

Abstract
Molecular genetic analysis indicates that the problematic human bacterial pathogen methicillin-resistant Staphylococcus aureus possesses more than 2000 open reading frames in its genome. This number of potential gene products, coupled with intrinsic mechanisms of posttranslational modification, endows methicillin-resistant Staphylococcus aureus with a highly complex biochemical repertoire. Recent proteomic and metabolomic advances have provided methodologies to better understand and characterize the biosynthetic factors released by microbial organisms. Here, the emerging tool of mass spectrometry-based molecular networking was used to visualize and map the repertoire of biosynthetic factors produced by a community-associated methicillin-resistant Staphylococcus aureus strain representative of the epidemic USA300 clone. In particular, the study focused on elucidating the complexity of the recently discovered phenol soluble modulin family of peptides when placed under various antibiotic treatment stresses. Novel PSM truncated variant peptides were captured, and the type of variants that were clustered by the molecular networks platform changed in response to the different antibiotic treatment conditions. After discovery, a group of the peptides were selected for functional analysis in vitro. The peptides displayed bioactive properties including the ability to induce proinflammatory responses in human THP-1 monocytes. Additionally, the tested peptides did not display antimicrobial activity as previously reported for other phenol soluble modulin truncated variants. Our findings reveal that the PSM family of peptides are quite structurally diverse, and suggest a single phenol soluble modulin parent peptide can functionally spawn differential bioactivities in response to various external stimuli.
A significant percentage of the human population harbors Staphylococcus aureus as a natural resident of their microbial flora (1). In non-infected individuals, S. aureus maintains a homeostatic coexistence with neighboring microbes and with the host (2). However, when equilibrium is disrupted, S. aureus is capable of establishing diseases ranging from minor superficial infections to potentially life-threatening invasive conditions (3, 4). Through the discovery and administration of antibiotics, clinicians have significantly reduced the morbidity and mortality associated with S. aureus (5, 6). In recent years, the consequences of persistent antibiotic exposure have become increasingly manifest through the emergence of resistant strains, including those left unperturbed by methicillin and related β-lactam antibiotics (MRSA)1 or those with intermediate or full resistance to vancomycin (VISA/VRSA) (7). Certain drug-resistant S. aureus strains, such as the USA300 clone, have spread beyond the hospital setting and into the community, producing serious infections even in previously healthy individuals (8, 9). Both community-associated (CA) and hospital-associated (HA) multi-drug resistant S. aureus are highly evolved, multifaceted pathogens that pose an increasing threat to the health of not only our own, but future generations as well.
The pathogenic mechanisms of S. aureus have been examined extensively through different stages of infection (10, 11), with an improved understanding of specific functions that various staphylococcal components contribute to virulence (12). The ability to outcompete other microflora for colonization, or to circumvent the host immune response during infectious spread, largely reflects a complex network of proteins and non-protein molecules that S. aureus secretes into its surroundings-which may collectively be termed biosynthetic release factors. One component of this network is the newly studied and multifunctional phenol soluble modulin (PSM) family of peptides. Associated with biofilm formation and possessing bioactivity toward host cells, PSMs are produced more abundantly by CA-MRSA compared with HA-MRSA strains (13, 14). PSMs are biosynthesized and released with formylated initiator methionine, allowing binding and activation of host neutrophils via the formyl peptide receptor 2 (FPR2) (15). A number of PSMs show cytolytic properties toward host cell lines, and supporting this, isogenic PSM-knockout mutants have decreased virulence in murine models of invasive infection (13). Recently, it was found that many in vitro PSMs phenotypes (e.g. neutrophil lysis, FPR2 binding) were strongly inhibited by serum lipoproteins (16). The same study showed up-regulation of PSM biosynthesis following phagocytosis, suggesting an important role in intracellular survival. Additionally, N-terminal truncated derivative forms of PSMs (dPSMs) with antibacterial properties were identified (17, 18). The finding that dPSMs possess antibacterial activities that are absent in the corresponding full-length parent PSMs, inspired a hypothesis that dPSMs endow CA-MRSA strains with a competitive advantage against resident microbiota upon colonizing host epithelia (i.e. once truncated PSMs gain antimicrobial properties).
The invaluable tool of MS has played a major role in characterizing biosynthetic release factors for a diverse number of organisms (19). Over the past decade, the field has spawned several innovative methods to optimize data collection, processing, and organization. The advancement of MS processing methods has improved the capabilities for extracting significant information regarding an organism's biochemical repertoire. One emerging tool to study biosynthetic release factor potential is the tandem MS data processing program, Spectral Networks. In conjunction with Cytoscape graphical mapping, Spectral Networks allows for the construction of a molecular network map that gives a visualization of tandem mass spectra grouping, based on similar scoring of overlapping ion fragmentation patterns (20, 21). The matching of spectra, termed spectral pairing, is unlike the conventional mode of processing MS data by matching against a reference library or specifying specific posttranslational modifications for identification (22). Instead, within a molecular network map, ion fragmentation patterns can be represented by correlation points, displayed as circular nodes, connected with a straight line to other nodes containing high similarity scores. The power of molecular networking is the information provided on tandem MS subpopulation clusters, which represent structurally matching but variant protein or non-protein biosynthetic release factors.
In this work, the MS-based molecular networking platform was applied to a representative strain of the epidemic CA-MRSA USA300 clone. The study sought to provide insight into the biosynthetic release factor map of this pre-eminent human bacterial pathogen under normal growth and antibiotic challenged conditions. Biologically relevant pharmacological stimuli studied included sub-inhibitory concentrations of commonly prescribed commercial antibiotics as well as the innate human defense peptide LL-37 (23–25). In particular, our study goal was to monitor any variations in the type of dPSMs clustered within the molecular network map in response to these stimuli. We show an enriched extract derived from CA-MRSA alone or under these specific challenge conditions provides a highly complex biosynthetic release factor map. Analysis of subpopulation node clusters associated with PSMs prompted the discovery of several previously unreported α, β, and γ type dPSMs. The antibiotic stimuli modulated the type of dPSMs clustered, allowing for differential subclusters to be visualized that were absent in the untreated sample; including nodes that did not sequence to proteogenic PSMs. A group of the newly discovered PSM variants were assessed for functional properties and showed the induction of proinflammatory responses in THP-1 cells. In addition, in contrast to previous reports (17, 18), the tested dPSMs did not display antimicrobial activity. We conclude MS-based molecular networking can be used as a targeted discovery tool with the ability to identify previously unreported bioactive peptides.
EXPERIMENTAL PROCEDURES
Bacterial Strain and Culture Conditions
S. aureus USA300 strain TCH1516 (26) alone or in the presence of (1) 0.125 mg/L daptomycin; (2) 0.125 mg/L vancomycin; or (3) 1.2 mg/L LL-37 was grown in Todd Hewitt broth (THB) and propagated at 37 °C. Growth curves were constructed on several occasions to ensure no significant changes were encountered between treated and untreated samples by monitoring the OD600 until reaching a robust cell density.
Sample Preparation for Nanocapillary Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS)
Samples at equivalent OD600, along with a THB media alone control sample, were incubated at a 1:1 ratio with 1-butanol, mixed and allowed to separate into polar and nonpolar phases overnight. The solvent 1-butanol was used to selectively capture small molecules and peptides. The samples were then centrifuged at 2,000 rpm for 5 min and the layer containing 1-butanol was collected in 1 ml portions. These were each centrifuged at 14,000 rpm, transferred to a new container, and brought to complete dryness by SpeedVac centrifuge. Two sample tubes for each experiment were then resuspended and pooled before passing samples through a size-exclusion column. Collected fractions were brought to complete dryness and stored. Synthetic PSMα4 (American Peptide Co, Sunnyvale, CA) of >95% purity was also processed as a control to ensure the method per se did not alter the peptide nor create variant forms.
Nanocapillary LC-MS/MS
Nanocapillary columns were prepared by splitting deactivated, fused silica tubing (Agilent, Santa Clara, CA) with a model P-2000 laser puller (Sutter Instruments, Novato, CA). Pulled 15 cm columns were packed with C18 reverse phase resin suspended in methanol. Thereafter, the column was mounted onto the LC-MS/MS apparatus and equilibrated with 95% solvent A (water, 0.1% formic acid) until a stable ion chromatogram was achieved. A gradient for loading and eluting extract contents was established with a time-varying mixture of solvent A and B (acetonitrile, 0.1% formic acid) and electrosprayed directly into a calibrated Thermo Finnigan LTQ-XL. Acquisition parameters included data-dependent ion selection targeting the first to fifth most abundant ions in the parent mass spectrum for fragmentation. Data sets for the extracted samples were collected in the following order: (1) ESI solvent; (2) THB media; (3) CA-MRSA untreated; (4) CA-MRSA + daptomycin; (5) CA-MRSA + vancomycin; and (6) CA-MRSA + LL-37. A water blank was run between samples to minimize overlapping contamination.
Construction of Molecular Networks
Molecular Networks were constructed as previously described (21). Briefly, collected .RAW files were converted to MZXML files by the program ReAdW. Converted files were then clustered at cosine scores of greater than or equal to 0.6. The graphical display program Cytoscape was used to view clustered spectra as molecular networks based on tandem MS fragmentation patterns. In each Cytoscape map, nodes that were found overlapping with both the CA-MRSA alone sample and also in the CA-MRSA plus antibiotic stimulus were colored gray. Nodes that were specific to CA-MRSA alone are colored gold. Nodes that were specific to the CA-MRSA plus daptomycin were colored aqua blue, those specific to CA-MRSA plus vancomycin colored green, and those specific to CA-MRSA plus LL-37 colored red. Nodes pertaining to components of THB media and LC-MS/MS solvents were filtered out from the networks to minimize false positive matching rates.
Identification of PSM Derivatives
To identify network clusters related to PSMs, a targeted approach was taken displaying the parent ion mass of each individual node and matching according to theoretical values for full-length PSMs and truncated dPSMs. Amino acid sequences pertaining to PSMs were obtained from previously published work (18). When an ion cluster was found that contained the mass indicative of PSMs or dPSMs and the specific node mass contained less than 450 ppm mass error (parent masses) relative to the theoretical value, de novo sequencing of the peptide was performed. PSMs are released extracellularly containing a formylated (+28 kDa) initiator methionine; when appropriate, the PTM was considered in the manual searches. Tandem mass spectra with a minimum of seven contingent ions of correct mass accuracy and fragment peaks with a mass tolerance of 0.5 Da were accepted as valid sequence tags. When a node was identified according to the set criteria, adjacent nodes were processed for identification purposes using the same criteria for validation. LC-MS/MS runs were performed in triplicate, with different growth experiments prepared on separate days using the same USA300 CA-MRSA (TCH1516) strain stock. All analyzed nodes including sequence tags for newly discovered PSMs are provided in the Online Supplemental Information.
PSMs Peptide Levels Monitored by Spectral Counts
To monitor the effect of relative PSM production in the presence of antibiotic stimulus, spectral counts for each identified dPSM were enumerated by the use of InsPecT (20). Relevant input search parameters included the following: (1) post-translational modification search, +28 Da (formylation); (2) database, modified TCH1516 USA300 genome, TCH1516 USA300 genome “reversed,” and common contaminants database; (3) PTM allowed per peptide, 2; (4) b- and y-ion mass offset tolerance, 0.5 Da; and (5) parent mass tolerance, 1.5 Da. Statistical significance was determined by one way ANOVA with Tukey's post-hoc test. ** indicates p < 0.01, *** indicates p < 0.001.
Hemolysis Activity
Defibrinated sheep blood was washed in PBS and diluted to a final concentration of 1:25. Washed blood was then placed into individual wells of a round-bottom 96-well microtiter plate (Costar, Corning). dPSM peptides at a final concentration of 20 μg/ml, 10 μg/ml, 5 μg/ml, or 1 μg/ml were added directly to the wells. Thereafter, the plate was sealed by parafilm and incubated for 360 min and overnight at 37 °C. After incubation, plates were centrifuged at 1600 rpm for 5 min, and 60 μl of supernatant (without disturbing pellet) was placed in a separate microtiter plate to measure hemoglobin absorbance at 450 nm on a microplate reader. Diluted Triton-X (0.0125%) was used as a positive control for hemolysis and 1 × PBS was used as a negative control. Hemolysis was normalized to the Triton-X positive control, which was considered 100% hemolysis.
Antimicrobial Activity
The representative skin and soft tissue microbes Staphylococcus epidermidis ATCC36981, Staphylococcus epidermidis Nizet lab strain, Group A Streptococcus (GAS) serotype M3, and methicillin-sensitive Staphylococcus aureus (MSSA) ATCC29213 were incubated with 10 μg/ml or 1 μg/ml of the dPSM test peptides. Growth curves were monitored at an OD600 every 30 min until late exponential-stationary phase (OD600 ∼0.8–1.0). Bacterial growth in medium without the dPSM peptides was used as control.
Immunostimulatory Effects Induced by the Truncated PSM Variants
THP-1 cells were grown in medium until confluent (RPMI, 10%FBS, 10 mm HEPES, 1 mm sodium pyruvate, D+glucose, and 0.35‰ Basal Medium Eagle). THP-1 cells were seeded in 24-well plates with a final concentration of 5 × 105 cells/well and differentiated overnight with 25 nm PMA. New medium without PMA was added for 24 h before usage in successive experiments. The dPSM peptides were added with a final concentration of 20 μg/ml, 10 μg/ml, 5 μg/ml, or 1 μg/ml respectively and cells were incubated at 37 °C in a 5% CO2 incubator for 6 h. Cell culture medium was used as negative control and 10 μg/ml LPS or 0.01% Triton-X respectively, were used as positive control. Supernatant was harvested and stored at −20 °C until used. dPSM peptide induced secretion of human IL-8 and IL-1β levels by THP-1 cells were measured by ELISA assay kits, according to manufacturer's instructions (R&D Systems, Minneapolis, MN).
RESULTS
Building CA-MRSA Spectral Networks
Files were compiled and processed as a group to obtain consolidated files in the following manner: (1) CA-MRSA ± daptomycin + THB media; (2) CA-MRSA ± vancomycin + THB media; and (3) CA-MRSA ± LL-37 + THB media. Molecular Network maps were displayed by the use of Cytoscape graphical mapping, which displayed overlays of CA-MRSA alone and CA-MRSA plus antibiotic stimuli. In each case, ion signals derived from either the LC-MS/MS solvents or the THB media were subtracted from each overlay map. The Cytoscape map architecture is comprised of ion signals displayed as individual nodes that are clustered according to ion fragmentation similarity based on cosine scoring (20). Ion signals that have identical parent mass values and tandem MS fingerprints were grouped and displayed together as one signal node. Ion signals that had off-set parent masses and similar fragmentation patterns were displayed as connected nodes and grouped into clusters. The graphical layout was based on the mathematical modeling algorithm Fast Multipole Multilevel Method (FM3), which arranges similar nodes as straight-line connections in space representing correlation by proximity for graphical interpretation. The width of the straight-line connection indicates the cosine value between nodes and thus, similarity scoring (Fig. 1). In each case throughout this manuscript, nodes specific to CA-MRSA alone were colored gold, nodes specific to the CA-MRSA plus daptomycin were colored aqua blue, nodes specific to the CA-MRSA + LL-37 were colored red, and nodes specific to the CA-MRSA + vancomycin were colored green. Overlapping nodes found in both CA-MRSA alone and when challenged with antibiotic were colored gray (Fig. 2A, 3A–3B, 4A, 5A).
Fig. 1.

Mass spectrometry-based molecular networking. CA-MRSA USA 300 strain TCH1516 was grown to late stationary phase alone or in the presence of antibiotic agents (1). The organic based solvent 1-butanol was used to extract small molecules and peptides from the CA-MRSA growth culture (2). The 1-butanol extract was brought to complete dryness by the use of a SpeedVac centrifuge (3). The solid extract was re-suspended in H2O and then components were separated by HPLC and directly electrosprayed into a Thermo Finnegan LTQ (4) for tandem MS analysis (5). The resulting data sets were processed by Spectral Networks (6), which compiles and clusters similar overlapping spectra based on similarly scores expressed as cosine values. A graphical representation of the clustered spectra population was displayed by the use of Cytoscape network maps. Individual tandem mass spectra are represented as circular nodes that are connected to other nodes with similar fragmentation patterns (7). Nodes within subpopulation clusters can be targeted based on theoretical parent ion masses. Once a validated node is identified (denoted as 1* in schematic), the structural information can be used as a building block to propagate and identify adjacent nodes in the cluster.
Fig. 2.

PSMα1 and related variants upon exposure to challenged conditions. A, The molecular network resulting from ion clusters of PSMα1 is shown. Nodes colored blue are ion clusters found only in daptomycin challenged conditions. Nodes colored gold are found only in MRSA alone samples. Nodes colored gray are found in MRSA + daptomycin challenged conditions. Nodes labeled with only a sequence tag are within 500 ppm of the accurate theoretical mass. Nodes labeled with sequence tags plus (+) numbers have their mass accuracy noted. A sub-nodal cluster of PSMα1 is also shown with clusters from daptomycin (colored blue) and MRSA + daptomycin (colored gray). B, Sequence of PSMα1 is shown in bold followed by its related variants. C, Spectral counts for the formylated 13-mer peptide under respective challenged conditions. PSMα1 variants were not detected (ND) in the vancomycin treated sample. Analysis of the all nodes in the different conditions containing dPSMs is provided in supplemental material.
Fig. 3.

PSMα2 and PSMα4 and related variants upon exposure to challenged conditions. A, The molecular network resulting from ion clusters of PSMα2 is shown, as well as the sequence for PSMα2 (shown in bold) and its variants. Nodes labeled with only a sequence tag are within 500 ppm of the theoretical mass. Nodes labeled with sequence tags plus (+) numbers have their mass accuracy noted. Nodes colored gray are found in MRSA alone and MRSA + daptomycin. The nodes colored red found in LL-37 treated samples. The nodes colored gold are found in MRSA alone samples. B, PSMα2 variant IIAGIIKFIKGLIEKFTGK peptide counts tested in replicates under respective conditions. PSMα2 variants were not observed in the vancomycin treated sample. C, The molecular network resulting from ion clusters of PSMα4 is shown, as well as the sequence for PSMα4 (shown in bold) and its variants. Nodes colored blue are ion clusters found only in daptomycin challenged conditions. Nodes labeled with only a sequence tag are within 500 ppm of the accurate mass. Nodes labeled with sequence tags plus numbers have their mass accuracy noted. D, PSMα4 variant TIIKIIKAIIDIFAK peptide counts tested in replicates under respective conditions. Analysis of the all nodes in the different conditions containing dPSMs is provided in supplemental material.
Fig. 4.
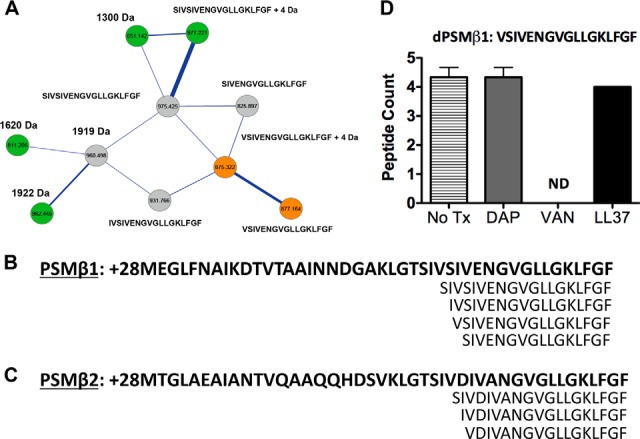
PSMβ1, PSMβ2 and their related variants upon exposure to challenged conditions. A, The molecular network resulting from ion clusters of PSMβ is shown. Nodes colored green are ion clusters found only in MRSA + vancomycin challenged conditions. Nodes colored gold are found only in MRSA samples. Nodes colored gray are found overlapping in MRSA alone samples and MRSA + daptomycin challenged conditions. Nodes labeled with only a sequence tag are within 500 ppm of the theoretical mass. Nodes labeled with sequence tags +numbers have their mass accuracy noted. B, Sequence of PSMβ1 (shown in bold) and related variants. Amino acid sequences for PSMβ1 are not shown in full form but are abbreviated at the N-terminal end. C, Sequence of PSMβ2 (shown in bold) and related variants. Amino acid sequences for PSMβ2 are not shown in full form but are abbreviated at the N-terminal end. Analysis of the all nodes in the different conditions containing dPSMs is provided in supplemental material. D, PSMβ1 variant VSIVENGVGLLGKLFGF and the peptide counts tested in 3 replicates under respective conditions.
Fig. 5.

PSMγ and related variants upon exposure to challenged conditions. A, The molecular network resulting from ion clusters of PSMγ is shown. Nodes colored blue are found in daptomycin challenged conditions. Nodes colored gold are found in MRSA only conditions. Nodes colored gray are found in MRSA + daptomycin challenged conditions. Nodes labeled with only a sequence tag are within 500 ppm of accurate mass. Nodes labeled with sequence tags plus numbers have their mass accuracy noted. B, The sequence for PSMγ is shown in bold followed by its variants. C, PSMγ variant ISTIGDLVKWIIDTVNKFTKK peptide counts tested in three replicates under respective conditions. Analysis of the all nodes in the different conditions containing dPSMs is provided in supplemental material.
CA-MRSA Phenotypes at sub-MIC Levels of Antibiotics
Several growth curves were generated to ensure that the treatment concentrations of antibiotics did not significantly alter normal growth of the CA-MRSA strain (supplemental Fig. S1A). Morphology and approximate counts of individual CA-MRSA cells were monitored by microscopy, with no distinguishable differences between the CA-MRSA control sample and samples grown with sub-MIC levels of antibiotics (supplemental Fig. S1B). Further, the 1-butanol extractions used to create the networks were examined by SDS-PAGE gel, with no observable large molecular bands on protein staining (gel not shown). Thus, all observed MS signals were predicted to be low molecular weight peptides and small molecules. Analysis of synthetic PSMα4 (> 95% pure) with the same methodology ± daptomycin did not produce any variant forms when checked by MS. Therefore, as previously reported (18), we hypothesize native proteases or other intrinsic mechanisms contribute to the processing of PSMs.
Spectral Networks Variants Captured in PSMα1
PSMα1 is a 21-mer formylated peptide that is biosynthetically produced and secreted in multi-variant forms by CA-MRSA. In vitro concentrations of secreted PSMα1 have been reported to vary depending on the S. aureus strain tested and media conditions (13). Prior experiments that monitored effects of sub-inhibitory antibiotic concentrations on PSMα1 production showed perturbations from baseline (27). To date, PSMα1 formylated and non-formylated forms have been detected in culture media, including two N-terminal truncated forms. The two variant forms were comparatively tested against full-length formylated PSMα1 and showed a gain of antibacterial activity (17, 18). In our study, we searched for the theoretical intact masses of truncated PSMs within node clusters. Clusters that had at least one node with a theoretical mass corresponding to dPSMα1 were separated and further analyzed. The node with correct theoretical mass accuracy was then sequenced de novo for identification. In those cases where a peptide sequence tag contained reasonable mass accuracy and greater than seven contingent amino acids, adjacent nodes were sequenced to complete the identification of the cluster. Fig. 2A shows two dPSMα1 nodal clusters constructed by merging the CA-MRSA untreated and daptomycin treated MS data sets. Within each cluster the gray individual nodes indicate overlapping spectra found in both settings, the blue nodes are represent spectra specific to daptomycin treatment and the gold nodes are spectra specific to the untreated sample. Each dPSMα1 node includes the acquired de novo sequence tags and mass off-sets from the theoretical peptide sequence. In the CA-MRSA untreated sample, six dPSMα1 variants were captured, including an N terminus truncated 16-mer, 17-mer, 18-mer, and a 19-mer. The 19-mer and 16-mer peptide variants have been previously described (17, 18). Two C-terminal truncated dPSMα1 peptides, a 13-mer and a 14-mer, each with conserved formyl groups on the initiator methionine were also identified (Fig. 2B). Treatment of CA-MRSA with sub-inhibitory daptomycin retained all captured variants except the previously described 16-mer formylated dPSMα1. Sub-inhibitory LL-37 and vancomycin treatment led to loss of the 13-mer dPSMα1 formylated peptide. In addition, vancomycin treatment had the most dramatic effect, as only the 19-mer, 18-mer, and 17-mer peptide dPSMα1 variants were detected (supplemental Fig. S2–S8). To assess false negatives as they pertain to the dPSMα1 detected by the MS-based molecular networking platform and to examine the relative production levels of dPSMα1 in response to antibiotics, spectral counts were enumerated. The overall trend for dPSMα1 when stimulated by each antibiotic treatment was an increase in peptide abundance; vancomycin showed the weakest stimulatory potential (supplemental Fig. S9). Consistent with the molecular networks observation, the 13-mer formylated peptide was only detected in the untreated and daptomycin sample extracts by a traditional database search. Treatment with daptomycin significantly increased the spectral counts for the formylated 13-mer peptide (Fig. 2C).
Spectral Networks Variants Captured in PSMα2
Like PSMα1, PSMα2 is structurally comprised of 21 amino acids that share high homology at terminal ends. Cumulative studies on the abundance of biosynthetically released PSMα2 indicated low secretion in vitro relative to other PSMα peptides among strains tested (13). Full-length PSMα2 triggers phagocyte production of superoxide and permeates cell membranes in its mode of action (28). Two dPSMα2, a 19-mer peptide and a 16-mer, have been detected in different CA-MRSA strains (17, 18). Fig. 3A shows two dPSMα2 nodal clusters constructed by merging MS data sets acquired from the CA-MRSA untreated and LL-37 treated extracts. Within each cluster the gray individual nodes indicate overlapping spectra found in both settings, the red nodes are represent spectra specific to LL-37 treatment and the gold nodes are spectra specific to the untreated sample. In the untreated CA-MRSA sample, four N-terminal truncated dPSMα2 were captured, including a 20-mer, the previously described 19-mer, a 17-mer, and a 12-mer (Fig. 3A). When treated with daptomycin, the molecular network map was able to group only the 20-mer and 19-mer peptides. The 17-mer, 16-mer, and 12-mer peptides were absent when treated with daptomycin (supplemental Fig. S10–S14). The dPSM variants captured in the LL-37 treated CA-MRSA sample mirrored the untreated sample, except the longer 20-mer and 19-mer were not present. Interestingly, the vancomycin treated sample abolished clustering of any dPSMα2 peptides. Peptide abundance analysis via spectral counts for the clustered dPSMα2 peptides showed only the 19-mer was produced by the untreated CA-MRSA sample. Spectral counts for the daptomycin and LL-37 treated samples showed a significant increase in the abundance of all four dPSMα2 peptides (Fig. 3B and supplemental Fig. S15). The spectral counts originating from the vancomycin treated sample only detected the 17-mer dPSMα2, consistent with previous reports on the effect of vancomycin treatment on PSMα type peptide production (29).
Spectral Networks Variants Captured in PSMα4
PSMα4 is a 20-mer peptide secreted with its initiator methionine in formylated and non-formylated forms. To date, two dPSMα4 have been discovered and tested for in vitro phenotypes. The variants of PSMα4, reminiscent of dPSMα1 and dPSMα2, showed increased antibacterial activity against Staphylococcus epidermidis and group A Streptococcus. Interestingly, dPSMα4 showed enhanced lytic activity against red blood cells. In addition, it was shown the S. aureus exoprotease aureolysin contributed to processing of PSMα4 into variant forms in vitro (18). When the theoretical masses for full-length PSMα4 formylated and non-formylated or dPSMs were sought in the CA-MRSA untreated molecular network, none were detected. Next, the daptomycin treated CA-MRSA molecular network was searched, resulting with the detection of full-length PSMα4 clustered with a 19-mer dPSMα4. Although, the 19-mer was clustered with PSMα4, the spectra quality did not allow de novo sequencing and therefore the peptide was not further considered. The daptomycin treated clusters also showed the presence of two additional dPSMα4, 17-mer and 15-mer, previously reported (Fig. 3C and supplemental Fig. S10, S12). Neither the LL-37, nor the vancomycin-treated CA-MRSA showed node clusters indicative of dPSMα4 when theoretical masses were searched. Spectral counts of dPSMα4 showed the untreated sample produced detectable amounts of the 17-mer, but not the 15-mer peptide. In each case where CA-MRSA was treated with antibiotics, the increase of both dPSMα4, identified through molecular networking, were significantly increased (Fig. 3D and supplemental Fig. S16).
Spectral Networks Variants Captured in PSMβ1 and PSMβ2
PSMβ1 and PSMβ2 belong to the PSM peptide family but are almost twice as long as PSMα-type peptides. PSMβ1 and PSMβ2 are 44 amino acids in length, share high homology particularly in their C-terminal regions, and are more highly prevalent in CA-MRSA compared with HA-MRSA. PSMβ type peptides do not contribute to hemolysis in vitro and genetic disruption of the genes had negligible contributions to disease progression in a mouse model compared with the PSMα type members (13, 30). The PSMβ group is least well understood with respect to virulence contributions, but is postulated to contribute to biofilm structure formation (14). Furthermore, in contrast to the PSMα family, PSMβ variants (dPSMβ) have not been described. The molecular network map for the CA-MRSA untreated sample did not contain nodes indicative of either full-length PSMβ1 or full-length PSMβ2. Instead, only node clusters with masses corresponding to dPSMβ1 were identified. Four peptides consisting of a 19-mer, 18-mer, 17-mer, and a 16-mer contained accurate masses and reliable peptide sequence tags corresponding to truncated variants of PSMβ1 (Fig. 4A and 4B). Daptomycin and LL-37 treatment of CA-MRSA showed the 16-mer, present in the untreated sample, not to be clustered within the network. Treatment with vancomycin restored the clustering of the 16-mer dPSMβ1; however, the 17-mer dPSMβ1 peptide found in the untreated sample was no longer detected. Interestingly, the network obtained from the vancomycin treated CA-MRSA was complex, dPSMβ1 variants with node masses that did not fit the known theoretical values or mass deviations because of common ion adducts were grouped together with sequenced peptides (Fig. 4A and supplemental Fig. S17–S23). PSMβ2 showed clustering of two variants truncated at similar regions as dPSMβ1 that included the 19-mer and 18-mer peptides. In each molecular network map, the treatment with sub-MIC levels of any of the three antibiotic agents resulted in the node loss of the 19-mer dPSMβ2 and revealed a 17-mer dPSMβ2 not present in the untreated molecular network (Fig. 4C and supplemental Fig. S17, S22–S26). Spectral counts showed all clustered dPSMβ1 were present in the sample. Daptomycin treatment retained all observed dPSMβ1 peptides and enhanced the number of peptides for the 19-mer. Vancomycin also greatly enhanced the number of spectral counts for the 19-mer and did not detect any of the 17-mer peptide in successive runs. LL-37 treatment mirrored that of daptomycin and vancomycin (Fig. 4D and supplemental Fig. S27).
Spectral Networks Variants Captured in PSMγ
Delta-toxin or PSMγ is a highly expressed 26-mer peptide exotoxin with the ability to lyse many cell types (31). PSMγ has been detected in its formylated and non-formylated forms in addition to one truncated dPSMγ peptide (18). PSMγ is of particular interest given the gene is embedded within the long studied RNAIII effector of the Agr quorum sensing system (32). In our work, PSMγ resulted in the most number of node clusters detected in the constructed molecular networks corresponding to dPSMγ. Fig. 5A shows a node cluster indicative of dPSMγ peptides derived from the molecular network map obtained by overlapping CA-MRSA ± daptomycin. After searching through the all three sample maps, twelve N-terminal truncated dPSMγ peptides were clustered that included a 24-mer, previously described 23-mer, 22-mer, 21-mer, 20-mer, 19-mer, 18-mer, 15-mer, 10-mer, 9-mer, 8-mer, and a 7-mer (Fig. 5B and supplemental Fig. S28–S41). Daptomycin treatment of CA-MRSA did not perturb the variants clustered in the network, whereas LL37 treatment reduced our ability to cluster nodes indicative of the 19-mer, 18-mer, 15-mer, and the 7-mer dPSMγ peptides. Vancomycin treatment had a pronounced effect on the type of variants captured in the molecular networks, as only the 24-mer, 23-mer, 22-mer, and 20-mer were present. In addition, the clusters derived from vancomycin treatment had additional nodes that included masses that did not correspond to theoretical dPSMγ values or the addition of common ion adducts; indicating potentially unknown posttranslational modifications (supplemental Fig. S41). dPSMγ spectral counts showed the 24-mer, 15-mer, 9-mer, 8-mer, and 7-mer were not detectable. Treatment with daptomycin restored all observed dPSMγ within the molecular network map and significantly increased the number of peptides in particular for the 21-mer and 20-mer peptides (Fig. 5C and supplemental Fig. S42). Vancomycin treatment significantly decreased the detection of the 21-mer dPSMγ peptide and did not match any of the 9-mer and 10-mer. LL-37 treatment yielded similar spectral counts to that of daptomycin, except the 9-mer peptide was not detected similar to the untreated sample.
Bioactivity Displayed by Discovered dPSM
Previous reports demonstrated dPSMα-type had antimicrobial properties against bacteria that colonize human skin (17, 18). Based on these published reports, it was suggested dPSMs contribute to CA-MRSA success in out competing host microflora en route to establishing a suitable niche toward causing an infection. To reinforce the notion that the molecular network platform can be used as a tool to discover bioactive molecules, we selected to further study, in a functional manner, a pair of the identified peptides: (1) +28MGIIAGIIKVIKS; formylated 13-mer dPSMα1; and (2) VSIVENGVGLLGKLFGF; nonformylated 17-mer dPSMβ1. Both peptides were of particular interest given C-terminally truncated dPSMs and dPSMβ-type peptides have not been reported nor studied for functional properties. We first sought to determine if the peptides contained antimicrobial activity against a library of bacteria known to colonize human skin; including strains that were previously shown to be susceptible to dPSMs (17, 18). The tested dPSM peptides did not alter the growth curves of the strains in successive experiments (Fig. 6A and supplemental Fig. S43A and S43B). In vitro, dPSMα-type has been shown to act as hemolysins and to synergize with the staphylococcal protein β-toxin, while in the in the same study, PSMβ-type did not contribute to hemolysis (30). To test if the 13-mer dPSMα1 retained hemolytic properties once C-terminally cleaved or if the dPSMβ1 gained hemolytic potential once cleaved, both peptides were separately incubated with erythrocytes. In a dose dependent manner, both peptides showed a weak ability to act as hemolysins (Fig. 6B). Next, we sought to determine if the human monocyte cell lines THP-1 recognized and, in turn, were stimulated by the dPSMs by monitoring cytokine release. It was observed that dPSMα1 readily stimulated the release of IL-8 in a dose dependent manner (Fig. 6C). When the dPSMβ1 peptide was tested for cytokine release, a similar release trend was observed when IL-1β was monitored (supplemental Fig. S43C).
Fig. 6.
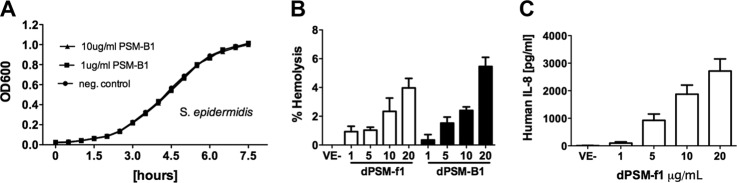
Bioactivity of the discovered dPSMα1 and dPSMβ1 peptides. A, Growth curve for Staphylococcus epidermis with and without dPSMβ1 measured at OD600 in 30 min increments over a 7.5 h period. B, Hemolytic assay performed with both dPSMα1 and dPSMβ1 and control (0.0125% Triton-X); results normalized to 0.0125% Triton-X and hemoglobin absorbance measured at 450 nm. Lane 1, 0.0125% Triton-X. Lane 2, 1 μg/ml dPSMα1. Lane 3, 5 μg/ml dPSMα1. Lane 4, 10 μg/ml dPSMα1. Lane 5, 20 μg/ml dPSMα1. Lane 6, 1 μg/ml dPSMβ1. Lane 7, 5 μg/ml dPSMβ1. Lane 8, 10 μg/ml dPSMβ1. Lane 9, 20 μg/ml dPSMβ1. C, Enzyme-linked immunosorbent assay (ELISA) of human monocyte IL-8 response to dPSMα1 with IL-8 values in pg/ml and dPSMα1 concentration in μg/ml, performed in a dose dependent manner.
DISCUSSION
The current discovery rate of novel anti-staphylococcal therapeutics has not kept pace with the evolution of multi-resistant S. aureus strains of bona fide virulence potential (33). For this reason and others in the United States, S. aureus related deaths have been reported to approach or surpass those associated with HIV/AIDS (34). The high level of complexity and complementary nature of the biosynthetic release factors produced by S. aureus make it challenging to tease out the molecular basis of S. aureus infections. One contributing factor to this hurdle is the lack of available tools with the ability to capture biosynthetic release factors in a cooperative manner on a system-wide scale. Using the emerging tool of MS-based molecular networking, we constructed and investigated the biosynthetic release factor map of the leading CA-MRSA lineage associated with skin infections and sepsis in children and adults - USA300 (35). Graphical maps of the constructed molecular networks indicate an abundance of biosynthetic release factors in the small molecule and peptide range (supplemental Fig. S44). Recent reports have shown that PSMs contribute to biofilm fiber formation (14) and that truncated variant forms of PSMs gain antibacterial properties and/or modify host cell activities (17, 18). Therefore, we hypothesized the structural diversity of the PSMs family of peptides was more plentiful than previously reported. Using the molecular networking strategy, we were able to discover 31 dPSMs variants clustered in the molecular networks that were valid according to our set standards, only seven of which had been previously described in the literature. The number of captured dPSM variants varied when S. aureus was exposed to different pharmaceutical or natural antibiotics. The change in the spectrum of dPSMs most likely reflects concentration changes induced in response to environmental factors, consistent with previous studies showing antibiotic perturbations of PSM baseline expression levels (28). In addition, because of the inherent nature of spectral pairing, which molecular networking is based on, some spectra will not be clustered even if present in the sample. To give a broader perspective of the effect the tested antibiotics had on PSM production, spectral counts were enumerated by a traditional database matching strategy. As expected, we observed molecular networking and traditional database searching for PSM identification did not always correlate. Therefore, it can be concluded that the two MS data processing platforms are complementary and when applied give a more thorough representation of an organism's biosynthetic potential. The recent literature on the topic of PSMs indicates this family of peptides has important, but not yet fully elucidated, roles during the course of S. aureus infection (16). We observed that clustered within the PSMs were additional masses that did not match to known theoretical PSMs, but had similar fragmentation patterns indicating related structural sequences. The clustering of inconclusive masses with PSMs identified with high confidence indicates PSMs may contain yet-to-be discovered post-translational modifications important for bioactivity or unidentified molecules exist that are structurally similar. Astoundingly, the discoveries describing the PSM family of peptides herein are based on mining < 1% of the total molecular network in each case. Stimulation of IL-8 and IL-1β release from THP-1 cells demonstrates the peptides are bioactive in nature. We conclude the further development and application of tools such as MS-based molecular networking could steer new lines of discovery in the context of bacterial physiology, ecology, or pathogenesis.
Supplementary Material
Footnotes
Author contributions: D.J.G. and V.N. designed research; D.J.G., L.V., I.S.G., J.H.H., and J.H. performed research; D.J.G., L.V., I.S.G., N.K., D.M., J.H.H., J.H., A.S.Z., J.E.D., P.C.D., and V.N. analyzed data; D.J.G. and V.N. wrote the paper.
* This work was supported by NIH grants HD071600 (V.N.), AI057153 (to V.N.) and GM086283 (to P.C.D.). D.J.G. was supported by the A.P. Giannini Foundation and by the NIH IRACDA K12 grant program (GM068524).
 This article contains supplemental Figs. S1 to S44.
This article contains supplemental Figs. S1 to S44.
1 The abbreviations used are:
- PSMs
- Phenol soluble modulins
- dPSMs
- Phenol soluble modulin variants or derivatives
- MRSA
- methicillin-resistant Staphylococcus aureus
- VISA/VRSA
- vancomycin-intermediate or -resistant Staphylococcus aureus
- CA
- community-associated
- HA
- hospital-associated
- MIC
- minimal inhibitory concentration.
REFERENCES
- 1. Human Microbiome Project Consortium (2012) Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Grice E. A., Kong H. H., Conlan S., Deming C. B., Davis J., Young A. C., Bouffard G. G., Blakesley R. W., Murray P. R., Green E. D., Turner M. L., Segre J. A. (2009) Topographical and temporal diversity of the human skin microbiome. Science 324, 1190–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. DeLeo F. R., Chambers H. F. (2009) Reemergence of antibiotic-resistant Staphylococcus aureus in the genomics era. J. Clin. Invest. 119, 2464–2474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Datta R., Huang S. S. (2008) Risk of infection and death due to methicillin-resistant Staphylococcus aureus in long-term carriers. Clin. Infect. Dis. 47, 176–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fischbach M. A., Walsh C. T. (2009) Antibiotics for emerging pathogens. Science 325, 1089–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moore C. L., Osaki-Kiyan P., Haque N. Z., Perri M. B., Donabedian S., Zervos M. J. (2012) Daptomycin versus vancomycin for bloodstream infections due to methicillin-resistant Staphylococcus aureus with a high vancomycin minimum inhibitory concentration: a case-control study. Clin. Infect. Dis. 54, 51–58 [DOI] [PubMed] [Google Scholar]
- 7. Sievert D. M., Rudrik J. T., Patel J. B., McDonald L. C., Wilkins M. J., Hageman J. C. (2008) Vancomycin-resistant Staphylococcus aureus in the United States, 2002–2006. Clin. Infect. Dis. 46, 668–674 [DOI] [PubMed] [Google Scholar]
- 8. Sakoulas G., Moellering R. C., Jr. (2008) Increasing antibiotic resistance among methicillin-resistant Staphylococcus aureus strains. Clin. Infect. Dis. 46 Suppl. 5, S360–S367 [DOI] [PubMed] [Google Scholar]
- 9. Harris S. R., Feil E. J., Holden M. T., Quail M. A., Nickerson E. K., Chantratita N., Gardete S., Tavares A., Day N., Lindsay J. A., Edgeworth J. D., de Lencastre H., Parkhill J., Peacock S. J., Bentley S. D. (2010) Evolution of MRSA during hospital transmission and intercontinental spread. Science 327, 469–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Traber K. E., Lee E., Benson S., Corrigan R., Cantera M., Shopsin B., Novick R. P. (2008) agr function in clinical Staphylococcus aureus isolates. Microbiology 154, 2265–2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cheng A. G., Kim H. K., Burts M. L., Krausz T., Schneewind O., Missiakas D. M. (2009) Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J. 23, 3393–3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dumont A. L., Nygaard T. K., Watkins R. L., Smith A., Kozhaya L., Kreiswirth B. N., Shopsin B., Unutmaz D., Voyich J. M., Torres V. J. (2011) Characterization of a new cytotoxin that contributes to Staphylococcus aureus pathogenesis. Mol. Microbiol. 79, 814–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang R., Braughton K. R., Kretschmer D., Bach T. H., Queck S. Y., Li M., Kennedy A. D., Dorward D. W., Klebanoff S. J., Peschel A., DeLeo F. R., Otto M. (2007) Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat. Med. 13, 1510–1514 [DOI] [PubMed] [Google Scholar]
- 14. Periasamy S., Joo H. S., Duong A. C., Bach T. H., Tan V. Y., Chatterjee S. S., Cheung G. Y., Otto M. (2012) How Staphylococcus aureus biofilms develop their characteristic structure. Proc. Natl. Acad. Sci. U.S.A. 109, 1281–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kretschmer D., Nikola N., Durr M., Otto M., Peschel A. (2012) The virulence regulator Agr controls the staphylococcal capacity to activate human neutrophils via the formyl peptide receptor 2. J. Innate Immun. 4, 201–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Surewaard B. G., Nijland R., Spaan A. N., Kruijtzer J. A., de Haas C. J., van Strijp J. A. (2012) Inactivation of staphylococcal phenol soluble modulins by serum lipoprotein particles. PLoS Pathog. 8, e1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Joo H. S., Cheung G. Y., Otto M. (2011) Antimicrobial activity of community-associated methicillin-resistant Staphylococcus aureus is caused by phenol-soluble modulin derivatives. J. Biol. Chem. 286, 8933–8940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gonzalez D. J., Okumura C. Y., Hollands A., Kersten R., Akong-Moore K., Pence M. A., Malone C. L., Derieux J., Moore B. S., Horswill A. R., Dixon J. E., Dorrestein P. C., Nizet V. (2012) Novel phenol-soluble modulin derivatives in community-associated methicillin-resistant Staphylococcus aureus identified through imaging mass spectrometry. J. Biol. Chem. 287, 13889–13898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gonzalez D. J., Xu Y., Yang Y. L., Esquenazi E., Liu W. T., Edlund A., Duong T., Du L., Molnar I., Gerwick W. H., Jensen P. R., Fischbach M., Liaw C. C., Straight P., Nizet V., Dorrestein P. C. (2012) Observing the invisible through imaging mass spectrometry, a window into the metabolic exchange patterns of microbes. J. Proteomics 75, 5069–5076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bandeira N., Tsur D., Frank A., Pevzner P. A. (2007) Protein identification by spectral networks analysis. Proc. Natl. Acad. Sci. U.S.A. 104, 6140–6145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Watrous J., Roach P., Alexandrov T., Heath B. S., Yang J. Y., Kersten R. D., van der Voort M., Pogliano K., Gross H., Raaijmakers J. M., Moore B. S., Laskin J., Bandeira N., Dorrestein P. C. (2012) Mass spectral molecular networking of living microbial colonies. Proc. Natl. Acad. Sci. U.S.A. 109, E1743–E1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guthals A., Watrous J. D., Dorrestein P. C., Bandeira N. (2012) The spectral networks paradigm in high throughput mass spectrometry. Mol. Biosyst. 8, 2535–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Horne K. C., Howden B. P., Grabsch E. A., Graham M., Ward P. B., Xie S., Mayall B. C., Johnson P. D., Grayson M. L. (2009) Prospective comparison of the clinical impacts of heterogeneous vancomycin-intermediate methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-susceptible MRSA. Antimicrob. Agents Chemother. 53, 3447–3452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dhand A., Bayer A. S., Pogliano J., Yang S. J., Bolaris M., Nizet V., Wang G., Sakoulas G. (2011) Use of antistaphylococcal beta-lactams to increase daptomycin activity in eradicating persistent bacteremia due to methicillin-resistant Staphylococcus aureus: role of enhanced daptomycin binding. Clin. Infect. Dis. 53, 158–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nizet V., Ohtake T., Lauth X., Trowbridge J., Rudisill J., Dorschner R. A., Pestonjamasp V., Piraino J., Huttner K., Gallo R. L. (2001) Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature 414, 454–457 [DOI] [PubMed] [Google Scholar]
- 26. Highlander S. K., Hulten K. G., Qin X., Jiang H., Yerrapragada S., Mason E. O., Jr., Shang Y., Williams T. M., Fortunov R. M., Liu Y., Igboeli O., Petrosino J., Tirumalai M., Uzman A., Fox G. E., Cardenas A. M., Muzny D. M., Hemphill L., Ding Y., Dugan S., Blyth P. R., Buhay C. J., Dinh H. H., Hawes A. C., Holder M., Kovar C. L., Lee S. L., Liu W., Nazareth L. V., Wang Q., Zhou J., Kaplan S. L., Weinstock G. M. (2007) Subtle genetic changes enhance virulence of methicillin resistant and sensitive Staphylococcus aureus. BMC Microbiol. 7, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yamaki J., Synold T., Wong-Beringer A. (2011) Antivirulence potential of TR-700 and clindamycin on clinical isolates of Staphylococcus aureus producing phenol-soluble modulins. Antimicrob. Agents Chemother. 55, 4432–4435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Forsman H., Christenson K., Bylund J., Dahlgren C. (2012) Receptor-dependent and -independent immunomodulatory effects of phenol-soluble modulin peptides from Staphylococcus aureus on human neutrophils are abrogated through peptide inactivation by reactive oxygen species. Infect. Immun. 80, 1987–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Joo H. S., Chan J. L., Cheung G. Y., Otto M. (2010) Subinhibitory concentrations of protein synthesis-inhibiting antibiotics promote increased expression of the agr virulence regulator and production of phenol-soluble modulin cytolysins in community-associated methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 54, 4942–4944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cheung G. Y., Duong A. C., Otto M. (2012) Direct and synergistic hemolysis caused by Staphylococcus phenol-soluble modulins: implications for diagnosis and pathogenesis. Microbes Infect. 14, 380–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kerr I. D., Doak D. G., Sankararamakrishnan R., Breed J., Sansom M. S. (1996) Molecular modelling of Staphylococcal delta-toxin ion channels by restrained molecular dynamics. Protein Eng. 9, 161–171 [DOI] [PubMed] [Google Scholar]
- 32. Montgomery C. P., Boyle-Vavra S., Daum R. S. (2010) Importance of the global regulators Agr and SaeRS in the pathogenesis of CA-MRSA USA300 infection. PLoS One 5, e15177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bahcall O. G. (2013) MRSA evolution and spread. Nat. Genet. 45, 123 [Google Scholar]
- 34. Klevens R. M., Morrison M. A., Nadle J., Petit S., Gershman K., Ray S., Harrison L. H., Lynfield R., Dumyati G., Townes J. M., Craig A. S., Zell E. R., Fosheim G. E., McDougal L. K., Carey R. B., Fridkin S. K. (2007) Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298, 1763–1771 [DOI] [PubMed] [Google Scholar]
- 35. Diep B. A., Gill S. R., Chang R. F., Phan T. H., Chen J. H., Davidson M. G., Lin F., Lin J., Carleton H. A., Mongodin E. F., Sensabaugh G. F., Perdreau-Remington F. (2006) Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367, 731–739 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.