kit (Life Technologies GmbH, Darmstadt, Germany). Products were prepared for emulsion PCR by diluting amplicon pools to a concentration of 2x106 molecules/μl. The following emulsion PCR was performed using the Lib-A emPCR kit (Roche) with 0.6 copies per bead inserting 5,000,000 beads per emulsion oil tube in accordance with the manufacturer’s instructions. Amplicon-based sequencing was then performed using the workflow as recommended by the manufacturer.
Sequencing data analysis
Sequencing runs were performed using the GS Junior Sequencer Software Version 2.7 (GType Leukemia 2.0) and sequencing data analysis was carried out with the GS Amplicon Variant Analyzer Version 2.7.
Variants detected with a frequency of 3% or higher on both strands were considered present. Regarding the patient cohort n=39, variants located outside the two evolutionarily conserved regions of the TET2 gene, as well as silent mutations or known single-nucleotide polymorphisms were excluded from further analysis [18]. Missense variations in exon 8 and 9 of CBL as well as in exon 2 and 3 of KRAS were considered to be of significance without regard for their specific location as these regions have been shown to be essential for protein function.
Comparative sanger sequencing
Comparative conventional Sanger sequencing was performed for all samples in order to confirm data obtained by next-generation sequencing. Information on allele burden of sequence variations as measured by NGS was collected to establish differential sensitivity of both methods. The cut-off for detecting low-level variants employing the classical chain-termination method was at an average allele frequency of 20%.
Statistical analysis
Dichotomous variables were compared between different groups using Fisher’s exact test and continuous variables were analyzed by the nonparametric Mann-Whitney U test. Wilcoxon signed rank test was applied for comparison of mutational allele burden of fresh mononuclear cells and dFFPE samples. All analyses were two-sided and the statistical significance level was set to 5% (p<0.05). All statistical data analyses were performed using GraphPad Prism 5.
Results
Comparative evaluation of fresh and dFFPE samples
Quantity and purity of extracted DNA were consistent between fresh and dFFPE samples. As expected, DNA from dFFPE samples revealed significantly elevated fragmentation as determined by a multiplex PCR approach.
Sequence analysis comparing fresh mononuclear cells and dFFPE bone marrow samples from four CMML patients revealed complete conformance in terms of sensitivity in the detection of sequence variants regardless of allele frequency. In all cases mutations and sequence variations found in the fresh material were confirmed in the corresponding dFFPE sample (see Table 2). Moreover, no significant difference in mutational allele burden between fresh and dFFPE samples was detectable employing the Wilcoxon signed rank test (p=0.3133).
Table 2.
Massively parallel pyrosequencing of TET2, CBL and KRAS in fresh mononuclear cell samples and decalcified, formalin-fixed, paraffin-embedded bone marrow trephine biopsies from four CMML patients
| Fresh mononuclear cell sample | dFFPE bone marrow trephine biopsy | ||||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| Sequence variation | Allele frequency (%) | Reads | Sanger | Allele frequency (%) | Reads | Sanger | |
| Case 1 | TET2 p.Q916X | 42.92 | 890 | pos. | 45.9 | 1011 | pos. |
| TET2 p.I1762V | 50.84 | 2026 | pos. | 49.13 | 2019 | pos. | |
| TET2 p.I1873T | 42.97 | 1145 | pos. | 43.48 | 1334 | pos. | |
| TET2 p.I1873MfsX14 | 6.2 | 1145 | neg. | 6.6 | 1334 | neg. | |
| Case 2 | CBL p.L380P | 30.5 | 2279 | pos. | 31.6 | 883 | pos. |
| TET2 p.Y1255LfsX13 | 49.1 | 1564 | pos. | 37.23 | 462 | pos. | |
| TET2 p.L1816X | 46.5 | 1688 | pos. | 41.9 | 432 | pos. | |
| Case 3 | CBL p.L380P | 51.91 | 836 | pos. | 45.71 | 862 | pos. |
| TET2 p.G355D | 50.68 | 876 | pos. | 50.52 | 1348 | pos. | |
| TET2 p.D527EfsX6 | 12.78 | 1221 | neg. | 11.37 | 1381 | neg. | |
| TET2 p.D527EfsX6 | 27.17 | 530 | pos. | 30.8 | 685 | pos. | |
| TET2 p.H1380Y | 50.59 | 597 | pos. | 42.23 | 753 | pos. | |
| TET2 p.I1762V | 48.84 | 1116 | pos. | 43.86 | 1058 | pos. | |
| Case 4 | TET2 p.N752KfsX59 | 34.59 | 743 | pos. | 38.51 | 1688 | pos. |
| TET2 p.D1704EfsX9 | 5.19 | 752 | neg. | 4.19 | 1741 | neg. | |
| TET2 p.I1762V | 48.94 | 1561 | pos. | 51.3 | 3302 | pos. | |
Run quality data in dFFPE specimen
Following these observations, we sought to validate NGS applicability to dFFPE samples on a larger cohort of CMML samples (n=39).
Seeking to assess performance of amplicon-based next-generation deep-sequencing using dFFPE specimen, we comparatively evaluated our run quality data with previously published results, obtained using fresh mononuclear cell samples [11,12].
Following DNA extraction from dFFPE bone marrow trephine biopsies, emulsion PCR and sequencing preparation procedures, NGS was performed on a GS Junior platform. Here we generated a median of 247,764 sequencing beads per run (82,588 per patient), resulting in a median of 137,097 high-quality sequencing reads per run (45,699 per patient).
A median of 49.69 Mb was sequenced per run, which is 16.57 Mb per patient.
In order to investigate the abovementioned genes a total of 31 amplicons per patient was prepared, followed by NGS. Merely, in one patient the amplicon covering TET2 exon 6 could not be amplified and subsequent sequencing analysis failed. This equals a drop out probability of 0.0827% per amplicon.
We obtained a median sequencing coverage of 2025 reads per amplicon (range 2550; 940-3490). The highest coverage was achieved for TET2 with a median of 2202 combined reads (for: 1149; rev: 1057), followed by CBL exons 8 and 9 with 1511 reads (for: 790; rev: 742,5) and KRAS exons 2 and 3 with 1121 reads (for: 586,5; rev: 527,5).
Both forward and reverse strands were successfully sequenced in nearly all cases. Nevertheless, we were able to detect a significant difference between forward and reverse reads favouring the A-sequencing-bead prepared forward strand when combining all sequenced amplicons (p=0.0027) or TET2 alone (p=0.0041). A similar trend was evaluated for both CBL exons 8 and 9 as well as KRAS exons 2 and 3 separately, but failed, however, to reach statistical significance. Coverage data is briefly summarized in Figure 1.
Figure 1.
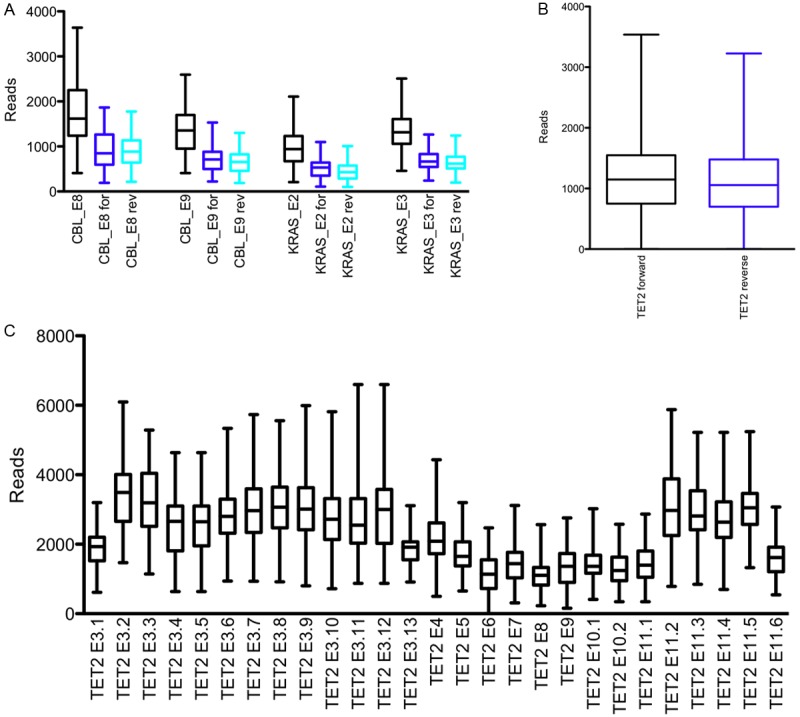
Combined coverage data of TET2, CBL and KRAS next-generation sequencing on dFFPE samples. Combined, forward and reverse reads for CBL and KRAS (A), overall forward and reverse TET2 reads per amplicon (B) and combined TET2 reads for all amplicons (C). Data reveal a consistent as well as high resolution coverage and summarized forward/reverse strand sequencing of TET2 illustrate a slight predominance of the A-sequencing-bead prepared forward strand.
TET2, CBL and KRAS mutations in a validation cohort of 39 CMML samples
Amplicon-based next-generation deep-sequencing of the TET2 gene coding regions as well as exon 8 and 9 of the CBL gene and exon 2 and 3 of the KRAS gene revealed sequence variations in 84.6% of patients. Five known single-nucleotide polymorphisms, ten silent mutations and ten previously unreported missense mutations located outside of evolutionarily conserved regions were excluded (Supplementary Table 1). In summary, sequence analysis revealed 67 putative protein damaging variances, including 26 missense mutations located within conserved regions, 14 nonsense mutations, 24 frameshift mutations and three potential splice site mutations (Table 3).
Table 3.
TET2, CBL and KRAS mutations in CMML
| Sample | Diagnosis | TET2 | CBL | KRAS |
|---|---|---|---|---|
| 1 | CMML I | WT | p.C419S | WT |
| 2 | CMML I | c.3409+1 G>T | p.R420Q | WT |
| p.V1864E | ||||
| 3 | CMML I | p.P1419LfsX29 | p.R420Q | WT |
| p.G1869E | ||||
| 4 | CMML I | WT | ||
| 5 | CMML II | c.3954+1 G>A | p.P417S | WT |
| p.R1366L | ||||
| 6 | CMML I | p.G1869E | WT | p.G13D |
| 7 | CMML I | p.P413HfsX14 | WT | WT |
| 8 | CMML II | c.4183-16_-6del11 | p.P417S | WT |
| p.G1519DfsX52 | p.D460del | |||
| c.3954+1 G>A | ||||
| p.R1366L | ||||
| 9 | CMML I | p.L1340R | WT | p.G12D |
| p.R1516X | ||||
| 10 | CMML II | p.Q1834X | p.K392N | WT |
| 11 | CMML I | WT | p.C401S | WT |
| 12 | CMML II | p.L655NfsX45 | WT | WT |
| p.Q705X | ||||
| 13 | CMML I | WT | p.Y371S | WT |
| 14 | CMML I | WT | p.C384Y | p.E37K |
| p.L370F | ||||
| 15 | CMML I | p.L199FfsX3 | WT | WT |
| p.E1178KfsX48 | ||||
| 16 | CMML I | p.L1081X | WT | p.D33E |
| 17 | CMML I | p.C1221VfsX5 | p.I383M | WT |
| p.M804WfsX9 | ||||
| 18 | CMML I | p.P1741HfxX4 | WT | WT |
| p.P399TfsX44 | ||||
| 19 | CMML I | WT | p.Q367P | WT |
| 20 | CMML II | WT | ||
| 21 | CMML II | p.Q635X | WT | WT |
| p.R1261H | ||||
| 22 | CMML I | p.Q403X | p.C381Y | WT |
| p.Q635X | ||||
| 23 | CMML II | p.H1386AfsX15 | WT | WT |
| p.L615SfsX24 | ||||
| 24 | CMML I | p.Q417X | WT | WT |
| 25 | CMML I | p.E294X | WT | WT |
| p.I873DfsX28 | ||||
| 26 | CMML II | p.H1881N | WT | WT |
| 27 | CMML I | p.Q769X | WT | WT |
| 28 | CMML I | p.K1299fsX1 | p.C401Y | WT |
| p.E576NfsX4 | ||||
| p.Q324X | ||||
| 29 | CMML II | WT | ||
| 30 | CMML I | p.L1120X | WT | WT |
| p.C1298MfsX2 | ||||
| 31 | CMML II | p.T1251KfsX15 | WT | WT |
| p.C1271WfsX29 | ||||
| p.K306fsX1 | ||||
| p.D551AfsX15 | ||||
| 32 | CMML II | WT | ||
| 33 | CMML I | p.S393LfsX34 | WT | p.E91K |
| p.G1754IfsX7 | ||||
| p.T1280I | ||||
| 34 | CMML I | p.Q904X | WT | WT |
| p.Y1245C | ||||
| 35 | CMML II | p.M1164K | WT | WT |
| p.E1279X | ||||
| p.P409FfsX33 | ||||
| p.R1366L | ||||
| 36 | CMML II | WT | ||
| 37 | CMML I | p.Y1569X | WT | WT |
| 38 | CMML I | WT | p.P417S | WT |
| 39 | CMML I | WT | ||
CMML – Chronic myelomonocytic leukemia, WT – wildtype.
In total, various mutations were detected in 33 of 39 patients (84.6%) at an average of 1.92 mutations per patient. Of these mutations 19 were previously described in malignant samples. In agreement with previous studies (Kohlmann 2010) TET2 was the most frequently mutated gene as we were able to detect nine missense mutations, 14 nonsense mutations, 23 frameshift mutations and three potential splice site mutations in 27 of 39 patients (69.2%) at an average of 1.38 mutations per patient (Figure 2). Four of these mutations could be detected independently in multiple patients (p.G1869E, p.Q635X, p.R1366L, c.3954+1G>A). As described, amplification of TET2 exon 6 failed in a single patient. However, conventional Sanger sequencing was successful for this amplicon and we found no sequence variation.
Figure 2.
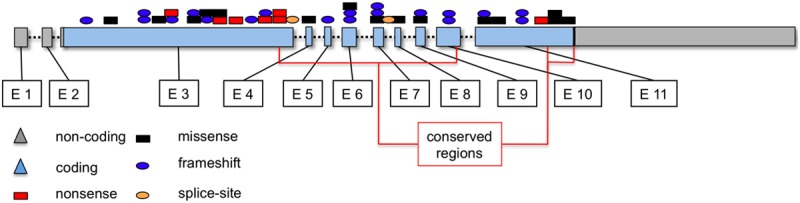
TET2 Mutation types and distribution in CMML samples. Nine missense mutations within the two evolutionarily conserved regions (black boxes), 14 nonsense mutations (red boxes), 23 frameshift mutations (blue ovals) and three potential splice site mutations (orange ovals) in 27 of 39 patients (69.2%) at an average of 1.38 mutations per patient. Four of these mutations could be detected independently in multiple patients.
In the CBL gene we found 13 distinct missense mutations in 14 (35.9%) of 39 patients at an average of 0.41 mutations per patient. The previously described missense mutation p.R420Q was detected in two patients.
Further we observed five missense mutations in KRAS exons 2 and 3 in five patients (12.8%) resulting in an average of 0.13 mutations per patient.
There was no statistically significant difference in mutation status between CMML I and CMML II (p=0.927).
Discussion
The introduction of consumer platforms, capable of massively parallel pyrosequencing has substantially enhanced the spectrum in which genomic sequencing is applicable in the routine diagnostic workup for clinical patients at affordable costs.
The molecular pathogenesis of CMML remains widely elusive, as do predictors of clinical course, outcome and response to therapy. To date, the diagnostic follow up for CMML patients focuses mainly on haematological and clinical parameters, such as anaemia, splenomegaly, or leukocytosis [5,7,9]. Despite ongoing discussions concerning prognostic implications, the recent identification of TET2, CBL and KRAS mutations as a highly recurrent event in MDS/MPS, particularly CMML, has greatly expanded the molecular understanding of these diseases and holds promise for upcoming targeted therapy approaches [11,18-23].
Here, we applied an amplicon based deep-sequencing approach on a GS Junior platform (454 Life Sciences, Branford, CT, USA) to comparatively analyze the mutation status of TET2, CBL and KRAS in four paired samples of fresh and dFFPE specimen. Next, we validated NGS applicability on an extended cohort of dFFPE bone marrow trephine biopsy samples from 39 CMML patients. The above mentioned genes were selected due to their well-defined role as mutational hot spots in MDS/MPS. Moreover, multiple studies were recently conducted employing the same commercially available primer set and reagents (Roche, Mannheim, Germany).
Further, TET2 mutations were recently identified to play an important role in numerous other proliferative and/or dysplastic malignancies of the myeloid linage, including MDS, polycythemia vera, essential thrombocytosis, primary myelofibrosis and systemic mastocytosis [18,24,25]. Moreover, TET2 mutations appear to be of prognostic and pathogenetic significance in a subset of acute myeloid leukaemia patients [26].
Comparative evaluation of fresh and dFFPE samples revealed no difference in run quality data or diagnostic sensitivity. Next, in order to further underline NGS applicability to dFFPE specimen, we extended our investigations on a larger cohort of 39 CMML patients. In this comprehensive approach, 85 pathogenic sequence variations were detected by NGS and subsequently evaluated by Sanger sequencing. Low-level variations (allele burden <20%), however, could not be reproducibly confirmed by Sanger sequencing. We were able to detect 44 missense mutations, 14 nonsense mutations leading to a truncated translation of the protein, 24 frame shift mutations and three potential splice site mutations in 33 of 39 patients.
Of the abovementioned 67 TET2 mutations, 19 mutations have previously been described in cancer samples adding to our understanding of potential mutation hot-spot regions of TET2. In accordance with previously published results, mutations were distributed throughout the entire coding regions of TET2, with missense mutations being predominantly found in evolutionarily conserved regions of the gene [22]. As proposed by Weissmann et al., we excluded ten novel single nucleotide variations located outside the two evolutionary conserved regions from further analysis [26], however, this does not exclude the possibility that these mutations might be of functional relevance in an as of yet unknown modality. The effect of substitutions leading to altered amino acid sequences outside of the conserved regions, proposed to be essential for protein function, still needs to be clarified. Moreover, little is known about TET2 protein function and about the functional implications of most of these mutations [27,28].
As dFFPE samples remain the most widely used material in both routine histopathological diagnostics as well as retrospective clinical studies, we and others have successfully focused on transferring novel investigative molecular methods from their initial application on fresh material to these more common, yet more demanding (in terms of nucleic acid quality and quantity) specimen [29].
Massively parallel pyrosequencing has been shown to be a robust, fast and highly reproducible method delivering excellent coverage and sensible detection of genetic aberrations. So far, however, no data exist about its utility using DNA extracted from FFPE samples. DNA from these samples is known, to be highly fragmented and its quality is often compromised but since hematopathological diagnostics mainly rely on these samples, their applicability is crucial to the implementation of any novel diagnostic method in the field of pathology [30]. In many instances bone marrow smears or aspirate specimen are not at hand and in addition bone marrow aspirate samples are often unavailable from patients who suffer from extensive marrow fibrosis (punctio sicca). Thus, any molecular method, which may be used with FFPE or even dFFPE, is of high value for the diagnostic process.
Overall sequencing coverage surpassed our expectations. The read quality data obtained in the present study on dFFPE samples were comparable to those of previously published data, applying similar approaches to fresh mononuclear cell samples from blood and/or bone marrow [11,12]. With reference to genomic coverage as well as to average read length our results partly even exceeded published data.
Up until now the classical chain-termination sequencing was the only method applicable to dFFPE samples in routine hematopathological practice. Here we demonstrate, in a systematical comparative analysis, that a significantly improved sensitivity in detecting sequence variations can be obtained employing NGS. In many cases this will have a major impact on diagnosis, risk stratification and follow-up for the individual patient.
The approach presented in this study was conducted in an average run-time from DNA extraction to final sequencing results of approximately five days, leaving room for further acceleration employing automated sample preparation approaches, thus rendering next-generation sequencing fit for diagnostic requirements in hematopathological practice.
The detection of mutations in TET2, KRAS and CBL genes may in many instances assist in patient management, e.g. planning revaluation of bone marrow or cytogenetic analysis or for the decision to initiate cytoreductive therapy as they ultimately aid in discriminating reactive and neoplastic bone marrow lesions.
Acknowledgements
We thank Tanja Oeltermann for her skilled and dedicated technical assistance.
Disclosure of conflict of interest
None declared.
Supporting Information
References
- 1.Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, Avezov E, Li J, Kollmann K, Kent DG, Aziz A, Godfrey AL, Hinton J, Martincorena I, Van Loo P, Jones AV, Guglielmelli P, Tarpey P, Harding HP, Fitzpatrick JD, Goudie CT, Ortmann CA, Loughran SJ, Raine K, Jones DR, Butler AP, Teague JW, O’Meara S, McLaren S, Bianchi M, Silber Y, Dimitropoulou D, Bloxham D, Mudie L, Maddison M, Robinson B, Keohane C, Maclean C, Hill K, Orchard K, Tauro S, Du MQ, Greaves M, Bowen D, Huntly BJ, Harrison CN, Cross NC, Ron D, Vannucchi AM, Papaemmanuil E, Campbell PJ, Green AR. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369:2391–2405. doi: 10.1056/NEJMoa1312542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, Them NC, Berg T, Gisslinger B, Pietra D, Chen D, Vladimer GI, Bagienski K, Milanesi C, Casetti IC, Sant’Antonio E, Ferretti V, Elena C, Schischlik F, Cleary C, Six M, Schalling M, Schonegger A, Bock C, Malcovati L, Pascutto C, Superti-Furga G, Cazzola M, Kralovics R. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369:2379–2390. doi: 10.1056/NEJMoa1311347. [DOI] [PubMed] [Google Scholar]
- 3.Swerdlow SH. WHO. 2008. WHO Classification of Tumors of Haematopoetic and Lymphoid Tissues. [Google Scholar]
- 4.Anon C. Chronic myelomonocytic leukemia: single entity or heterogeneous disorder? A prospective multicenter study of 100 patients. Groupe Francais de Cytogenetique Hematologique. Cancer Genet Cytogenet. 1991;55:57–65. doi: 10.1016/0165-4608(91)90235-m. [DOI] [PubMed] [Google Scholar]
- 5.Cortes J. CMML: a biologically distinct myeloproliferative disease. Curr Hematol Rep. 2003;2:202–208. [PubMed] [Google Scholar]
- 6.Germing U, Strupp C, Knipp S, Kuendgen A, Giagounidis A, Hildebrandt B, Aul C, Haas R, Gattermann N, Bennett JM. Chronic myelomonocytic leukemia in the light of the WHO proposals. Haematologica. 2007;92:974–977. doi: 10.3324/haematol.11051. [DOI] [PubMed] [Google Scholar]
- 7.Beran M, Wen S, Shen Y, Onida F, Jelinek J, Cortes J, Giles F, Kantarjian H. Prognostic factors and risk assessment in chronic myelomonocytic leukemia: validation study of the M. D. Anderson Prognostic Scoring System. Leuk Lymphoma. 2007;48:1150–1160. doi: 10.1080/10428190701216386. [DOI] [PubMed] [Google Scholar]
- 8.Onida F, Kantarjian HM, Smith TL, Ball G, Keating MJ, Estey EH, Glassman AB, Albitar M, Kwari MI, Beran M. Prognostic factors and scoring systems in chronic myelomonocytic leukemia: a retrospective analysis of 213 patients. Blood. 2002;99:840–849. doi: 10.1182/blood.v99.3.840. [DOI] [PubMed] [Google Scholar]
- 9.Beran M, Shen Y, Onida F, Wen S, Kantarjian H, Estey E. Prognostic significance of monocytosis in patients with myeloproliferative disorders. Leuk Lymphoma. 2006;47:417–423. doi: 10.1080/10428190500305448. [DOI] [PubMed] [Google Scholar]
- 10.Fenaux P, Jouet JP, Zandecki M, Lai JL, Simon M, Pollet JP, Bauters F. Chronic and subacute myelomonocytic leukaemia in the adult: a report of 60 cases with special reference to prognostic factors. Br J Haematol. 1987;65:101–106. doi: 10.1111/j.1365-2141.1987.tb06142.x. [DOI] [PubMed] [Google Scholar]
- 11.Kohlmann A, Grossmann V, Klein HU, Schindela S, Weiss T, Kazak B, Dicker F, Schnittger S, Dugas M, Kern W, Haferlach C, Haferlach T. Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J. Clin. Oncol. 2010;28:3858–3865. doi: 10.1200/JCO.2009.27.1361. [DOI] [PubMed] [Google Scholar]
- 12.Kohlmann A, Klein HU, Weissmann S, Bresolin S, Chaplin T, Cuppens H, Haschke-Becher E, Garicochea B, Grossmann V, Hanczaruk B, Hebestreit K, Gabriel C, Iacobucci I, Jansen JH, te Kronnie G, van de Locht L, Martinelli G, McGowan K, Schweiger MR, Timmermann B, Vandenberghe P, Young BD, Dugas M, Haferlach T. The Interlaboratory RObustness of Next-generation sequencing (IRON) study: a deep sequencing investigation of TET2, CBL and KRAS mutations by an international consortium involving 10 laboratories. Leukemia. 2011;25:1840–1848. doi: 10.1038/leu.2011.155. [DOI] [PubMed] [Google Scholar]
- 13.Li Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, Yang FC, Xu M. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011;118:4509–4518. doi: 10.1182/blood-2010-12-325241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grand FH, Hidalgo-Curtis CE, Ernst T, Zoi K, Zoi C, McGuire C, Kreil S, Jones A, Score J, Metzgeroth G, Oscier D, Hall A, Brandts C, Serve H, Reiter A, Chase AJ, Cross NC. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood. 2009;113:6182–6192. doi: 10.1182/blood-2008-12-194548. [DOI] [PubMed] [Google Scholar]
- 15.Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 16.Tyner JW, Erickson H, Deininger MW, Willis SG, Eide CA, Levine RL, Heinrich MC, Gattermann N, Gilliland DG, Druker BJ, Loriaux MM. High-throughput sequencing screen reveals novel, transforming RAS mutations in myeloid leukemia patients. Blood. 2009;113:1749–1755. doi: 10.1182/blood-2008-04-152157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gebauer N, Bernard V, Feller AC, Merz H. ID3 Mutations Are Recurrent Events in Double-hit B-Cell Lymphomas. Anticancer Res. 2013;33:4771–4778. [PubMed] [Google Scholar]
- 18.Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, Stevens-Linders E, van Hoogen P, van Kessel AG, Raymakers RA, Kamping EJ, Verhoef GE, Verburgh E, Hagemeijer A, Vandenberghe P, de Witte T, van der Reijden BA, Jansen JH. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41:838–842. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- 19.Kosmider O, Gelsi-Boyer V, Cheok M, Grabar S, Della-Valle V, Picard F, Viguie F, Quesnel B, Beyne-Rauzy O, Solary E, Vey N, Hunault-Berger M, Fenaux P, Mansat-De Mas V, Delabesse E, Guardiola P, Lacombe C, Vainchenker W, Preudhomme C, Dreyfus F, Bernard OA, Birnbaum D, Fontenay M. TET2 mutation is an independent favorable prognostic factor in myelodysplastic syndromes (MDSs) Blood. 2009;114:3285–3291. doi: 10.1182/blood-2009-04-215814. [DOI] [PubMed] [Google Scholar]
- 20.Bacher U, Weissmann S, Kohlmann A, Schindela S, Alpermann T, Schnittger S, Kern W, Haferlach T, Haferlach C. TET2 deletions are a recurrent but rare phenomenon in myeloid malignancies and are frequently accompanied by TET2 mutations on the remaining allele. Br J Haematol. 2012;156:67–75. doi: 10.1111/j.1365-2141.2011.08911.x. [DOI] [PubMed] [Google Scholar]
- 21.Grossmann V, Kohlmann A, Eder C, Haferlach C, Kern W, Cross NC, Haferlach T, Schnittger S. Molecular profiling of chronic myelomonocytic leukemia reveals diverse mutations in >80% of patients with TET2 and EZH2 being of high prognostic relevance. Leukemia. 2011;25:877–879. doi: 10.1038/leu.2011.10. [DOI] [PubMed] [Google Scholar]
- 22.Kosmider O, Gelsi-Boyer V, Ciudad M, Racoeur C, Jooste V, Vey N, Quesnel B, Fenaux P, Bastie JN, Beyne-Rauzy O, Stamatoulas A, Dreyfus F, Ifrah N, de Botton S, Vainchenker W, Bernard OA, Birnbaum D, Fontenay M, Solary E. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica. 2009;94:1676–1681. doi: 10.3324/haematol.2009.011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith AE, Mohamedali AM, Kulasekararaj A, Lim Z, Gaken J, Lea NC, Przychodzen B, Mian SA, Nasser EE, Shooter C, Westwood NB, Strupp C, Gattermann N, Maciejewski JP, Germing U, Mufti GJ. Next-generation sequencing of the TET2 gene in 355 MDS and CMML patients reveals low-abundance mutant clones with early origins, but indicates no definite prognostic value. Blood. 2010;116:3923–3932. doi: 10.1182/blood-2010-03-274704. [DOI] [PubMed] [Google Scholar]
- 24.Tefferi A, Levine RL, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Finke CM, Mullally A, Li CY, Pardanani A, Gilliland DG. Frequent TET2 mutations in systemic mastocytosis: clinical, KITD816V and FIP1L1-PDGFRA correlates. Leukemia. 2009;23:900–904. doi: 10.1038/leu.2009.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tefferi A, Pardanani A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Gangat N, Finke CM, Schwager S, Mullally A, Li CY, Hanson CA, Mesa R, Bernard O, Delhommeau F, Vainchenker W, Gilliland DG, Levine RL. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia. 2009;23:905–911. doi: 10.1038/leu.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weissmann S, Alpermann T, Grossmann V, Kowarsch A, Nadarajah N, Eder C, Dicker F, Fasan A, Haferlach C, Haferlach T, Kern W, Schnittger S, Kohlmann A. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia. 2012;26:934–942. doi: 10.1038/leu.2011.326. [DOI] [PubMed] [Google Scholar]
- 27.Metzeler KH, Maharry K, Radmacher MD, Mrozek K, Margeson D, Becker H, Curfman J, Holland KB, Schwind S, Whitman SP, Wu YZ, Blum W, Powell BL, Carter TH, Wetzler M, Moore JO, Kolitz JE, Baer MR, Carroll AJ, Larson RA, Caligiuri MA, Marcucci G, Bloomfield CD. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. J. Clin. Oncol. 2011;29:1373–1381. doi: 10.1200/JCO.2010.32.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, Malinge S, Yao J, Kilpivaara O, Bhat R, Huberman K, Thomas S, Dolgalev I, Heguy A, Paietta E, Le Beau MM, Beran M, Tallman MS, Ebert BL, Kantarjian HM, Stone RM, Gilliland DG, Crispino JD, Levine RL. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114:144–147. doi: 10.1182/blood-2009-03-210039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoefig KP, Thorns C, Roehle A, Kaehler C, Wesche KO, Repsilber D, Branke B, Thiere M, Feller AC, Merz H. Unlocking pathology archives for microRNA-profiling. Anticancer Res. 2008;28:119–123. [PubMed] [Google Scholar]
- 30.Hosein AN, Song S, McCart Reed AE, Jayanthan J, Reid LE, Kutasovic JR, Cummings MC, Waddell N, Lakhani SR, Chenevix-Trench G, Simpson PT. Evaluating the repair of DNA derived from formalin-fixed paraffin-embedded tissues prior to genomic profiling by SNP-CGH analysis. Lab Invest. 2013;93:701–10. doi: 10.1038/labinvest.2013.54. [DOI] [PubMed] [Google Scholar]
- 31.Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick H, Sultan C, Cox C. The chronic myeloid leukaemias: guidelines for distinguishing chronic granulocytic, atypical chronic myeloid, and chronic myelomonocytic leukaemia. Proposals by the French-American-British Cooperative Leukaemia Group. Br J Haematol. 1994;87:746–754. doi: 10.1111/j.1365-2141.1994.tb06734.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.