

Abstract
An approach to proteomic analysis that combines bioorthogonal noncanonical amino acid tagging (BONCAT) and pulsed stable isotope labeling with amino acids in cell culture (pSILAC) provides accurate quantitative information about rates of cellular protein synthesis on time scales of minutes. The method is capable of quantifying 1400 proteins produced by HeLa cells during a 30 min interval, a time scale that is inaccessible to isotope labeling techniques alone. Potential artifacts in protein quantification can be reduced to insignificant levels by limiting the extent of noncanonical amino acid tagging. We find no evidence for artifacts in protein identification in experiments that combine the BONCAT and pSILAC methods.
Methods for the analysis of cellular protein synthesis should be quantitative and fast. In 2006, Dieterich and coworkers introduced a proteomics discovery tool called bioorthogonal noncanonical amino acid tagging (BONCAT),1 in which noncanonical amino acids (ncAAs) with bioorthogonal functional groups (e.g. azides or alkynes) are used as metabolic labels to distinguish new proteins from old (1, 2). Labeled proteins can be conjugated to fluorescent reporters for visualization or affinity tags for purification and subsequent identification by mass spectrometry (3). Because the ncAA probe can be introduced to cells in a well-defined “pulse,” affinity purification removes pre-existing proteins and provides both reduced sample complexity and excellent time resolution.
The methionine (Met) surrogate l-azidohomoalanine (Aha) has become standard in the application of BONCAT methodologies. Using Aha and fluorescent tagging, Tcherkezian et al. observed co-localization of the DCC receptor with sites of protein synthesis, providing support for the role of netrin as a stimulant of extranuclear protein production in neurons (4). Combining Aha labeling and 2D gel electrophoresis, Yoon et al. discovered that the protein lamin B2 is synthesized in axons and crucial to mitochondrial function and axon maintenance in Xenopus retinal glial cells (5). Aha has also been used to study histone turnover (6), protein palmitoylation (7), pathogen amino acid uptake (8), inflammatory response (9), and local translation in neuronal dendrites and axons (10). These labeling techniques have been expanded to tissue and animal culture, where Aha has been used to profile protein synthesis in rat hippocampal brain slices (11, 12) and zebrafish embryos (13).
The development of fast, reliable, quantitative BONCAT methods will enable new insights into proteome dynamics in response to biological stimuli. Recent work by Eichelbaum et al. combined Aha labeling with stable isotope labeling to measure lipopolysaccharide-stimulated protein secretion by macrophages (14). Using similar approaches, Somasekharan et al. identified a set of proteins that are translationally regulated by the Y-box binding protein-1 (YB-1) in TC-32 Ewing sarcoma cells (15), and Howden et al. monitored changes in protein expression following stimulation of primary T cells with phorbol 12-myristate 13-acetate and ionomycin (16).
A concern that arises in the use of Aha (as it does for all chemical probes of biological processes) is that the protocols used for Aha labeling might perturb cellular protein synthesis. The development of ncAAs as reliable analytic tools hinges on our ability to understand and minimize such unintended effects. For Aha, previous work has shown that protein labeling does not visibly alter cellular morphology in dissociated hippocampal neurons or HEK293 cells, and 1D gels reveal no discrepancies between the proteomes of Aha- and Met-treated cells (1). These experiments, however, offer only coarse measures of effects on protein synthesis, and as Aha labeling is frequently coupled to mass spectrometry-based proteomic analysis, the biological effects of Aha treatment must be investigated with equivalent sensitivity and resolution.
Here we report sound methods for fast, reliable measurement of proteome dynamics via noncanonical amino acid tagging. First, we use the quantitative proteomics technique pulsed stable isotope labeling with amino acids in cell culture (pSILAC) to investigate potential unintended effects of Aha labeling on protein abundance in HeLa cell cultures, and we develop a strategy for minimizing these effects. Second, we show that a combined BONCAT-pSILAC approach, capable of both enriching and quantifying newly synthesized proteins, yields detailed proteomic information on time scales that are inaccessible to isotope labeling techniques alone.
EXPERIMENTAL PROCEDURES
pSILAC in HeLa Cell Culture
HeLa cells were maintained in DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% FBS (Invitrogen) and 1% penicillin/streptomycin (Invitrogen) in a humidified incubator at 37 °C and 5% CO2. For each pSILAC experiment, 2.1 million cells were seeded in 2 T-75 flasks and grown for 24 h. Cultures were washed with warm PBS twice and resuspended in custom lysine-free and Met-free DMEM (Invitrogen) supplemented with either “medium” lysine (D4 l-lysine, Cambridge Isotope Laboratories) or “heavy” lysine (U-13C6 U-15N2 l-lysine, Cambridge Isotope Laboratories, Andover, MA) at 1 mm. Cultures were also supplemented with either Met (1 mm), Aha (1 mm), or Aha30:1 (1 mm Aha, 33 μm Met) as indicated for each experiment. Aha was synthesized as previously described (17). pSILAC experiments measuring changes in protein abundance upon treatment with Aha or Aha30:1 and Met were conducted with four biological replicates, two of which were arranged as label swap experiments. pSILAC experiments with pulse durations of 4 h and 30 min were performed with three biological replicates. After the desired labeling time, cells were removed from the flask by trypsinization and pelleted at 4 °C. Cells were lysed in 2% SDS in PBS by heating to 90 °C for 10 min. DNA was digested with Benzonase (Sigma) and lysates were cleared by centrifugation. Protein concentrations were measured with the BCA protein quantitation kit (Thermo Scientific).
BONCAT
BONCAT experiments were carried out as described in the HeLa cell pSILAC protocol, with a few modifications. T-150 flasks were seeded with 4 million cells prior to each experiment. The larger culture size compensates for the relatively small amounts of protein that are produced during short pulses. During the pulse, both medium and heavy cultures were supplemented with either Aha or Aha30:1. Each BONCAT experiment was conducted with three biological replicates. Protein synthesis was halted prior to cell lysis by addition of cycloheximide (Sigma) to 100 μg/ml. Cells were lysed in freshly prepared 2% SDS in PBS with 100 mm chloroacetamide (Sigma) to alkylate free cysteines in proteins. Cysteine alkylation reduces thiol addition of cyclooctyne reagents, and increases the specificity of tagging of Aha-labeled proteins (18). After mixing heavy and medium lysates, Aha-labeled proteins were conjugated to a biotin tag by strain-promoted azide-alkyne click chemistry (19). DBCO-sulfo-biotin tag (Click Chemistry Tools (Scottsdale, AZ)) was added to 1 mg of mixed lysates to a final concentration of 12 μm and allowed to react for 15 min, after which the reaction was quenched with excess Aha. Tagged proteins were captured with Streptavidin Plus UltraLink Resin (Thermo Scientific), washed with 64 column volumes of 1% SDS in PBS, and eluted by boiling the resin in 1 mm biotin in 1% SDS in PBS for 15 min. Eluted proteins were concentrated on a 3 kDa molecular weight cut-off centrifugation filter (Amicon, Bilerica, MA) prior to SDS-PAGE. Separation of excess or unreacted biotin tag prior to streptavidin capture was unnecessary because of the small quantity of tag used in the click reaction.
GeLC-MS
Proteins were separated on precast 4–12% polyacrylamide gels (Invitrogen) and visualized with the colloidal blue staining kit (Invitrogen). Lanes were cut into 8 gel pieces and destained by iterative washing with 50 mm ammonium bicarbonate (Baker) and 1:1 (v/v) 50 mm ammonium bicarbonate and acetonitrile (LC-MS grade, Fluka, Ronkonkoma, NY). Proteins were reduced in 7 mm DTT (Research Products International, Mt. Prospect, IL) in 50 mm ammonium bicarbonate at 50 °C for 30 min. After removing the DTT solution, proteins were alkylated with freshly prepared 40 mm chloroacetamide (Sigma) in 50 mm ammonium bicarbonate for 20 min in the dark. Gel pieces were washed with 50 mm ammonium bicarbonate and acetonitrile for 5 min each. Proteins were digested with endoproteinase LysC (Mako) at 3 ng/μl in 50 mm Tris (Sigma), pH 8.5, overnight at 37 °C. Digested peptides were extracted from gel pieces by washing in 1% formic acid/2% acetonitrile for 5 min, 1:1 acetonitrile/water for 5 min, and 1% formic acid in acetonitrile for 5 min. Extracted peptides were desalted with custom packed C18 columns as described in Rappsilber et al. (20), concentrated by lyophilization, and resuspended in 0.1% formic acid (Sigma) prior to LC-MS/MS.
NanoLC-Mass Spectrometry Analysis
All liquid chromatography-mass spectrometry experiments were performed on an EASY-nLC (Proxeon Biosystems, now Thermo Scientific, Waltham, MA) connected to a hybrid LTQ-Orbitrap or LTQ-FT (Thermo Scientific) equipped with a nano-electrospray ion source (Proxeon Biosystems, now Thermo Scientific). pSILAC experiments with 24 h pulse times were analyzed on the LTQ-FT, and experiments comparing pSILAC and BONCAT with 4 h and 30 min pulse times were analyzed on the LTQ-Orbitrap. Peptides on the Orbitrap were separated on a 15 cm reversed phase analytical column (75 μm ID) packed in-house with 3 μm C18AQ beads (ReproSil-Pur C18AQ) using a 60 min elution from 0% to 30% solvent B at a flow rate of 350 nL/min. Solvent A was 0.2% formic acid, 2% acetonitrile, and 97.8% water. Solvent B was 0.2% formic acid, 19.8% water, and 80% acetonitrile. For the LTQ-FT, a 90 min gradient from 0% to 40% solvent B was used. The mass spectrometers were operated in data-dependent mode to automatically switch between MS and MS/MS scans, essentially as described (21). Survey full scan mass spectra were acquired in the Orbitrap (300–1700 m/z), following accumulation of 500,000 ions, with a resolution of 60,000 at 400 m/z. The top ten most intense ions from the survey scan were isolated and, after the accumulation of 5000 ions, fragmented in the linear ion trap by collisionally induced dissociation (collisional energy 35% and isolation width 2 Da). Precursor ion charge state screening was enabled and all singly charged and unassigned charge states were rejected. A reject mass list with the major streptavidin contaminants was used. The dynamic exclusion list was set with a maximum retention time of 90 s and a relative mass window of 10 ppm.
Survey full scan mass spectra were acquired in the LTQ-FT with an m/z of 400–1600, following accumulation of 1,000,000 ions, with a resolution of 50,000 at 400 m/z. The top seven most intense ions from the survey scan were isolated and, after the accumulation of 5000 ions, fragmented in the linear ion trap by collisionally induced dissociation (collisional energy 35% and isolation width 3 Da). Precursor ion charge state screening was enabled and all singly charged and unassigned charge states were rejected. The dynamic exclusion list was set with a maximum retention time of 60 s and a relative mass window of 10 ppm.
Protein Identification and Quantification
MaxQuant (v. 1.3.0.5) was used to process the Thermo RAW files. All default parameters were used, except LysC was specified as the enzyme and requantify was disabled. Up to two missed cleavages were allowed. Met oxidation (+15.9949) and N-terminal acetylation (+42.0106) were specified as variable modifications, and carbamidomethyl cysteine (+57.0125) was specified as a fixed modification. In all Aha labeling experiments, Aha (-4.9863) and l-2,4-diaminobutanoate (-30.9768), a product of reduction of Aha, were specified as variable modifications for Met. In BONCAT experiments, DBCO-sulfo-biotin (+648.2115) was specified as an additional variable modification for Met. Medium (+4.0251) and heavy (+8.0142) lysine labels were specified in all experiments. Mass tolerance for precursor ions and fragment ions were 7 ppm and 0.5 Da, respectively. In pSILAC experiments, multiplicity was set to three and light, medium, and heavy peptides were specified. In BONCAT experiments, multiplicity was set to two, specifying only medium and heavy lysine labels. The human database searched (IPI v 3.54) consisted of 75,710 sequences, 262 of which were common contaminants. The database was appended to a decoy database of equal size in MaxQuant. Protein and peptide false discovery rates were fixed at 1% using a target decoy approach. All members of a protein family with shared identifications were reported in one protein group. Individual evidence ratios were calculated by MaxQuant as the ratio of peak areas. Overall protein ratios were reported as the mean of the individual experiment ratios. Only ratios associated with unique or razor peptides were used for quantification. Annotated spectra for proteins identified by single peptides and detailed information on peptide and protein identifications are available in the supplementary information. We report only proteins quantified by at least two evidences in each set of experiments. In pSILAC experiments comparing Met versus Met, Aha versus Met, and Aha30:1 versus Met-treated cultures, we report only proteins quantified in both arrangements of the label swap experiments, and peptides that contained Met were discarded when calculating protein ratios to avoid unreliable quantifications because of unmodified counterpart peptides.
Protein Ratio Statistics
Overall protein ratios and their standard errors were calculated using a hierarchical model combined with bootstrap estimates and pooled variance estimates at the peptide level. Briefly, a global estimate of measurement error is calculated using pooled variance from the protein ratio in each replicate. Next, a hierarchical model of the overall protein ratio is calculated by first calculating the protein replicate ratio as the median of the peptide ratios in each replicate and then calculating the overall protein ratio as the mean of the protein replicate ratios. Finally, the standard error of the overall protein ratio is calculated using a bootstrap procedure where resampling with replacement occurs within the hierarchical model at both the replicate and peptide level and each peptide ratio in the bootstrap procedure is augmented by adding a random “noise” effect drawn from a normal distribution with mean zero and standard deviation equal to the previously calculated global estimate of measurement error. In total, 1000 bootstrap iterations are performed. The standard error of the overall protein ratio is then calculated as the standard deviation of the bootstrapped overall protein ratios. Z-tests can then be used to calculate p values of overall protein ratios with respect to a 1-to-1 ratio.
RESULTS
In pSILAC, metabolic labeling with amino acid isotopologs is used to determine relative rates of protein translation in different cell populations (22). To measure the effect of Aha treatment on protein abundance, cells grown in standard “light” media were split and shifted into isotopically “medium” and “heavy” culture media containing either Met, Aha, or a mixture of Aha and Met (Fig. 1A). After labeling times of 24 h in HeLa cell culture, paired medium and heavy cultures were lysed and mixed in a 1:1 ratio. Separate experiments in which both medium and heavy populations were treated with Met served as controls. Proteins were prepared for LC-MS/MS by standard gel separation and in-gel digestion protocols. Raw MS data were processed with the MaxQuant quantitative proteomics software (23).
Fig. 1.
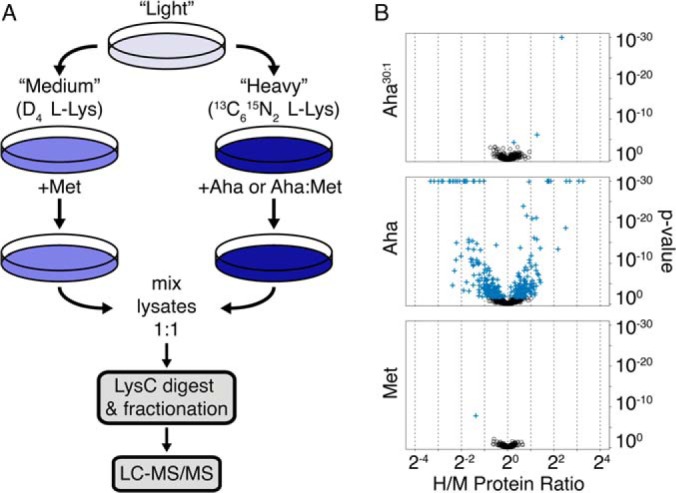
Quantifying the effects of Aha labeling on protein abundance in HeLa cell cultures. A, We used pSILAC to determine the effects of Aha labeling on protein abundance. Incorporation of “medium” and “heavy” isotope-labeled lysine allowed quantification of proteins from cultures treated with Aha or with mixtures of Aha and Met, relative to cultures treated with Met only. B, Global proteomic effects of Aha labeling in HeLa cell cultures. Quantified proteins are compared for cultures treated with Aha30:1 versus Met (top), Aha versus Met (middle), and Met versus Met (bottom). Proteins that show statistically significant differences from an H/M ratio of 1 (Benjamini-Hochberg FDR < 0.05) are marked by the blue cross symbol. H/M ratios greater than 1 indicate increased expression of proteins in the presence of Aha. Proteins with p values less than 10−30 were plotted at 10−30.
The extent of replacement of Met by Aha can be controlled by adjusting the relative concentrations of the two amino acids in the culture medium (24). A set of scouting experiments in E. coli suggested that treating cells with a 30:1 ratio of Aha to Met might limit the extent of replacement to levels that would allow enrichment of newly synthesized proteins with minimal effects on protein abundance (supplemental Fig. S1 and S2). We then investigated similar conditions, hereafter referred to as Aha30:1, for quantitative, time-resolved proteomic analysis of HeLa cells in culture.
We performed pSILAC experiments with 24 h labeling times to determine whether Aha30:1 labeling causes differences in protein abundance relative to Met-labeled controls. Of the 1257 quantified proteins produced during the 24 h pulse, only three exhibited H/M ratios significantly different from 1 (Fig. 1B). The 78-kDa glucose-regulated protein (Grp78), an ER-localized chaperone, was up-regulated 1.2-fold, whereas the mannose-P-dolichol utilization defect 1 protein (MPDU1) and S-adenosylmethionine (SAM) synthase isoform type 2 (MAT2A) were up-regulated 2.4- and 5.1-fold, respectively. MPDU1 is an ER membrane protein required for use of mannose-P-dolichol in the synthesis of GPI anchors and lipid-linked oligosaccharides (25). SAM synthase catalyzes the conversion of methionine to SAM, an important methyl donor in the cell (26). In these experiments the extent of replacement of Met by Aha was 6% (supplemental Table S1). The ratio of kcat/Km values for activation of Aha and Met by the E. coli methionyl-tRNA synthetase is 1:390, which for the Aha30:1 labeling condition predicts a replacement rate of 7.7%, in close agreement with the measured rate (27).
In contrast to the results just described, treatment of HeLa cells with Aha alone (i.e. without added Met) for a 24 h period caused substantial changes in the abundance of many proteins. Statistically significant differences were noted for 362 of the 1001 quantified proteins, 101 of which showed differences greater than 2-fold (Fig. 1B, supplemental Table S2).
Gene ontology (GO) analysis suggests that protein folding and translation are among the cellular processes affected most significantly by Aha labeling in the absence of added Met (Fig. 2A). The heat shock 70 kDa protein 1A/1B (HSPA1A), a member of the protein folding group, showed the largest increase (9.6-fold) in the Aha-treated samples. Other up-regulated heat shock proteins and chaperones included the heat shock 105 kDa protein (HSPH1), endoplasmin (HSP90B1), calnexin (CANX), and several DnaJ homologs (DNAJB11, DNAJA1, and DNAJB1), which were up-regulated 2–3.5-fold. In the translation group, most of the strongly down-regulated proteins were ribosomal proteins, which also constituted 17 of the 19 most down-regulated proteins overall.
Fig. 2.
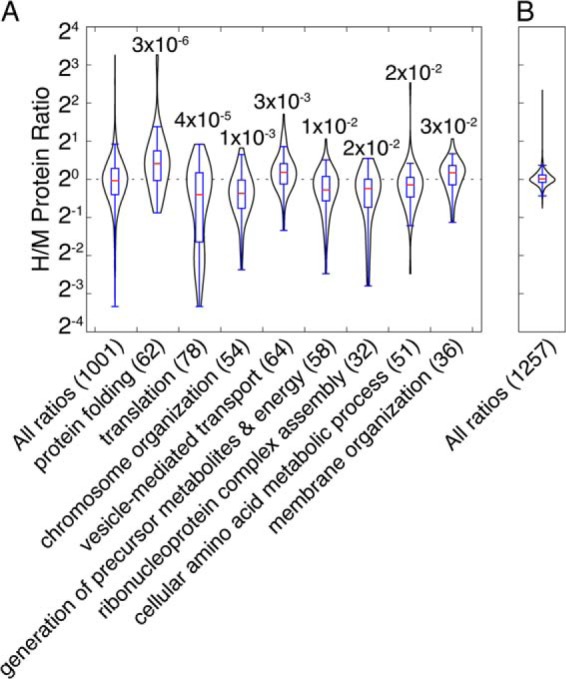
Gene ontology analysis of proteins affected by Aha treatment. A, Quantified proteins from Aha versus Met experiments were mapped to gene ontology groups, and the ratio distributions of the groups were compared with the total protein distribution by a two-sample K-S test (p < 0.05). Violin plots of the distributions are shown with p values and the number of proteins in each group. B, A similar violin plot for total quantified proteins from Aha30:1 experiments confirms the reliability of relative protein abundances measured in such experiments.
Among proteins in other GO groups, fatty acid synthase (FASN) showed the largest decrease (-5.6-fold) in Aha-treated cells. FASN produces long-chain fatty acids and is sensitive to amino acid levels in the cell; deprivation of single essential amino acids, including Met, diminishes FASN abundance (28). Regulation of SAM synthase is also linked to Met abundance, and the type 2 isoform identified here is up-regulated in the absence of Met (29). These examples suggest that Met starvation, independent of Aha incorporation, probably contributes to the changes in protein abundance observed in Met-depleted cultures. The extent to which the observed changes are because of Aha incorporation, Met depletion, or a combination of the two, however, is unknown.
A similar representation of the results of labeling of HeLa cells with a 30:1 mixture of Aha and Met (Fig. 2B) confirms that light labeling yields reliable measurements of relative protein abundances. The full lists of proteins from Aha30:1 and Aha-treated cultures and their quantitative analyses are available in supplemental Table S2.
Because BONCAT provides a convenient means of identifying newly synthesized cellular proteins, we examined the pSILAC experiments for evidence that Aha labeling might give rise to artifacts in protein identification. Peptides containing medium and heavy lysine labels allowed for direct comparison of proteins identified in either Aha- or Met-pulsed cultures from individual pSILAC experiments. For each experiment we determined the total number of proteins identified, the number shared between the sample treated with either Aha or Aha30:1 and the Met control, and the number of proteins found only in the sample culture (supplemental Table S1). In each case the number of proteins found only in the sample culture constituted between 0.5% and 6.1% of the total, and no increase was observed in Aha-treated cultures compared with those treated with Met (Fig. 3). Thus we find no evidence that Aha labeling, even under conditions of Met depletion, gives rise to artifacts in protein identification.
Fig. 3.
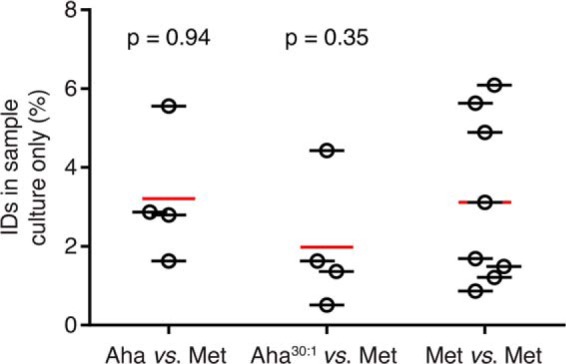
Aha labeling does not cause artifacts in protein identification. Proteins identified in sample cultures treated with Aha, Aha30:1, or Met are compared with proteins identified in their respective control cultures treated with Met only. Comparisons of protein identifications are made between medium and heavy-labeled proteins from individual pSILAC experiments. The percentage of identifications in the sample culture only are plotted for each of the four replicate experiments, and the average of these values is marked by a red line. Unpaired, two-sample t-tests comparing Aha versus Met and Aha30:1 versus Met to the Met versus Met control failed to reject the null hypothesis that average values are identical (p values are shown). Because of the symmetry of Met versus Met control experiments, the number of identification comparisons is effectively doubled.
Both BONCAT and pSILAC can be used to measure changes in the cellular proteome that occur during time windows defined by amino acid pulse-labeling protocols. The key difference between the two methods is that BONCAT physically separates newly synthesized proteins prior to MS analysis, whereas pSILAC distinguishes and quantifies new proteins informatically. By combining the BONCAT and pSILAC approaches, newly synthesized proteins produced during a pulse can be both enriched and quantified (Fig. 4A). Here we show that quantitative BONCAT can be used as a general approach for monitoring rapid proteomic changes, and we compare the performance of BONCAT and pSILAC for short labeling times.
Fig. 4.
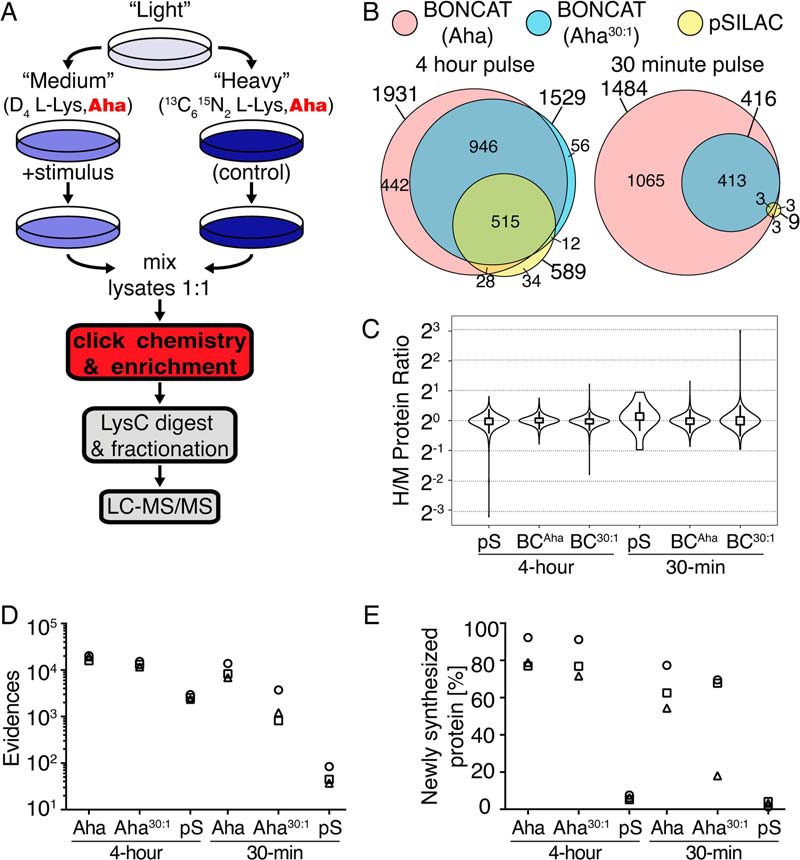
Comparison of the BONCAT and pSILAC approaches in HeLa cell culture. A, Combining BONCAT with pulsed isotope labeling allows enrichment, identification and quantification of newly synthesized proteins. Deviations from a standard pSILAC protocol are shown in red. In a quantitative BONCAT experiment, cultures are treated during the pulse with both lysine isotopologs and Aha. After mixing lysates, Aha-labeled proteins are conjugated to affinity tags, enriched by affinity purification, and analyzed by standard MS protocols. B, Quantified proteins from the BONCAT and pSILAC methods are compared for experiments performed during 4 h and 30 min pulses. Venn diagrams show total proteins quantified in each experiment and proteins shared between experiments. C, BONCAT experiments provide accurate protein quantification. Each violin plot shows a density distribution of H/M protein ratios, a box plot that indicates the span from the 25th to the 75th percentile, and whiskers that extend to 1.5-fold times the inner quartile range from the box edges. D, The total number of peptide evidences and E, the percent of newly synthesized protein are plotted for individual BONCAT and pSILAC experiments. BONCAT (BC), pSILAC (pS).
For short pulse-labeling times, newly synthesized proteins make up a small fraction of the total proteome. We expected that enriching newly synthesized proteins by pulse-labeling with Aha would aid in their detection and quantification by mass spectrometry for short pulse times. We compared the performance of the combined BONCAT-pSILAC approach to that of pSILAC alone at pulse times of 4 h and 30 min in HeLa cell culture. In BONCAT experiments, Aha-labeled proteins were enriched by conjugation to a DBCO-biotin tag via strain-promoted azide-alkyne click chemistry and purification on a streptavidin resin (30). Enriched proteins were prepared for LC-MS/MS as described in the experimental methods.
Using the combined BONCAT-pSILAC method in triplicate experiments, we quantified 1931 and 1529 newly synthesized proteins in HeLa cell cultures within a 4 h window with the Aha and Aha30:1 labeling strategies, respectively. Similar pSILAC experiments quantified 589 proteins within the 4 h pulse (Fig. 4B). For a pulse time of 30 min, pSILAC quantified only nine newly synthesized proteins, whereas BONCAT quantified 1484 and 416 proteins with the Aha and Aha30:1 labeling strategies, respectively. Comparisons of protein identifications produced similar results (supplemental Fig. S3). BONCAT-pSILAC experiments yielded H/M protein ratios that were accurate and consistent, showing narrow distributions centered about a value of 1 (Fig. 4C). BONCAT replicates were reproducible, sharing an average of 89% of quantified proteins between replicates (supplemental Table S3). Individual BONCAT experiments provided 5–7 times more peptide evidences than parallel pSILAC experiments in the 4 h pulse, and 35–175 times more evidences in the 30 m pulse experiments (Fig. 4D, supplemental Fig. S4). Prior to enrichment, newly synthesized proteins in the 4 h and 30 min experiments made up ∼6 and 3% of the total proteome, respectively. After BONCAT enrichment, newly synthesized proteins constituted an average of 80–83% and 52–65% of the total protein in the 4 h and 30 min experiments, respectively (Fig 4E). The list of quantified proteins from the 4 h and 30 min pSILAC and BONCAT experiments is available in supplemental Table S4.
DISCUSSION
Pulse-labeling with Aha allows fast, accurate and sensitive detection of changes in the cellular proteome. Complete replacement of Met by Aha, even for pulses of 24 h in HeLa cell culture, does not significantly change the identities of proteins detected by mass spectrometry, although differences in protein abundance are observed under such conditions. To mitigate the latter effect, we describe an Aha30:1 labeling strategy that minimizes perturbations in protein abundance while maintaining a level of labeling that is sufficient for modification by click chemistry and affinity enrichment. The Aha30:1 labeling approach is recommended for use in studies in which preservation of natural protein abundances is of the utmost importance. Alternatively, full Aha labeling increases the yield of affinity enrichment and can be used to identify larger numbers of newly synthesized proteins, especially under conditions in which enrichment is more challenging, as in experiments that use short pulse times.
A comparison of the BONCAT and pSILAC methods shows that affinity enrichment of newly synthesized proteins enhances the time resolution of proteomic analysis. A combined BONCAT-pSILAC approach enables acquisition of extensive, quantitative proteomic information within 30 min in HeLa cell culture, a time scale that is inaccessible to isotope labeling techniques alone. We expect quantitative BONCAT-pSILAC experiments to be especially useful for monitoring proteome dynamics, that is, for identifying early, middle, and late changes in protein production in response to biological cues.
Supplementary Material
Acknowledgments
We thank Kai Yuet for providing the E. coli KY2 strain and the PEL staff for technical support.
Footnotes
Author contributions: J.D.B., E.M.S., and D.A.T. designed research; J.D.B., Y.J.X., and Y.Q. performed research; M.J.S. and S.H. contributed new reagents or analytic tools; J.D.B. and M.J.S. analyzed data; J.D.B., Y.Q., S.H., and D.A.T. wrote the paper.
* This work was supported by National Institutes of Health grant NIH RO1 GM062523, the Institute for Collaborative Biotechnologies through grant W911NF-09–0001 from U.S. Army Research Office, the Joseph J. Jacobs Institute for Molecular Engineering for Medicine, the Betty and Gordon Moore Foundation through Grant GBMF775, and the Beckman Institute. Y.J.X. acknowledges funding from the Caltech Summer Undergraduate Research Fellowships (SURF) program.
 This article contains supplemental Figs. S1 to S4 and Tables S1 to S4.
This article contains supplemental Figs. S1 to S4 and Tables S1 to S4.
1 The abbreviations used are:
- BONCAT
- Bioorthogonal noncanonical amino acid tagging
- pSILAC
- Pulsed stable isotope labeling with amino acids in cell culture
- Aha
- L-azidohomoalanine
- Aha30:1
- A mixture of Aha (1 mM) and Met (33 μM)
- ncAA
- Noncanonical amino acid
- H/M
- Heavy to medium ratio
- GO
- Gene ontology.
REFERENCES
- 1. Dieterich D. C., Link A. J., Graumann J., Tirrell D. A., Schuman E. M. (2006) Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proc. Natl. Acad. Sci. U.S.A. 103, 9482–9487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dieterich D. C., Lee J. J., Link A. J., Graumann J., Tirrell D. A., Schuman E. M. (2007) Labeling, detection and identification of newly synthesized proteomes with bioorthogonal non-canonical amino-acid tagging. Nat. Protoc. 2, 532–540 [DOI] [PubMed] [Google Scholar]
- 3. Szychowski J., Mahdavi A., Hodas J. J. L., Bagert J. D., Ngo J. T., Landgraf P., Dieterich D. C., Schuman E. M., Tirrell D. A. (2010) Cleavable biotin probes for labeling of biomolecules via azide-alkyne cycloaddition. J. Am. Chem. Soc. 132, 18351–18360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tcherkezian J., Brittis P. A., Thomas F., Roux P. P., Flanagan J. G. (2010) Transmembrane receptor DCC associates with protein synthesis machinery and regulates translation. Cell 141, 632–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yoon B. C., Jung H., Dwivedy A., Hare C. M. O., Zivraj K. H., Holt C. E. (2011) Local translation of extranuclear lamin B promotes axon maintenance. Cell 148, 752–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Deal R. B., Henikoff J. G., Henikoff S. (2010) Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science 328, 1161–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang M. M., Tsou L. K., Charron G., Raghavan A. S., Hang H. C. (2010) Tandem fluorescence imaging of dynamic S-acylation and protein turnover. Proc. Natl. Acad. Sci. U.S.A. 107, 8627–8632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ouellette S. P., Dorsey F. C., Moshiach S., Cleveland J. L., Carabeo R. A. (2011) Chlamydia species-dependent differences in the growth requirement for lysosomes. PLoS One 6, e16783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Choi K.-Y. G., Lippert D. N. D., Ezzatti P., Mookherjee N. (2012) Defining TNF-α and IL-1β induced nascent proteins: Combining bio-orthogonal non-canonical amino acid tagging and proteomics. J. Immunol. Methods 382, 189–195 [DOI] [PubMed] [Google Scholar]
- 10. Melemedjian O. K., Asiedu M. N., Tillu D. V., Peebles K. A., Yan J., Ertz N., Dussor G. O., Price T. J. (2010) IL-6- and NGF-induced rapid control of protein synthesis and nociceptive plasticity via convergent signaling to the eIF4F complex. J. Neurosci. 30, 15113–15123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dieterich D. C., Hodas J. J. L., Gouzer G., Shadrin I. Y., Ngo J. T., Triller A., Tirrell D. A., Schuman E. M. (2010) In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat. Neurosci. 13, 897–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hodas J. J. L., Nehring A., Höche N., Sweredoski M. J., Pielot R., Hess S., Tirrell D. A., Dieterich D. C., Schuman E. M. (2012) Dopaminergic modulation of the hippocampal neuropil proteome identified by bio-orthogonal non-canonical amino-acid tagging (BONCAT). Proteomics 12, 2464–2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hinz F. I., Dieterich D. C., Tirrell D. A., Schuman E. M. (2012) Noncanonical amino acid labeling in vivo to visualize and affinity purify newly synthesized proteins in larval zebrafish. ACS Chem. Neurosci. 3, 40–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eichelbaum K., Winter M., Diaz M. B., Herzig S., Krijgsveld J. (2012) Selective enrichment of newly synthesized proteins for quantitative secretome analysis. Nat. Biotechnol. 30, 984–990 [DOI] [PubMed] [Google Scholar]
- 15. Somasekharan S. P., Stoynov N., Rotblat B., Leprivier G., Galpin J. D., Ahern C. a, Foster L. J., Sorensen P. H. B. (2012) Identification and quantification of newly synthesized proteins translationally regulated by YB-1 using a novel Click-SILAC approach. J. Proteomics 77, e1–e10 [DOI] [PubMed] [Google Scholar]
- 16. Howden A. J. M., Geoghegan V., Katsch K., Efstathiou G., Bhushan B., Boutureira O., Thomas B., Trudgian D. C., Kessler B. M., Dieterich D. C., Davis B. G., Acuto O. (2013) QuaNCAT: quantitating proteome dynamics in primary cells. Nat. Methods 10, 343–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Link A. J., Vink M. K. S., Tirrell D. A. (2007) Synthesis of the functionalizable methionine surrogate azidohomoalanine using Boc-homoserine as precursor. Nat. Protoc. 2, 1884–1887 [DOI] [PubMed] [Google Scholar]
- 18. Van Geel R., Pruijn G. J. M., van Delft F. L., Boelens W. C. (2012) Preventing thiol-yne addition improves the specificity of strain-promoted azide-alkyne cycloaddition. Bioconjugate Chem. 23, 392–398 [DOI] [PubMed] [Google Scholar]
- 19. Debets M. F., van Berkel S. S., Schoffelen S., Rutjes F. P. J. T., van Hest J. C. M., van Delft F. L. (2010) Aza-dibenzocyclooctynes for fast and efficient enzyme PEGylation via copper-free (3+2) cycloaddition. Chem. Commun. 46, 97–99 [DOI] [PubMed] [Google Scholar]
- 20. Rappsilber J., Mann M., Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 21. Kalli A., Hess S. (2012) Effect of mass spectrometric parameters on peptide and protein identification rates for shotgun proteomic experiments on an LTQ-orbitrap mass analyzer. Proteomics 12, 21–31 [DOI] [PubMed] [Google Scholar]
- 22. Schwanhäusser B., Gossen M., Dittmar G., Selbach M. (2009) Global analysis of cellular protein translation by pulsed SILAC. Proteomics 9, 205–209 [DOI] [PubMed] [Google Scholar]
- 23. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 24. Nowatzki P. J., Franck C., Maskarinec S. A., Ravichandran G., Tirrell D. A. (2008) Mechanically tunable thin films of photosensitive artificial proteins: preparation and characterization by nanoindentation. Macromolecules 41, 1839–1845 [Google Scholar]
- 25. Hirata T., Fujita M., Kanzawa N., Murakami Y., Maeda Y., Kinoshita T. (2013) Glycosylphosphatidylinositol mannosyltransferase II is the rate-limiting enzyme in glycosylphosphatidylinositol biosynthesis under limited dolichol-phosphate mannose availability. J. Biochem. 154, 257–264 [DOI] [PubMed] [Google Scholar]
- 26. Markham G. D., Hafner E. W., Tabor C. W., Tabor H. (1980) S-Adenosylmethionine synthetase from Escherichia coli. J. Biol. Chem. 255, 9082–9092 [PubMed] [Google Scholar]
- 27. Kiick K. L., Saxon E., Tirrell D. A., Bertozzi C. R. (2002) Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation. Proc. Natl. Acad. Sci. U.S.A. 99, 19–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dudek S. M., Semenkovich C. F. (1995) Essential amino acids regulate fatty acid synthase expression through an uncharged transfer RNA-dependent mechanism. J. Biol. Chem. 270, 29323–29329 [DOI] [PubMed] [Google Scholar]
- 29. Martínez-Chantar M. L., Latasa M. U., Varela-Rey M., Lu S. C., García-Trevijano E. R., Mato J. M., Avila M. a. (2003) L-methionine availability regulates expression of the methionine adenosyltransferase 2A gene in human hepatocarcinoma cells. J. Biol. Chem. 278, 19885–19890 [DOI] [PubMed] [Google Scholar]
- 30. Sletten E. M., Bertozzi C. R. (2011) Bioorthogonal reactions. Acc. Chem. Res. 44, 666–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.