

Abstract
Sphingosine-1-phosphate (S1P) signaling regulates lymphocyte egress from lymphoid organs into systemic circulation. Sphingosine phosphate receptor 1 (S1P1) agonist, FTY-720 (Gilenya™) arrests immune trafficking and prevents multiple sclerosis (MS) relapses. However, alternative mechanisms of S1P-S1P1 signaling have been reported. Phosphoproteomic analysis of MS brain lesions revealed S1P1 phosphorylation on S351, a residue crucial for receptor internalization. Mutant mice harboring a S1pr1 gene encoding phosphorylation-deficient receptors [S1P1(S5A)] developed severe experimental autoimmune encephalomyelitis (EAE) due to T helper (TH) 17-mediated autoimmunity in the peripheral immune and nervous system. S1P1 directly activated Janus-like kinase–signal transducer and activator of transcription 3 (JAK-STAT3) pathway via interleukin 6 (IL-6). Impaired S1P1 phosphorylation enhances TH17 polarization and exacerbates autoimmune neuroinflammation. These mechanisms may be pathogenic in MS.
Introduction
The development of FTY-720 (Fingolimod, Gilenya™) as a first-line oral therapy in multiple sclerosis (MS) has illuminated sphingosine-1-phosphate (S1P) signaling as a targetable pathway in autoimmune neuroinflammation1–4. One of the best-characterized functions of S1P pathway is the regulation of lymphocyte trafficking from secondary lymphoid organs into the systemic circulation5–7. Interaction of the sphingolipid ligand, S1P in the blood or lymph with the G protein-coupled receptor (GPCR) S1P receptor 1 (S1P1), on lymphocytes is necessary for their egress from the spleen and lymph nodes into the systemic circulation8–10. The critical role played by the S1P-S1P1 in trafficking is perturbed by FTY-720, a functional antagonist of S1P1 (refs. 5,11,12). FTY-720 sequesters lymphocytes in the secondary lymphoid organs by inducing receptor internalization and degradation, thus sparing the central nervous system (CNS) from immune attack by autoreactive lymphocytes13–15. FTY-720 effectively decreases relapse rate up to 50% and is superior to interferon- (IFN-β) therapy16–18. However, a subset of relapsing remitting MS (RRMS) patients on FTY-720 therapy developed severe relapses and even tumorfactive MS lesions despite severe lymphopenia19–21. This finding suggests that S1P signaling may participate in immune regulatory functions other than lymphocyte trafficking.
S1P1 receptor internalization is a critical step in initiating S1P signaling22,23. This process is dependent on post-translational modification of the C-terminal domain of the receptor24–26. Binding of S1P to S1P1 promotes the phosphorylation of C-terminal domain serine residues of S1P1 by protein kinase GRK2 (ref. 24). This covalent addition of phosphate residue modifies the physicochemical properties of S1P1 leading to internalization of the ligand-receptor complex. Impaired internalization of S1P1 has been associated with arrested lymphocyte egress into the circulation and delayed lymphopenia in response to FTY720 treatment25,27. However, the physiological function of receptor internalization, subsequent effects on intracellular signaling pathways and how it modulates autoimmune neuroinflammation are yet to be determined.
Here, an unbiased, phosphoproteomic analysis of MS patient brain samples during active inflammation revealed that S1P1 was phosphorylated in vivo on S351. S1P1 expression was also observed in brain-infiltrating T lymphocytes in MS lesions demonstrated by immunohistochemistry. Complementary to our findings in the human disease, induction of experimental autoimmune encephalomyelitis (EAE) in mice carrying the phosphorylation-defective S1P1 receptor [S1P1(S5A)mice] resulted in severe degree of paralysis, more interleukin 17 (IL-17) mediated inflammation in the peripheral immune system and higher numbers of IL-17–expressing CD4+ T cells infiltrating in the CNS. We also demonstrated that the severe autoimmune neuroinflammation in the S1P1(S5A) mice was due to the activation of Janus-like kinase–signal transducer and activator of transcription 3 (JAK–STAT3)–IL-17 pathway and that signaling via S1P1 was directly responsible for this effect. Finally, we demonstrated that STAT3-mediated T helper 17 (TH17) polarization in S1P1(S5A) mice was dependent on IL-6 signaling. Collectively, these data suggest that S1P1 signaling is crucial for TH17 polarization and the clinical outcome in MS.
Results
S1P1 was phosphorylated in MS brain lesions
We performed phosphoproteomic analysis of fresh-frozen brain tissue from autopsy samples of MS patients to identify dysregulated pathways during MS pathogenesis. We first characterized the histopathological and cellular features of MS brain lesion samples by conventional staining methods (Hematoxylin and Eosin, Luxol Fast Blue and Bielschowsky) and immunohistochemistry. The MS lesions included in our study were classically characterized as chronic active lesions, the most common lesion type observed in RRMS patients. There was evidence of active inflammation (infiltration of T cells and macrophages in the peri-venular region and the brain parenchyma), myelin loss, axonal damage, astrocytosis and microglia activation (Supplementary Fig. 1 a–d)28,29. Tissue containing six individual MS brain lesions (from three MS patients) was then pooled and subjected to phosphoproteomic analysis by mass spectrometry30. MS lesions and control brain samples were homogenized separately and respective protein extracts were digested with protease trypsin. Peptide pools were then fractionated, phosphopeptides were enriched, and fractions containing phosphopeptide mixtures were then analyzed by nanoflow liquid chromatography and mass spectrometry (Supplementary Fig. 2). Identified phosphopeptides were selected by stringent criteria based on Xcorr (the observed to theoretical mass spectrum cross-correlation), ΔCn (the difference of normalized cross-correlation scored between the first and second peptide search hits), and a false-positive rate (FDR) less than 0.6% to ascertain the reliability of sequence identification and assignment of modifications31. This analysis identified a total of 7,404 unique phosphorylation sites, 6,035 from MS samples and 3,802 from controls (Supplementary Table 1, page 1). MS samples contained more phosphorylated proteins and the number of phosphorylation sites within an individual protein. Serine phosphorylation sites were most abundant (4,766 for MS, 2,998 for control and a total of 5,785 unique sites), followed by threonine (1,101 MS, 696 control, and 1,401 total) and tyrosine sites (168 MS, 108 control, and 218 total) (Supplementary Table 1, page 2). This analysis identified a total of 2,633 phosphoproteins; 2,243 were found in MS samples and 1,639 in control brains (Supplementary Table 1, page 3). Many of these proteins were phosphorylated on multiple sites.
We further explored the functional roles of these phosphoproteins to identify dysregulated pathways. Phosphoproteins from MS and control samples were selected based on the spectral count of phosphorylation sites (Control > MS and MS > Control). These data were then uploaded into the Database for Annotation, Visualization and Integrated Discovery (DAVID) bioinformatic analysis system and clustered32,33. We observed a differential functional enrichment pattern in MS and control samples based on the benjamini P values (Supplementary Fig. 3). Among these were several of the pathways involved in regulating immune response (such as T cell receptor signaling or chemokine signaling) and trafficking (including VEGF signaling, leukocyte transmigration, adherens or tight junction formation) were significantly up regulated in samples from active MS lesions.
Among the identified phosphopeptides from MS samples were four unique phosphopeptides belonging to the GPCR S1P1 (S1P1 342–352), a key regulator of immune cell egress (Fig. 1a). The fragmentation pattern and b and y ions from the mass spectra of S1P1 342–352 peptide unequivocally assigned the addition of a phosphate group on S351, on the carboxyl terminal tail of S1P1. The carboxyl terminus of S1P1 has previously been shown as a crucial region for S1P signaling by mediating phosphorylation-dependent receptor internalization24. This phosphopeptide was identified in the phosphopeptide fractions from the active MS brain lesions, suggesting that phosphorylation of S351 may play an important role in MS pathogenesis.
Figure 1. S1P1 phosphorylation in active MS brain lesions.
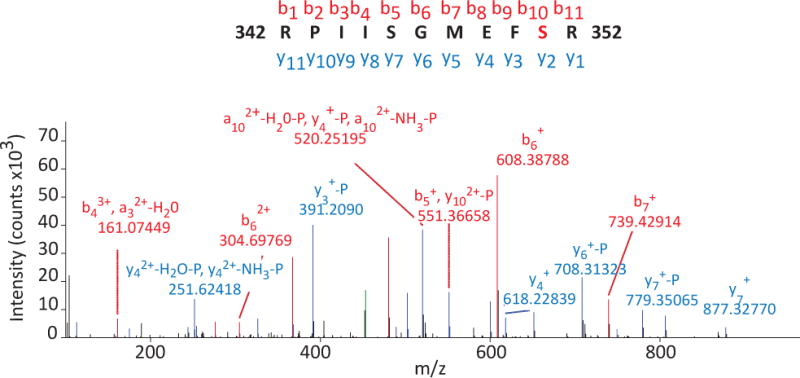
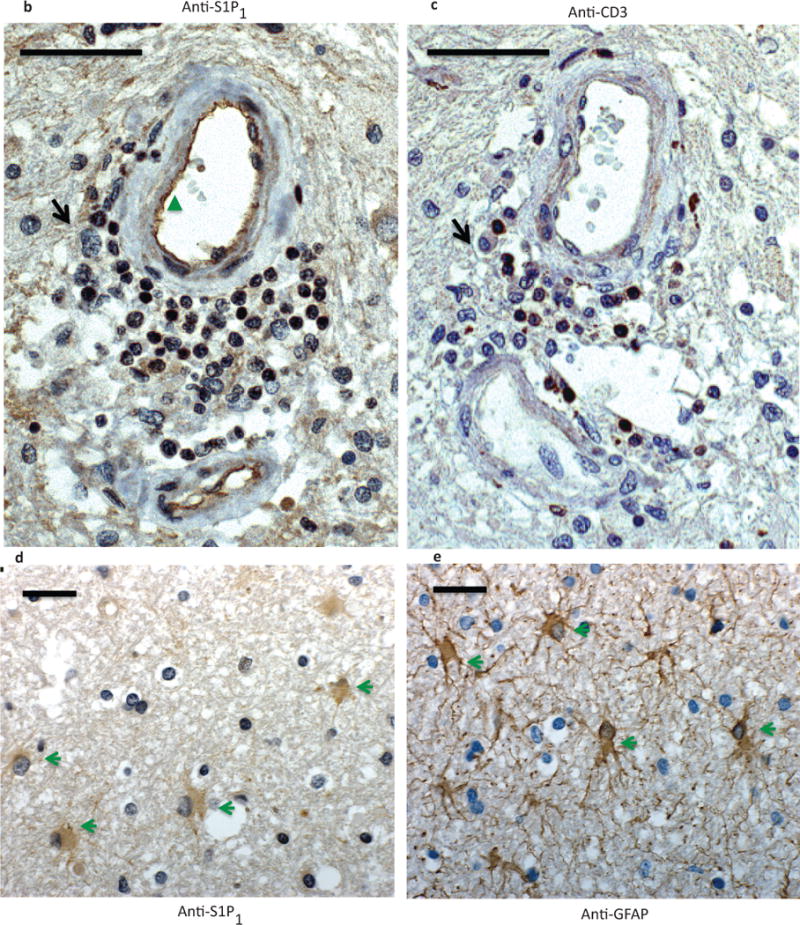
a) A representative mass spectra of S1P1 342–352 peptide depicting mass/charge (m/z) of identified amino-terminal (b, red) and carboxyl-terminal (y, blue) fragmented ions. Phosphorylation of S351 was assigned by the presence or loss of phosphate group among identified ions. Four unique S1P1 342–352 peptides were identified from two independent mass spectrometric analyses. Immunohistochemistry of formalin-fixed, paraffin-embedded sections from a MS brain lesion labeled with antibodies against S1P1 (anti-S1P1)(b and d), CD3 (anti-CD3)(c) and glia fibrillary acidic protein (GFAP)(anti-GFAP) (e). Perivascular immune cells (black arrow), endothelial cells (green triangle) and astrocytes (green arrowheads) expressed S1P1. Scale bar = 50 μm. Representative data from analysis of three MS brain samples.
We then investigated which cells expressed S1P1 in active MS brain lesions. We performed immunohistochemistry on formalin-fixed, paraffin-embedded sections on the same brain samples we utilized in our phosphoproteomic analysis. We utilized antibodies against S1P1, T cells (anti-CD3) and glia [astrocytes, anti-glia fibrillary acidic protein (GFAP)] on serial sections from several MS brain samples containing active lesions to localize expression of S1P1 on different cell types in the CNS. We observed that S1P1 was strongly expressed in the infiltrating CD3+ T cells in the active MS brain lesions (Fig. 1b, c and Supplementary Fig. 4). S1P1 expression was also observed in GFAP-positive glia cells (Fig. 1d, e and Supplementary Fig. 5). We could not, however, investigate the state of S1P1 phosphorylation in these cells utilizing this method due to the lack phospho-specific S1P1 antibody. These findings suggest that S1P1 is expressed in T cells and astrocytes in MS during active CNS inflammation.
S1P1(S5A) mice developed more severe EAE
Phosphorylation events serve as molecular switches to propagate cellular signaling events under physiological and pathological conditions30,34,35. Phosphorylation on the C terminal serines of S1P1 has previously been shown to initiate receptor internalization and regulate lymphocyte egress from lymphoid organs into the circulation25,27. Mice harboring a mutated S1pr1 gene encoding a phosphorylation-defective S1P1 receptor with five serines in the C terminal peptide were modified to alanines, [S1P1(S5A)mice] had normal immune cell counts and receptor function under physiological conditions, but exhibited delayed lymphopenia in response to treatment with FTY-720 (ref. 25). The findings of S1P1 phosphorylation in active MS brain lesions and S1P1 expression in T cells led us to investigate the effects of S1P1 phosphorylation in autoimmune neuroinflammation utilizing the S1P1(S5A) mice. We induced EAE in S1P1(S5A)and wild-type C57BL/6J mice with complete Freund’s adjuvant (CFA) and MOG35–55 peptide. We observed that S1P1(S5A) mice developed earlier onset of EAE and more severe degree of paralysis compared to their wild-type counterparts (Fig. 2a and Supplementary Table 2).
Figure 2. S1P1(S5A) mice developed severe EAE.
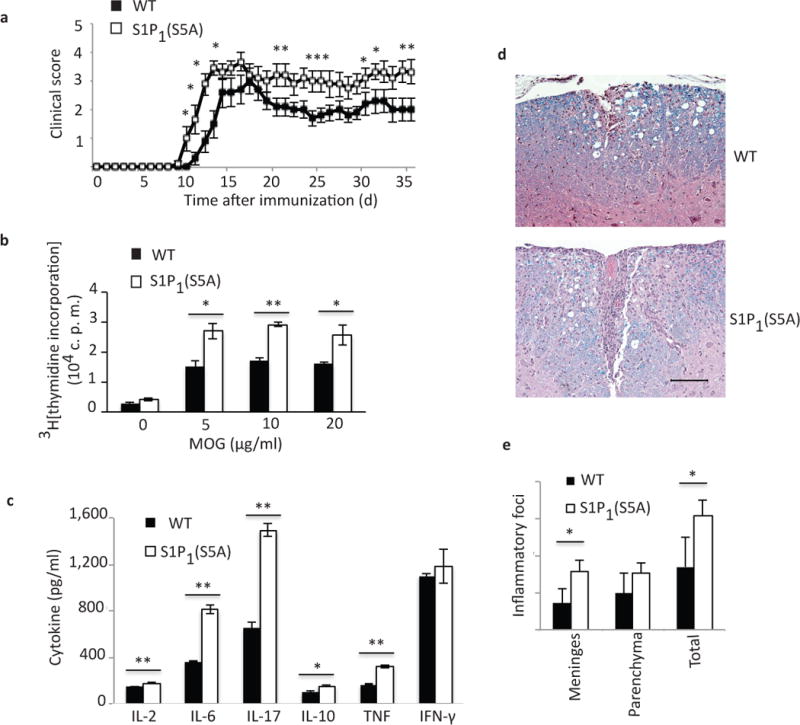
(a) Mean clinical score (± S. E. M.) of MOG35–55–immunized S1P1(S5A)(mice carrying phosphorylation defective S1pr1 gene) and WT (C57BL/6J) EAE mice (females, 8–9 weeks old). Immune cell proliferation (measured by 3H[thymidine] incorporation) (b) and cytokine expression (measured by ELISA) (c) of ex vivo recall assay from MOG35–55-immunized S1P1(S5A)and WT EAE splenocytes (day 8 post-immunization). MOG; myelin oligodendrocyte glycoprotein, c.p.m.; counts per minute. (d) Photomicrograph (Luxol Fast Blue, Hematoxylin stain) and (e) quantification of CNS inflammation in WT (top) and S1P1 (S5A) (bottom) EAE mice from (a). *p<0.05, **p<0.01, Mann-Whitney U-test (a) and Student’s t-test (b, c and e). n=9–10/arm (a) and 3–5/arm (b–e). These experiments were repeated 3 times.
We also observed a more robust proliferation of splenocytes from S1P1(S5A) EAE mice (day 8 post-immunization) in response to ex vivo reactivation with MOG35–55 peptide (Fig. 2b). Analysis of culture supernatants by enzyme-linked immunosorbent assay (ELISA) also revealed that splenocytes from S1P1(S5A) mice expressed higher concentrations of inflammatory cytokines, IL-2, IL-6, TNF and IL-17 (Fig. 2c). Similar findings were also observed in the lymph node cell cultures of S1P1(S5A) EAE mice (Supplementary Fig. 6a,b). Histopathological analysis revealed that the CNS of S1P1(S5A) EAE mice contained more numerous inflammatory foci compared to their wild-type counterparts (Fig. 2d,e). These data suggest that defective phosphorylation of S1P1 resulted in more severe autoimmune inflammation in the peripheral immune system and the CNS of S1P1(S5A) EAE mice.
Enhanced TH17-mediated neuroinflammation in S1P1(S5A) mice
S1P1 is ubiquitously expressed and participates in diverse cellular processes including maintaining vascular and immune functions in health and disease36–39. In order to determine if the increase EAE disease severity in the S1P1(S5A) mice was lymphocytes intrinsic, we performed an adoptive transfer experiment where we transferred encephalitogenic cells (MOG35–55-reactivated, expanded T cells) from wild-type C57BL/6J or S1P1(S5A) EAE mice into naïve Rag1−/− recipients. We observed that the recipients of cells from S1P1(S5A) EAE mice developed more severe paralysis, similar to the more severe disease course that we observed (Fig. 3a and Supplementary Table 3). These findings suggest that the primary determinant of the EAE phenotype in S1P1(S5A) mice was due to the T cell-intrinsic impaired S1P1 signaling.
Figure 3. Enhanced STAT3-mediated Th17 polarization in S1P1(S5A) EAE mice.
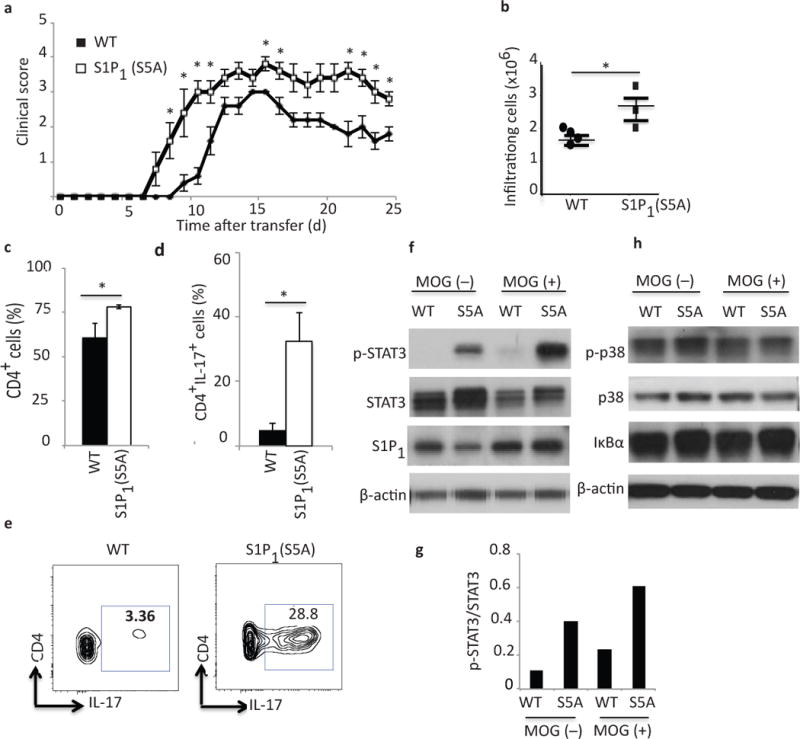
(a) Mean clinical scores ((±S. E. M.) of Rag1−/− adoptive transfer recipients of encephalitogenic cells from MOG35–55-immunized S1P1(S5A)and WT (C57BL/6J) EAE mice. Donors, n=10, females, 8–9 weeks old; recipients, n=5, females, 5–6 weeks old. (b) Quantification of total CNS infiltrating immune cells, (c) CD4+ and (d, e) CD4+IL-17+ cells from CNS of S1P1(S5A)and WT EAE mice (at peak disease, days 13–15). Immunoblot depicting p-STAT3 (f), p-p38, p38 and IκBα (h) expression in splenocytes of S1Ps1(S5A) and WT EAE mice (day8, post-immunization). (g) Quantification of relative p-STAT3 expression [form (f)]. (b-g) n=3–5 mice/arm. *p<0.05, Mann-Whitney U-test (a) and Student’s t-test (b–d). These experiments were repeated twice (a) and at least 3 times (b–g).
We then investigated the cellular mechanism of immune activation in S1P1(S5A) mice by profiling the immune cells in the peripheral immune system and CNS during EAE disease course. There was a trend towards higher splenocyte counts in S1P1(S5A) EAE mice compared to their wild-type counterparts (Supplementary Fig. 7a). However, the CNS of MOG-immunized S1P1(S5A) EAE mice contained significantly more infiltrating immune cells compared to the CNS of wild-type EAE mice (Fig. 3b) throughout the EAE disease course [pre-symptomatic (day 8), peak of disease (day 13) and plateau stage (day 16)](Supplementary Fig. 7b). Characterization by flow cytometric analysis utilizing CD45 staining suggested that these were indeed immune cells (Supplementary Fig. 7c). There were also higher number of myeloid lineage (CD45+CD11b+) cells suggesting enhanced immune cell trafficking and CNS inflammation in the S1P1(S5A) EAE mice (Supplementary Fig. 7d).
Further characterization of CNS-infiltrating immune cells demonstrated that there were significantly higher numbers of CD4+ T cells in the CNS of S1P1(S5A) mice (Fig. 3c). Many of these cells expressed IL-17 demonstrated by intracellular cytokine staining compared to their wild-type counter parts (Fig. 3d,e). However, there was no significant difference in IFN-γ production in the CD4+ T cells in the CNS of S1P1(S5A) and wild-type EAE mice, suggesting selective activation of pathways involved in TH17 differentiation (data not shown).
Nuclear factor kappa B (NF-κB), p38 mitogen-activated protein kinase (MAPK) and JAK-STAT pathways contribute to the proinflammatory responses in autoimmune neuroinflammation40,41. To investigate which of these pathways were activated in S1P1(S5A) EAE mice, we performed immunoblot analysis of splenocytes from MOG35–55-immunized, pre-symptomatic (day 8 post-immunization) wild-type and S1P1(S5A) EAE mice. We observed that there was significantly more STAT3 phosphorylation in the splenocytes of S1P1(S5A) EAE mice (Fig. 3f, top, Fig. 3g and Supplementary Fig. 8a, b).
We then investigated whether the surface residency of S1P1 had effects on other proinflammatory pathways involved in autoimmune neuroinflammation, such as the NF-κB and the p38 MAPK pathways. Immunoblot analysis of splenocytes from wild-type and S1P1(S5A) EAE mice revealed that neither p38, phosho-p38 nor IκBα expression were significantly altered in S1P1(S5A) EAE mice compared to their wild-type counterparts (Fig. 3h and Supplementary Fig. 8c–e). These findings strongly suggest that enhanced inflammatory response in S1P1(S5A) mice is likely due to the activation of IL-17–STAT3 signaling pathway.
S1P-S1P1 signaling directly activated TH17 polarization
We next addressed whether signaling via S1P1 was directly responsible for enhanced TH17 polarization in S1P1(S5A) EAE mice. We utilized several different approaches including in vitro activation with the physiological ligand, S1P;in vivo treatment of EAE mice with the sphingosine lyase inhibitor, 2-Acetyl-5-tetrahydroxybutyl Imidazole (THI); suppression of IL-17 expression by S1P1-specific inhibitor W146;S1pr1–specific siRNA knockdown experiments; and in vitro activation assays utilizing S1P1-deficient T cells26,42–44
First, we cultured splenocytes from MOG35–55-immunized wild-type and S1P1(S5A) EAE mice (day 8 post-immunization) in the presence or absence of the physiological ligand S1P and quantified STAT3 phosphorylation by immunoblot analysis. We observed substantial STAT3 phosphorylation in both S1P1(S5A) and wild-type EAE splenocytes (Fig. 4a,b), an effect decreased by incubation in the presence of S1P1 inhibitor W146.
Figure 4. S1P–S1P1 signaling activates STAT3 phosphorylation.
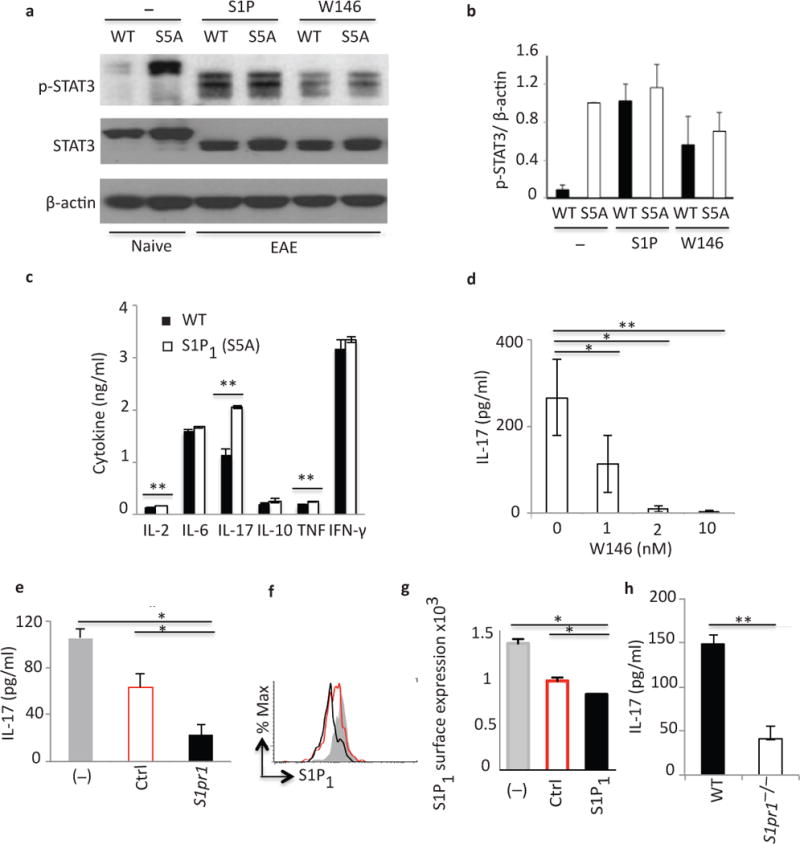
(a) Immunoblot depicting p-STAT3 expression in WT and S5A [S1P1(S5A)] EAE splenocytes (day 8 post-immunization) following in vitro activation with S1P (100nM) or W146 (S1P1 antagonist) (20nM). (b) Quantification of normalized p-STAT3 expression from (a). (c) Cytokine expression in splenocyte cultures of MOG35–55-immunized WT EAE mice treated in vivo with S1P lyase inhibitor, THI (6.25mg/kg, daily intraperitoneal injections). IL-17 expression in CD3+ cells (activated with anti-CD3/anti-CD28) from (d) S1P1(S5A) naive mice treated in vitro with W146 (0–10nM), (e) WT naïve mice, treated in vitro with scrambled control (Ctrl) or S1pr1-specific (S1pr1) siRNA, or (h) naïve S1pr1+/+(WT) or S1pr1−/− (S1pr1f/fRosa26-CreERT2) mice. (f and g) Flow cytometric analysis of S1P1 expression following treatment with S1pr1 or Ctrl siRNA treatment. *p<0.05, **p<0.01, Student’s t-test. c–e and h; analyzed by ELISA. (a, b, d–g) were performed 3–5 times, (c and h) twice. These experiments were performed with n=3–5 mice/arm. S1P; sphingosine-1-phosphate, W146; S1P1-specific antagonist, THI; 2-Acetyl-5-tetrahydroxybutyl Imidazole.
Next, we investigated whether enhanced S1P signaling promoted IL-17 production in vivo. S1P is metabolized into phosphatidylethanolamine and hexadecenal by S1P lyase (SL) under physiological conditions45. The SL inhibitor THI blocks this process, resulting in increased tissue concentrations of S1P and enhanced S1P1 signaling43. We treated MOG35–55-immunized wild-type EAE mice with THI or vehicle control in vivo, isolated splenocytes on day 8, cultured in the presence of MOG35–55 peptide and analyzed the culture supernatants by ELISA. We observed that splenocytes from THI-treated mice expressed high concentrations of inflammatory cytokines, especially IL-17, suggesting that exposure to high concentrations of S1P in vivo enhanced IL-17 expression (Fig. 4c). Since W146 treatment decreased STAT3 phosphorylation, we next determined whether W146 antagonism of S1P1 function could abrogate enhanced IL-17 expression in S1P1(S5A) mice. Activation of naïve S1P1(S5A) splenocytes in vitro with anti-CD3 and anti-CD28 in the presence of W146 significantly decreased IL-17 expression in a dose-dependent manner (Fig. 4d). Similar results were also obtained with wild-type CD3+ cells (data not shown).
Finally, we utilized genetic tools to investigate the connection between S1P1 signaling and the TH17 polarization. CD3+ T cells from naïve S1P1(S5A) mice were activated in vitro with anti-CD3 and anti-CD28 in the presence of S1pr1–specific siRNA. There was significantly less IL-17 expression in the culture supernatants when cells were incubated in the presence of S1pr1–specific siRNA (Fig. 4e–g). These findings were confirmed by experiments utilizing S1P1−/− T cells. CD3+ T cells from S1P1 conditional knock out mice (S1pr1f/f Rosa26-CreERT2; S1pr1f/f crossed with Rosa26-CreERT2 following tamoxifen treatment) activated in vitro with anti-CD3 and anti-CD28, expressed less IL-17 compared to CD3+ cells from their wild-type counterparts (Fig. 4h). Collectively, these data support the premise that S1P1 signaling directly modulates IL-17 expression.
S1P1-mediated TH17 polarization was IL-6-dependent
We next investigated whether T cells or antigen-presenting cells (APCs) were crucial for S1P1-mediated TH17 polarization. We performed a mixed-lymphocyte culture experiment where we co-cultured CD3+ cells from MOG35–55-immunized (day 8, pre-symptomatic) wild-type and S1P1(S5A) mice with naïve irradiated-wild-type or S1P1(S5A) APCs in the presence of MOG35–55 antigen. We then quantified IL-17 expression in the culture supernatant by ELISA. We observed that cultures containing either T cells, APCs or both belonging to S1P1(S5A) mice expressed significantly more IL-17 than those containing wild-type T cells and APCs (Fig. 5a). These findings suggest that S1P1(S5A) T cells were crucial, however, S1P1(S5A) APCs also acted synergistically to enhance IL-17 expression.
Figure 5. IL-6 is crucial for S1P1-mediated STAT3 activation.
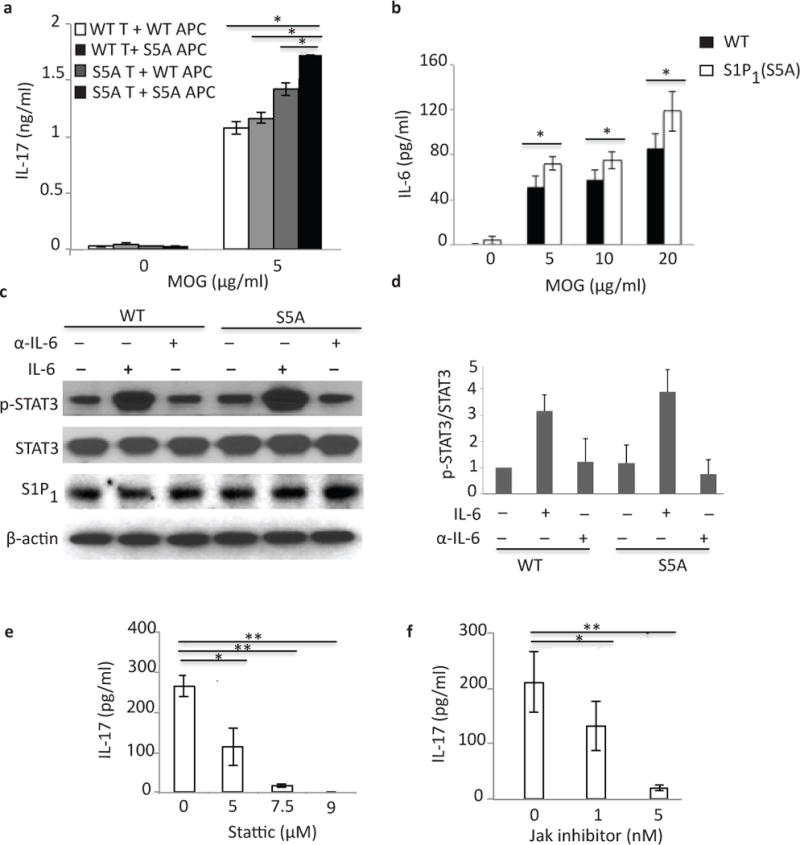
(a) IL-17 expression in culture supernatants of mixed lymphocyte cultures [CD3+ cells from MOG35–55-immunized S1P1(S5A) (S5A T) or C57BL/6J WT (WT T) EAE mice co-cultured with naïve, irradiated antigen presenting cells (APCs)from S1P1(S5A) (S5A APC) or WT (WT APC) in the presence of MOG35–55 peptide] measured by ELISA. (b) IL-6 expression in an ex vivo recall assay of splenocyte cultures from MOG35–55-imunized S5A [S1P1(S5A)]and WT EAE mice (day 8 post-immunization). (c) Immunoblot depicting p-STAT3 expression in splenocytes from WT and S1P1(S5A) EAE mice (day 8 post-immunization) treated in vitro with recombinant IL-6 (IL-6) or anti-IL-6 (α-IL-6) (20ng/ml, respectively). (d) Quantification of normalized p-STAT3 expression from (c). IL-17 expression in the culture supernatants of splenocyte cultures from MOG35–55-immunized S1P1(S5A) mice treated in vitro with Stattic (STAT3 inhibitor) (0–9 μM) (e) or Jak inhibitor (0–5nM)(f). **p<0.01, *p<0.05 by Students t-test. Experiments were performed 3–5 times with n=3–5 mice.
We then investigated which cytokines expressed by APCs might promote Th17 differentiation. We measured the elicited IL-1β, IL-6, IL-10 and TNF concentrations in the splenocyte cultures of S1P1(S5A) and wild-type EAE mice. We observed that IL-6 concentrations were significantly higher in the cultures from S1P1(S5A) mice compared to the wild-type counterpart (Fig. 5b). Expression of the other cytokines was not significantly different between the mutant and wild-type cells (data not shown).
IL-6 is a critical factor in TH17 polarization. To determine the role of IL-6 signaling S1P1(S5A) EAE mice, we treated splenocytes from MOG35–55-immunized wild-type and S1P1(S5A) pre-symptomatic EAE mice in vitro with recombinant IL-6 or IL-6 neutralizing antibody and performed immunoblot analysis. We observed that IL-6 treatment enhanced STAT3 phosphorylation, especially in the S1P1(S5A) splenocytes, which was reversed by the addition of anti-IL-6 (Fig. 5c,d). Finally, we observed that IL-17 expression from the splenocytes of S1P1(S5A) pre-symptomatic EAE mice were abolished by direct inhibition of STAT3 (with STAT3 inhibitor Stattic) or by upstream regulator of STAT3, JAK (Jak inhibitor) (Fig. 5e,f). Similar results were also obtained from wild-type splenocytes (data not shown). These data suggest that S1P1-mediated TH17 polarization was dependent on the IL-6-JAK-STAT3 pathway.
Discussion
Here, we characterize the MS brain lesion phosphoproteome from histologically defined brain tissue samples from MS patients. To the best of our knowledge, this is the first time such approach has been undertaken to illustrate dysregulated pathways in diseased tissue during MS pathogenesis. This unbiased approach has shed mechanistic insights on how S1P signaling modulates TH17 differentiation, a key determinant of disease severity in MS. This exemplifies how system-wide analysis of pathological samples could lead to the discovery of mechanistic underpinnings that could potentially guide patient care.
The question of whether S1P1 phosphorylation is detrimental or beneficial in the context of autoimmune demyelinating disease remains to be addressed. We observed enhanced S1P1 phosphorylation in the human MS brain lesions suggesting S1P1 phosphorylation correlates with worse CNS inflammation. However, worse EAE clinical phenotype and severe inflammation in the peripheral immune system and CNS in S1P1(S5A) EAE mice implies receptor phosphorylation is crucial for down regulating autoimmune neuroinflammation. One possible explanation is that phosphorylation of S1P1 is an attempt to down regulate TH17 differentiation as a compensatory mechanism to shut down neuroinflammation. Reciprocal activation of S1P and STAT3 pathways resulting in aggressive tumor progression has previously been reported but how this applies in the context of MS needs to be further investigated46. The downstream effects of S1P1 phosphorylation may also be cell type-specific. We observed S1P1 expression in the brain infiltrating T cells and astrocytes in active MS lesions. Our current study mainly focuses on the role of S1P signaling in the immune cells. Future studies will investigate the effects of S1P signaling in the CNS cells including astrocytes where S1P1 was also abundantly expressed.
Our findings suggest that the S1P1-STAT3 connection is at least in part dependent on IL-6 signaling. We have not yet explored how S1P signaling is linked to IL-6 expression. Our preliminary data suggest that S1P1 signaling does not seem to have major effects on the transcriptional regulation of IL-6 expression in T cells. Whether this is contributed by GPCR signaling and how S1P pathway cross talks with IL-6–JAK–STAT3 pathway remains to be investigated.
In summary, phosphoproteomic analysis of MS brain lesions unraveled the S1P1-STAT3 connection and that impaired S1P1 receptor internalization function may worsen autoimmune CNS inflammation. These findings beg several clinically relevant questions such as whether FTY-720 therapy modulates TH17 polarization and if there is synergism with arresting lymphocyte trafficking. Since an amino acid change (serine to alanine) in S1P1 leads to worse CNS inflammation in EAE, whether MS patients carrying variations in S1PR1 gene develop aggressive forms of MS and their response to S1P receptor modulators should also be investigated to prognosticate disease course and response to immune modulatory therapies.
Methods
Materials
All solvents and reagents were mass spectrometry or ACS grade from Fisher. The generation of S1P1(S5A) mice (C57BL/6J background) was previously described in25. Anti-CD3 (clone 145-2C11) and anti-CD28 (clone 37.51) were from eBioscience. Mouse recombinant IL-12 (419-ML) was from R&D Systems. Liberase enzyme cocktail was from Roche Biosciences. For flow cytometry, monoclonal antibodies against CD45 (clone 30-F11), CD3 (clone 145-2c11), CD8 (clone 53-6.7), CD11b (clone M1/70)and FoxP3 (clone MF23)were from BD Biosciences, and IL-17 (clone ebio17B7), CD4 (clone RM 4-5) and IFN-γ (clone XMG-1.2) were from eBioscience. Antibodies used for immunoblotting were S1P1/EDG-1 (sc25489) from Santa Cruz Biotechnology, pSTAT3 (#9131), STAT3 (#9132), IκBα (#9242), p38MAPK (#9212) from Cell Signaling, and β-actin (clone AC-74) from Sigma-Aldrich. Antibodies against CD3 (clone f.2.38) and GFAP (#z0334) for immunohistochemistry were from DakoCytomation28.
MS brain lesion phosphoproteome
MS brain lesion phosphoproteome was performed as previously described28,30. De-identified autopsy brain samples were obtained and archived according to the guidelines approved by the Stanford Institutional Review Board. Briefly, frozen MS (pooled from three MS samples) and control (pooled from two age-, sex-, and brain region-matched) samples (each with a wet weight of ~1g) were pulverized on dry ice in a dounce homogenizer in lysis buffer containing 8 M urea, 25 mM Tris-hydrochloride, pH8, 100 mM sodium chloride, 25 mM sodium fluoride, 10 mM sodium pyrophosphate, 50 mM β-glycerophosphate, 1 mM sodium orthovanadate, 1 mM EDTA-free protease inhibitor cocktail. The high-speed supernatant from tissue extract containing 20 mg total protein was subjected to reduction, alkylation followed by protease digestion with trypsin. Tryptic peptides were desalted with C18 SepPack extraction cartridges and phosphopeptide enrichment was performed. Peptides were subjected to offline HPLC separation on a strong cation exchange (SCX) column (polySULFEOTHYL A, 9.4 mm inner diameter, 200-mm length, 5 μM particle size, 200 Å pore size), desalted with C18 reverse phase SepPak cartridges, enriched on IMAC Select Affinity Gel (Sigma-Aldrich) and analyzed by nanoLC-MS/MS on an LTQ velos-Orbitrap (Thermo Fisher Scientific). Mass spectra were searched against a human IPI protein database (v 3.75) concatenated with a reversed database by the SEQUEST algorithm (Proteome Discoverer software v1.2, Thermo Scientific). Mass tolerance was setup as 10 ppm for precursor ions, 0.8 Da for fragment ions activated by CID, and 0.02 Da for fragment ions activated by HCD, allowing dynamic modification of Oxidation (15.995 Da) on methionine and phosphorylation (79.966 Da) on Ser, Thr and Tyr and static modification of carbamidomethyl (57.021 Da) on Cys. A maximum of four modifications per peptide was tolerated. Peptides identified were filtered based on Xcorr, ΔCn, peptide charge, peptide length and peptide rank to achieve a false positive rate (FDR) less than 0.6% for unique phosphopeptides.
Immunohistochemistry of MS brain lesions
Immunohistochemistry of formalin-fixed, paraffin-embedded section from active MS brain lesions and control brain samples were performed as previously described in Han et al. utilizing the following primary antibodies; anti-S1P1 (1:50), anti-CD3 (1:25) and anti-GFAP (1:500)47.
EAE, treatment with THI, immune cell proliferation and cytokine analysis
S1P1(S5A) mice and wild-type (WT) C57BL/6J mice were housed in the Research Animal Facility at Stanford University. Animal experiments were approved by, and performed in compliance with the National Institute of Health guidelines of the Institutional Animal Care and Use Committee at Stanford University.
EAE was induced in 8–9 weeks-old female WT or S1P1(S5A) mice (n = 10 per group) immunized with Complete Freund’s Adjuvant (CFA) and Myelin Oligoglycoprotein (MOG) peptide35–55 and Bordetella pertussis toxin (List Biological Laboratories) 400 ng per mouse on days 0 and 2 (ref. 28). For treatment with 2-Acetyl-5-tetrahydroxybutyl Imidazole (THI), the compound was dissolved in acidic aqueous solution, pH 3, and diluted 1:100 in PBS for dosing. C57BL/6J mice were administered 6.25 mg/kg THI intraperitoneal (i.p.) daily from the time of immunization up to day 8. EAE mice were scored according to: 0, normal; 1, tail paralysis; 2, hind limb weakness; 3, complete hind limb paralysis; 4, hind limb paralysis with forelimb weakness and 5, moribund or death28. Immune cell proliferation and cytokine analysis were performed as previously described28. Briefly, splenocytes and lymph node cells from WT and S1P1(S5A) mice were incubated in 96-well flat bottom plates in stimulation media and activated with increasing concentration of MOG35–55 peptide. For proliferation assays, cells were pulsed with 3[H]thymidine at 48 h and harvested 16 h later on a filter using a cell harvester. Incorporated radioactivity was counted using a beta scintillation counter. For cytokine analysis immune cells were incubated for 24, 48, 72, 96 and 120 h. and cytokine concentrations were measured at different time points utilizing anti-mouse ELISA kits (IL-2, IL-4, IL-6, IL-10, IL-12p40, IFN-γ; OptEIA, BD Pharmigen, IL-17, TNF; R&D Systems).
Adoptive transfer
WT C57BL/6J or S1P1(S5A) mice (female, 8–9 weeks old) were immunized with CFA and MOG35–55, splenocytes and lymph node cells were harvested on day 9 post-immunization and expanded in vitro with MOG 10 μg/ml and IL-12 (20 ng/ml)(R&D Systems) for 72 h47. Cells were then harvested, washed once with pre-warmed PBS, counted and injected into 5–6 weeks old Rag1−/− naïve recipient mice (1×107 cells per mouse) intraperitoneally and followed clinically up to at least day 30. Pertussis toxin (400 ng/dose) was administered by intraperitoneal injections on days 0 and 2.
Quantification of inflammatory foci in CNS tissue of EAE mice
To assess the degree of CNS inflammation, brain and spinal cords from WT C57BL/6J and S1P1(S5A) mice were harvested and fixed in formaldehyde. Paraffin-embedded sections were prepared as previously described28. Tissue sections were stained with Hematoxylin and Eosin and Luxol Fast Blue. An investigator blinded to the experimental groups quantified the numbers of CNS inflammatory foci.
Characterization of CNS immune cells in S1P1(S5A) EAE mice
Brain and spinal cord from S1P1(S5A) and WT C57BL/6J EAE mice (n = 3–5) were harvested on days 8, 13 and 16. Immune cells were isolated from CNS tissue according to methods outlined in Arac et al.48. Briefly, mice were perfused with 30 ml of cold saline after removal of the spleen. Brain and spinal cord were homogenized through a 70 μm filter, and treated with the liberase enzyme cocktail for 1 h at 37°C. Immune cells were harvested on a 30% Percoll gradient by centrifugation, and residual red blood cells were lysed by hypotonic solution. Cells were washed once in PBS and labeled with monoclonal antibodies against CD45, CD3, CD4, CD8b, and CD11b for flow cytometry48. Immune cells harvested from inflamed CNS tissue were also stimulated in vitro with 10 ng/ml phorbol 12-myristate, 13-acetate (PMA) and 1 μg/ml ionomycin in the presence of Golgi-stop (BD Biosciences) for 4 h. Cells were then labeled utilizing the BD Biosciences FoxP3 Staining Kit (BD Biosciences), anti-IL-17 and anti-IFN-γ (eBioscience). For flow cytometric analysis, 0.5–3 × 106 cells were washed twice with PBS, and resuspended in staining buffer [containing 3% Fetal Bovine Serum (FBS) in PBS] and labeled with respective antibodies for 30 min on ice. Cells were then washed twice with staining buffer and analyzed by LSRII Flow Cytometer (BD) at the Stanford Shared Flow Facility. BD FACSDiva 6.0 software was utilized to acquire data and FlowJo 7.6.2 was utilized for data analysis.
Immunoblot analysis
For immunoblot analysis, splenocytes from pre-symptomatic (day 8 post-immunization) S1P1(S5A) or C57BL/6J mice were harvested and activated in vitro under different conditions at 37 °C for 30 min. Cells were then washed once with PBS and harvested in lysis buffer supplemented with protease inhibitors (Roche Applied Sciences). The protein homogenate was quantified by using BCA assay (Thermo Scientific). A total of 20 μg protein was loaded on a 12% Novex NuPAGE pre-cast Bis-Tris Gel (Life Technologies) and resolved at 84 volts for 1.5 h. The gel was transferred onto a PVDF membrane (Sigma-Aldrich), blocked with 3% BSA in TBST at 25 °C for 1 h and probed with primary antibodies for p-STAT3, STAT3, p-p38, p38, IκBα (Cell Signaling), S1P1 (Abcam), and β-actin (Sigma-Aldrich) at 4 °C overnight. The blot was then incubated with respective horseradish peroxidase linked secondary antibodies at 25 °C for 1 h and developed using ECL Chemiluminescence kit (GE Healthcare).
For quantification of protein expression, bands on the X-ray film corresponding to the respective proteins were scanned utilizing a HP laser scanner. Protein expression was normalized by dividing with the densitometric units corresponding to the protein of interest with that of β-actin from the same sample and plotted in a bar graph. The experiment was repeated at least three times for each analysis.
S1pr1 siRNA gene knockdown assay
For gene knockdown assays, S1pr1 siRNA was purchased from Dharmacon. CD3+ cells were enriched from naïve S1P1(S5A) or WT C57BL/6J splenocytes and electroporated using the Neon transfection system (Life Technologies) in the presence of S1pr1 siRNA (20 nM). Cells were activated with anti-CD3 plus anti-CD28 (0.5 μg/ml and 0.25 μg/ml, respectively) at 37 °C for 24 h. Down regulation of S1pr1 transcript abundance was verified by reverse transcriptase and polymerase chain reaction after normalization with actin transcripts. Cell supernatants from transfected cultures transfected were harvested and assayed for IL-17 titers by ELISA at 96 h.
In vitro activation assay utilizing S1pr1−/− T cells for IL-17 expression
S1pr1f/f mice were crossed with Rosa26-CreERT2 mice to generate S1pr1f/f Rosa26-CreERT2 mice44. S1pr1f/f ± Rosa26-CreERT2 mice were treated with 4 mg tamoxifen per oral (p. o.) in 200 μl of 100% corn oil for two consecutive days. Eight to ten days after the second tamoxifen treatment, mice were sacrificed. The presence of Cre recombinase and deletion of the floxed S1pr1 gene were verified by polymerase chain reaction. 96-well plates were coated with 50 μl of 1 mg/ml anti-CD3 and 0.5 mg/ml anti-CD28. Control lanes were filled with 50 μl of PBS alone. Plates were kept at 4°C until use. Anti-CD3 and anti-CD28 cocktail was removed and wells washed twice with 100 μl PBS, then blocked at 25 °C for 20 min with complete medium: 100 μl 10% FBS in DMEM.
CD3+ T cells were isolated from splenocytes by negative selection utilizing the Stem Cell Technologies Easy Sep mouse T cell enrichment kit and plated at 1 × 106/ml in total volume of 200 μl complete medium/well in duplicate wells. Cells were harvested at 48, 72 and 96 h, in 0.5 ml tubes, spun at 800g for 5 min at 4°C. Supernatants were removed to new tubes and stored at −80°C until ready for analysis by ELISA.
Stattic, Jak inhibitor and W146 treatment
For in vitro inhibition assays, splenocytes from MOG-immunized, pre-symptomatic (day 8) S1P1(S5A) and WT EAE mice were activated with MOG35–55 (10 μg/ml) in the presence of Stattic (1–9 μM) or Jak inhibitor I (1–5 nM) for 24–96 h. For W146 antagonism, CD3+ cells from S1P1(S5A) and WT were activated with anti-CD3 and anti-CD28 (1 mg/ml and 0.5 mg/ml respectively) in the presence of W146 (0–10nM) for 24–96 h. Culture supernatants were assayed for IL-17 concentrations by ELISA.
Mixed lymphocyte assay
For lymphocyte mixing experiments, CD3+ T lymphocytes from WT C57BL/6J or S1P1(S5A) mice immunized with CFA and MOG35–55 were harvested on day 8 post-immunization following negative affinity selections by anti-CD3 affinity purification47. WT or S1P1(S5A) CD3+ cells were incubated in 96-well round-bottom plates (1 × 106 cells per well) with WT or S1P1(S5A) irradiated splenocytes (3,000 RADS, 5 × 106 cells per well) for 24–72 h and IL-17 expression in the culture supernatants was measured by ELISA.
Statistical Analysis
Data represent means +/− s.e.m. Mann-Whitney U-test for EAE and Student’s t-test were performed to detect between group-differences. A p value of <0.05 was rendered statistically significant.
Supplementary Material
Acknowledgments
We thank J. Saba (Children’s Hospital Oakland Research Institute) for providing 2-Acetyl-5-tetrahydroxybutyl Imidazole (THI). This work was supported by Neurology Department Startup Funds, Guthy-Jackson Charitable Foundation for Neuromyelitis Optica Research (M. H. H), NIH R37-HL R37-HL67330, PO1-HL70694 and RO1HL89934 (T. H.).
Footnotes
Author contributions
C.S. G. and M.H. H. formulated the hypothesis and designed all experiments. L. W., M. P. S. and D. K. H contributed to the phosphoproteomic analysis. V. A. B. performed S1P1−/− in vitro experiment and T. H. contributed to experiments related to S1P signaling. S. A. and D. B. L. assisted with siRNA experiments, R. A. S. with the histopathological studies, and A. A. and G. K. S. with intracellular cytokine staining and flow cytometry. R. C. A., P. P. H. and L. S. contributed to the EAE-related experiments. Y. H. and B. S. M. performed immunoblots and in vitro assays.
Competing financial interests
Authors declare no competing financial interests related to this manuscript.
References
- 1.Baumruker T, Billich A, Brinkmann V. FTY720, an immunomodulatory sphingolipid mimetic: translation of a novel mechanism into clinical benefit in multiple sclerosis. Expert Opin Investig Drugs. 2007;16:283–289. doi: 10.1517/13543784.16.3.283. [DOI] [PubMed] [Google Scholar]
- 2.Kappos L, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- 3.Cohen JA, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362:402–415. doi: 10.1056/NEJMoa0907839. [DOI] [PubMed] [Google Scholar]
- 4.Devonshire V, et al. Relapse and disability outcomes in patients with multiple sclerosis treated with fingolimod: subgroup analyses of the double-blind, randomised, placebo-controlled FREEDOMS study. Lancet Neurol. 2012;11:420–428. doi: 10.1016/S1474-4422(12)70056-X. [DOI] [PubMed] [Google Scholar]
- 5.Matloubian M, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 6.Chi H, Flavell RA. Cutting edge: regulation of T cell trafficking and primary immune responses by sphingosine 1-phosphate receptor 1. J Immunol. 2005;174:2485–2488. doi: 10.4049/jimmunol.174.5.2485. [DOI] [PubMed] [Google Scholar]
- 7.Rosen H, Goetzl EJ. Sphingosine 1-phosphate and its receptors: an autocrine and paracrine network. Nat Rev Immunol. 2005;5:560–570. doi: 10.1038/nri1650. [DOI] [PubMed] [Google Scholar]
- 8.Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol. 2012;30:69–94. doi: 10.1146/annurev-immunol-020711-075011. [DOI] [PubMed] [Google Scholar]
- 9.Zhi L, et al. FTY720 blocks egress of T cells in part by abrogation of their adhesion on the lymph node sinus. J Immunol. 2011;187:2244–2251. doi: 10.4049/jimmunol.1100670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rivera J, Proia RL, Olivera A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat Rev Immunol. 2008;8:753–763. doi: 10.1038/nri2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allende ML, et al. Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J Biol Chem. 2004;279:52487–52492. doi: 10.1074/jbc.M406512200. [DOI] [PubMed] [Google Scholar]
- 12.Halin C, et al. The S1P-analog FTY720 differentially modulates T-cell homing via HEV: T-cell-expressed S1P1 amplifies integrin activation in peripheral lymph nodes but not in Peyer patches. Blood. 2005;106:1314–1322. doi: 10.1182/blood-2004-09-3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oo ML, et al. Immunosuppressive and anti-angiogenic sphingosine 1-phosphate receptor-1 agonists induce ubiquitinylation and proteasomal degradation of the receptor. J Biol Chem. 2007;282:9082–9089. doi: 10.1074/jbc.M610318200. [DOI] [PubMed] [Google Scholar]
- 14.Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol. 2010;33:91–101. doi: 10.1097/WNF.0b013e3181cbf825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ingwersen J, et al. Fingolimod in multiple sclerosis: mechanisms of action and clinical efficacy. Clin Immunol. 2012;142:15–24. doi: 10.1016/j.clim.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 16.Cohen JA, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362:402–415. doi: 10.1056/NEJMoa0907839. [DOI] [PubMed] [Google Scholar]
- 17.Waubant E. Emerging therapies for MS. Rev Neurol (Paris) 2007;163:688–696. doi: 10.1016/s0035-3787(07)90481-6. [DOI] [PubMed] [Google Scholar]
- 18.Kieseier BC, Wiendl H, Hemmer B, Hartung HP. Treatment and treatment trials in multiple sclerosis. Curr Opin Neurol. 2007;20:286–293. doi: 10.1097/WCO.0b013e3281568b80. [DOI] [PubMed] [Google Scholar]
- 19.Jander S, Turowski B, Kieseier BC, Hartung HP. Emerging tumefactive multiple sclerosis after switching therapy from natalizumab to fingolimod. Mult Scler. 2012;18:1650–1652. doi: 10.1177/1352458512463768. [DOI] [PubMed] [Google Scholar]
- 20.Bourdette D, Gilden D. Fingolimod and multiple sclerosis: four cautionary tales. Neurology. 2012;79:1942–1943. doi: 10.1212/WNL.0b013e3182735edf. [DOI] [PubMed] [Google Scholar]
- 21.Visser F, Wattjes MP, Pouwels PJ, Linssen WH, van Oosten BW. Tumefactive multiple sclerosis lesions under fingolimod treatment. Neurology. 2012;79:2000–2003. doi: 10.1212/WNL.0b013e3182735cb3. [DOI] [PubMed] [Google Scholar]
- 22.Liu CH, et al. Ligand-induced trafficking of the sphingosine-1-phosphate receptor EDG-1. Mol Biol Cell. 1999;10:1179–1190. doi: 10.1091/mbc.10.4.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kohno T, Igarashi Y. N-glycosylation of sphingosine 1-phosphate receptor, Edg-1, and its role on receptor internalization through membrane microdomain. Tanpakushitsu Kakusan Koso. 2002;47:503–508. [PubMed] [Google Scholar]
- 24.Watterson KR, et al. Dual regulation of EDG1/S1P(1) receptor phosphorylation and internalization by protein kinase C and G-protein-coupled receptor kinase 2. J Biol Chem. 2002;277:5767–5777. doi: 10.1074/jbc.M110647200. [DOI] [PubMed] [Google Scholar]
- 25.Thangada S, et al. Cell-surface residence of sphingosine 1-phosphate receptor 1 on lymphocytes determines lymphocyte egress kinetics. J Exp Med. 2010;207:1475–1483. doi: 10.1084/jem.20091343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oo ML, et al. Engagement of S1P(1)-degradative mechanisms leads to vascular leak in mice. J Clin Invest. 2011;121:2290–2300. doi: 10.1172/JCI45403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arnon TI, et al. GRK2-dependent S1P1 desensitization is required for lymphocytes to overcome their attraction to blood. Science. 2011;333:1898–1903. doi: 10.1126/science.1208248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han MH, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. 2008;451:1076–1081. doi: 10.1038/nature06559. [DOI] [PubMed] [Google Scholar]
- 29.Lock C, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–508. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- 30.Huttlin EL, et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng J, Elias JE, Thoreen CC, Licklider LJ, Gygi SP. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J Proteome Res. 2003;2:43–50. doi: 10.1021/pr025556v. [DOI] [PubMed] [Google Scholar]
- 32.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 33.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krebs EG, Kent AB, Fischer EH. The muscle phosphorylase b kinase reaction. J Biol Chem. 1958;231:73–83. [PubMed] [Google Scholar]
- 35.Mayya V, Rezual K, Wu L, Fong MB, Han DK. Absolute quantification of multisite phosphorylation by selective reaction monitoring mass spectrometry: determination of inhibitory phosphorylation status of cyclin-dependent kinases. Mol Cell Proteomics. 2006;5:1146–1157. doi: 10.1074/mcp.T500029-MCP200. [DOI] [PubMed] [Google Scholar]
- 36.Chun J, Brinkmann V. A mechanistically novel, first oral therapy for multiple sclerosis: the development of fingolimod (FTY720, Gilenya) Discov Med. 2011;12:213–228. [PMC free article] [PubMed] [Google Scholar]
- 37.Hla T, Brinkmann V. Sphingosine 1-phosphate (S1P): Physiology and the effects of S1P receptor modulation. Neurology. 2011;76:S3–8. doi: 10.1212/WNL.0b013e31820d5ec1. [DOI] [PubMed] [Google Scholar]
- 38.Loh KC, et al. Sphingosine-1-phosphate enhances satellite cell activation in dystrophic muscles through a S1PR2/STAT3 signaling pathway. PLoS One. 2012;7:e37218. doi: 10.1371/journal.pone.0037218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosen H, Sanna MG, Cahalan SM, Gonzalez-Cabrera PJ. Tipping the gatekeeper: S1P regulation of endothelial barrier function. Trends Immunol. 2007;28:102–107. doi: 10.1016/j.it.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 40.Lovett-Racke AE, Yang Y, Racke MK. Th1 versus Th17: are T cell cytokines relevant in multiple sclerosis? Biochim Biophys Acta. 2011;1812:246–251. doi: 10.1016/j.bbadis.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Youssef S, Steinman L. At once harmful and beneficial: the dual properties of NF-kappaB. Nat Immunol. 2006;7:901–902. doi: 10.1038/ni0906-901. [DOI] [PubMed] [Google Scholar]
- 42.Lee MJ, et al. Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science. 1998;279:1552–1555. doi: 10.1126/science.279.5356.1552. [DOI] [PubMed] [Google Scholar]
- 43.Schwab SR, et al. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science. 2005;309:1735–1739. doi: 10.1126/science.1113640. [DOI] [PubMed] [Google Scholar]
- 44.Allende ML, Dreier JL, Mandala S, Proia RL. Expression of the sphingosine 1-phosphate receptor, S1P1, on T-cells controls thymic emigration. J Biol Chem. 2004;279:15396–15401. doi: 10.1074/jbc.M314291200. [DOI] [PubMed] [Google Scholar]
- 45.Zhou J, Saba JD. Identification of the first mammalian sphingosine phosphate lyase gene and its functional expression in yeast. Biochem Biophys Res Commun. 1998;242:502–507. doi: 10.1006/bbrc.1997.7993. [DOI] [PubMed] [Google Scholar]
- 46.Lee H, et al. STAT3-induced S1P1 expression is crucial for persistent STAT3 activation in tumors. Nat Med. 2010;16:1421–1428. doi: 10.1038/nm.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Han MH, et al. Janus-like opposing roles of CD47 in autoimmune brain inflammation in humans and mice. J Exp Med. 2012;209:1325–1334. doi: 10.1084/jem.20101974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arac A, et al. Systemic augmentation of alphaB-crystallin provides therapeutic benefit twelve hours post-stroke onset via immune modulation. Proc Natl Acad Sci USA. 2011;108:13287–13292. doi: 10.1073/pnas.1107368108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.