

The 1.60 Å resolution crystal structure of the aldo-keto reductase Rv2971, an isoniazid drug target in M. tuberculosis, reveals unique characteristics of the isoniazid–NADPH binding pocket.
Keywords: Rv2971, AKR5H1, Mycobacterium tuberculosis, isoniazid
Abstract
Aldo-keto reductases (AKR) are a large superfamily of NADPH-dependent oxidoreductases and play a role in detoxification of toxic metabolites. Rv2971, an AKR in Mycobacterium tuberculosis, has recently been identified as a target of isoniazid, a key first-line drug against tuberculosis. Here, the cloning, expression, purification, crystallization and structural characterization of Rv2971 are described. To gain insight into its function, the crystal structure of Rv2971 was successfully determined to 1.60 Å resolution in its unliganded form. The structure exhibits a TIM-barrel fold typical of AKRs, revealing structural characteristics essential for function and substrate specificities, allowing a structural comparison between Rv2971 and other mycobacterial AKRs.
1. Introduction
Mycobacterium tuberculosis, the aetiological agent of tuberculosis (TB), remains a major health burden globally, resulting in approximately 1.4 million deaths annually (World Health Organization, 2013 ▶). The emergence of drug-resistant strains underlines the urgent need for the development of new anti-mycobacterial therapeutics. Multiple drug resistant strains of TB (MDR-TB), and more recently extensively drug-resistant strains of TB (XDR-TB), are classified as expressing resistance to at least two of the first-line anti-TB drugs and a number of second-line anti-TB drugs, resulting in a limited means of treating diseased patients.
The key first-line drug in TB treatment is isoniazid (INH), a hydrazide prodrug activated by the catalase-peroxidase KatG to produce INH–NAD(P) adducts that exhibit potent anti-mycobacterial activity (Rozwarski et al., 1998 ▶; Timmins & Deretic, 2006 ▶). The primary target of the INH–NAD(P) adduct is InhA, an enoyl-acyl carrier protein reductase that plays an essential role in mycolic acid biosynthesis (Dessen et al., 1995 ▶). In addition to InhA, the drug acts against 17 recently identified targets within M. tuberculosis (Argyrou et al., 2006 ▶). These targets exhibit a variety of essential roles implemented in the fitness and survival of the mycobacterium, and provide a variety of potential candidates for the development of new anti-TB therapeutics.
One of the targets identified is the essential aldo-keto reductase (AKR) AKR5H1 encoded by the gene rv2971. rv2971 has previously been characterized as an essential gene for growth and survival in M. tuberculosis (Sassetti et al., 2003 ▶) with a potential role in the detoxification of toxic metabolites (Grimshaw, 1992 ▶; Grant et al., 2003 ▶; Penning & Drury, 2007 ▶). Recently, the catalytic activity of Rv2971 and its M. smegmatis orthologue MSMEG_2407 (67% sequence identity) has been characterized, revealing differences in dicarbonyl substrate specificities and affinities (Scoble et al., 2010 ▶). The potency of INH was characterized against both enzymes, revealing a higher affinity against MSMEG_2407 compared with Rv2971, with apparent K i values of 6.0 ± 1.2 and 31.0 ± 1.4 µM reported, respectively (Scoble et al., 2010 ▶).
To gain functional insight into the role of AKR5H1, the crystal structure of MSMEG_2407 has been determined in its apo form and in its holoenzyme form in the presence of NADPH (Scoble et al., 2010 ▶). The crystal structure reveals an (α/β)8-barrel topology, or a TIM-barrel fold, typical of other AKRs as previously described (Banner et al., 1975 ▶), and enabled residues essential for enzyme kinetic activity and cofactor binding to be ascertained. While the crystal structure of MSMEG_2407 provides invaluable information on its functionality, Rv2971 possesses a number of differences in these binding pockets.
Here, we determined the crystal structure of Rv2971 in its unliganded form to a resolution of 1.60 Å. The structure enables the characterization of the substrate and NADPH binding pockets, revealing the subtle architectural differences between Rv2971 and MSMEG_2407, and providing insight into the differences in substrate specificities and INH inhibition levels. In addition, the Rv2971 structure may provide an initial template for the development of new anti-mycobacterial therapeutics.
2. Materials and methods
2.1. Cloning, expression and purification of recombinant protein
The gene encoding Rv2971 was cloned, overexpressed and purified as soluble recombinant protein as previously described (Scoble et al., 2010 ▶). For crystallization experiments, the purified recombinant protein was buffer-exchanged into 10 mM Tris–HCl pH 8.0, 200 mM NaCl and concentrated to 7 mg ml−1. The concentration of purified Rv2971 was determined spectrophotometrically (NanoDrop 1000, Thermo Scientific) at 280 nm and was calculated using an extinction coefficient of 28 545 M −1 cm−1. The molecular weight, purity and identity of the protein were confirmed by SDS–PAGE and Western blotting with anti-hexahistidine antibody (R&D Systems).
2.2. Crystallization
The initial crystallization experiments involved screening 192 conditions from commercially available kits from Hampton Research (Crystal Screen HT and PEG/Ion HT) using a CrystalMation (Rigaku) integrated robotic workstation and the sitting-drop vapour-diffusion technique. Rod-shaped crystals were obtained initially at a protein concentration of 7 mg ml−1 at 277 K after 24 h of equilibration against a crystallization condition comprised of 0.2 M sodium malonate pH 5.0, 20%(w/v) polyethylene glycol (PEG) 3350. Larger crystals of high diffraction quality were obtained by optimizing the crystal condition in 24-well Linbro plates (Hampton Research). The crystallization condition was optimized by fine-tuning the pH, protein concentration and precipitant concentration using a hanging drop consisting of 1 µl protein solution and 1 µl precipitant solution and a 500 µl reservoir volume (Fig. 1 ▶ a). The best crystals appeared after 24 h of equilibration against a crystallization condition comprised of 0.2 M sodium malonate pH 5.0, 19%(w/v) PEG 3350 at a protein concentration of 7 mg ml−1 and grew to full size in 2 d.
Figure 1.

Crystallization and data collection of Rv2971. (a) Crystal of Rv2971 obtained from 7 mg ml−1 protein solution (in 10 mM Tris–HCl pH 8.0, 200 mM NaCl) using 0.2 M sodium malonate pH 5.0, 19%(w/v) PEG 3350. (b) A typical 0.50° oscillation image from an Rv2971 crystal flash-cooled in 15%(v/v) glycerol.
2.3. X-ray data collection
For X-ray diffraction data collection, crystals of Rv2971 were transferred to a CryoLoop and soaked in a cryoprotectant consisting of 19%(v/v) PEG 3350, 0.2 M sodium malonate pH 5.0, 15%(w/v) glycerol before cooling to 100 K in a nitrogen-gas stream. A complete data set was collected from a single crystal on the MX2 beamline at the Australian Synchrotron using an ADSC Quantum 315rr CCD detector. A total of 360 frames of 0.5° were recorded with an exposure time of 1 s per frame (Fig. 1 ▶ b). The data were processed using iMosflm (Battye et al., 2011 ▶) and various programs from the CCP4 suite such as SCALA and POINTLESS (Winn et al., 2011 ▶). The final statistics of data collection and processing are summarized in Table 1 ▶.
Table 1. Data-collection and refinement statistics for Rv2971.
Values in parentheses are indicative of the highest resolution shell.
| Data collection | |
| Diffraction source | MX2, Australian Synchrotron |
| Detector | ADSC Quantum 315r CCD |
| Space group | P3221 |
| Unit-cell parameters (Å, °) | a = b = 86.27, c = 86.29, α = 90.0 β = 90.0 γ = 120.0 |
| Wavelength (Å) | 0.95370 |
| Resolution range (Å) | 38.58–1.60 (1.66–1.60) |
| Total No. of reflections | 525473 (76789) |
| No. of unique reflections | 49373 (4857) |
| Completeness (%) | 100.0 (100.0) |
| Mean I/σ(I)† | 12.72 (2.35) |
| R merge ‡ (%) | 11.4 (91.7) |
| R p.i.m. ‡ (%) | 3.7 (29.1) |
| Multiplicity | 10.6 (10.8) |
| Structural refinement | |
| Resolution range (Å) | 38.59–1.60 (1.63–1.60) |
| R work § (%) | 13.60 (22.67) |
| R free § (%) | 16.90 (27.59) |
| R.m.s.d.,¶ bond lengths (Å) | 0.006 |
| R.m.s.d.,¶ bond angles (°) | 1.05 |
| Ramachandran plot | |
| Favoured (%) | 98.20 |
| Allowed (%) | 1.80 |
| Outliers (%) | 0.00 |
| No. of modelled non-H atoms | |
| Macromolecules | 2128 |
| Ligands | 15 |
| Water | 311 |
| Protein residues | 276 |
| Wilson B factor (Å2) | 18.51 |
| Average B factors (Å2) | |
| Protein atoms | 20.40 |
| Ligand molecules | 20.40 |
| Water molecules | 33.70 |
| PDB code | 4otk |
I is the integrated intensity and σ(I) is the estimated standard deviation of that intensity.
R
merge = 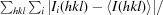
 where Ii(hkl) is the scaled intensity of the ith measurement and 〈I(hkl)〉 is the mean intensity for that reflection. R
p.i.m. = R
merge divided by the multiplicity.
where Ii(hkl) is the scaled intensity of the ith measurement and 〈I(hkl)〉 is the mean intensity for that reflection. R
p.i.m. = R
merge divided by the multiplicity.
R
work = 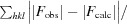
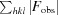 for all data excluding the 5% that comprise the R
free used for cross-validation.
for all data excluding the 5% that comprise the R
free used for cross-validation.
Root-mean-square deviation from ideal values (Engh & Huber, 1991 ▶).
2.4. Structural determination
The crystal structure of Rv2971 was determined by the molecular-replacement method with Phaser-MR as part of the PHENIX program suite (Adams et al., 2010 ▶). The coordinates of M. smegmatis AKR5H1 MSMEG_2407 (67% sequence identity; PDB entry 2wzm; Scoble et al., 2010 ▶) were used as the search model. The resultant LLG score and TZF score obtained were 2010.15 and 43.8, respectively. For cross-validation, a random set of 5% of the total reflections were kept aside from the refinement and used for the calculation of R free (Brünger, 1992 ▶). The initial model was refined as a rigid body followed by TLS refinement using TLS groups as suggested by the TLSMD server (Painter & Merritt, 2006 ▶). The TLS refinement was performed according to four TLS groups, which are A8–A171, A172–A188, A189–A257 and A258–A283. The model obtained was manually adjusted using the interactive graphics program Coot (Emsley et al., 2010 ▶) and refined using phenix.refine (Adams et al., 2010 ▶) until no further improvement of the model could be made. Atomic coordinates and structure factors have been deposited in the Protein Data Bank under accession code 4otk. The final statistics of structural refinement are summarized in Table 1 ▶.
2.5. Structural analysis
Refinement validation was conducted using MolProbity and the POLYGON tools in the PHENIX program suite (Adams et al., 2010 ▶; Chen et al., 2010 ▶). Secondary structure was confirmed by the STRIDE plugin in PyMOL (Zhu, 2011 ▶). All structural superpositions were achieved using the SSM superpose feature of Coot (Emsley et al., 2010 ▶). Electrostatic surface distribution calculations were performed using the APBS plugin for PyMOL (Baker et al., 2001 ▶). The M. smegmatis AKRH5 MSMEG_2407 holoenzyme crystal structure (PDB entry 2wzm) and the apo crystal structure (PDB entry 2wzt; Scoble et al., 2010 ▶) were used for structural alignments.
3. Results and discussion
3.1. Structural determination of Rv2971
To gain structural insight into the isoniazid drug target Rv2971, the crystal structure of the M. tuberculosis protein was determined. Recombinant M. tuberculosis Rv2971 was expressed, purified and crystallized using vapour-diffusion methods (Fig. 1 ▶ a). Rv2971 was purified as a monomer as determined by size-exclusion chromatography, and routinely produced yields of 2 mg of pure protein per litre. Crystals belonged to the trigonal space group P3221, with unit-cell parameters a = b = c = 86.3 Å, α = β = 90.0°, γ = 120.0°. A complete data set was collected to a resolution of 1.60 Å from a single crystal at 100 K (Fig. 1 ▶ b), with data collection and processing summarized in Table 1 ▶. Based on Matthews coefficient calculations, one polypeptide chain (59.96% solvent content) could be accommodated in the asymmetric unit, with a calculated V M value of 3.07 Å3 Da−1 (Matthews, 1968 ▶).
The crystal structure of Rv2971 was determined to 1.60 Å resolution via molecular replacement using the M. smegmatis orthologue, MSMEG_2407, as a search model (PDB entry 2wzm), with final R work and R free statistics of 13.90 and 16.90%, respectively. Refinement statistics are summarized in Table 1 ▶. Of the construct utilized in crystallization, the large vector-derived N-terminal domain (MGSSH6SSGLVPRGSHMASMTGGQQMGRGSEF) was unstructured and residues 7–282 of Rv2971 were modelled into the electron density. The final model consisted of 276 amino-acid residues and 311 water molecules. The final model also included two malonate ions and two chloride ions originating from the crystallization condition and crystallization buffer, respectively. A Ramachandran plot analysis of the final model by Cα geometry validation (Lovell et al., 2003 ▶) showed that 98.2% of the residues are in the most favoured regions, while 1.8% of residues are within the allowed regions.
3.2. Overall crystal structure of Rv2971
The overall 1.60 Å resolution crystal structure of Rv2971 in its unliganded form (Figs. 2 ▶ a and 2 ▶ b) adopts an (α/β)8-barrel topology, or a TIM-barrel fold, as previously described (Banner et al., 1975 ▶). The structural fold exhibits typical characteristics of AKR substrates, where the TIM barrel is comprised of eight β-strands (β1–β8), flanked by eight α-helices (α1–α8) interwoven in an antiparallel manner between each β-strand. Situated at the N-terminus of the structure are two antiparallel β-stands (βA and βB) that cover the base of the protein structure (Fig. 2 ▶ b). The TIM-barrel structure is punctuated by two α-helices (αA and αB) on the C-terminal side of the protein structure (Fig. 2 ▶ a). Dispersed through the C-terminal side, or ‘open rim’ face, of the protein structure are a series of 310-helices (h1, h3, h4 and hA) that contribute to the overall α-helical content of the Rv2971 structure by forming the NADPH binding pocket.
Figure 2.

Overall crystal structure of Rv2971. Cartoon representation of the (a) top view and (b) side view rotated 90° of the overall structure. The C- and N-termini of the structure are labelled, with secondary-structure elements β-strands, α-helices and 310-helices, as calculated by STRIDE (Zhu, 2011 ▶), coloured magenta, blue and cyan, respectively. All figures were prepared in PyMOL.
3.3. Substrate and NADPH binding pockets of Rv2971
The crystal structure of Rv2971 was determined in the presence of two malonate ions bound within the substrate binding pocket and NADPH binding pocket. Attempts to soak NADPH into the crystals were unsuccessful because of the presence of the malonate ions blocking the binding pockets. Within the substrate binding pocket, the malonate ion is bound to Rv2971 via hydrogen bonding with Asp52 and His115 at a bond distance of 2.6 and 2.9 Å, respectively (Fig. 3 ▶ a). Residues Asp52 and His115 belong to the Asp-Tyr-Lys-His catalytic tetrad typical of AKRs. In the case of Rv2971, the catalytic tetrad is comprised of residues Asp52-Tyr57-Lys82-His115. A single chloride ion is bound between the substrate and NADPH binding pockets, and is bound to residue Leu197 via hydrogen bonding (Fig. 3 ▶ b). A second malonate ion is bound within the NADPH pocket, binding residues involved in contacts with NADPH via hydrogen-bond formation, with bond distances ranging between 3.0 and 3.6 Å (Fig. 3 ▶ b). Both the substrate and NADPH binding pockets are electrostatically positively charged (Figs. 3 ▶ c and 3 ▶ d) which, as well as being highly surface exposed, facilitates the substrate and NADPH binding capabilities of Rv2971.
Figure 3.

Rv2971 substrate and cofactor binding pocket architecture. Cartoon representation of (a) the substrate binding pocket and (b) the NADPH binding pocket. Two malonate ions and a single chloride ion bound during crystallization are represented as cyan sticks and magenta spheres, respectively. Electron density of maps is shown as F o − F c simulated-annealing OMIT maps contoured at 3σ. Amino-acid residues involved in binding to the malonate and chloride ions are represented as green sticks. Hydrogen-bond formation between contact residues and ions is represented as black dashes, with bond distances ranging between 2.6 and 3.6 Å. The Asp52-Tyr57-Lys82-His115 catalytic tetrad is represented in wheat. The solvent-accessible surface representation coloured by electrostatic potential of the (c) substrate binding pocket and (d) NADPH binding pocket was calculated by APBS (Baker et al., 2001 ▶). The potential contours are shown on a scale from +5.0 (blue) to −5.0 k B T e−1 (red); white indicates a value close to 0 k B T e−1. Indicated are the positions of the Asp52-Tyr57-Lys82-His115 catalytic tetrad.
3.4. Structural comparison with the M. smegmatis orthologue MSMEG_2407
With the successful determination of the crystal structure of Rv2971, a structural comparison was conducted with MSMEG_2407 to characterize the previously observed differences in enzyme kinetic activity (Scoble et al., 2010 ▶). The crystal structures of Rv2971 and the apo form of MSMEG_2407 overlaid tightly, with an overall r.m.s.d. value of 0.85 Å observed. While Rv2971 and MSMEG_2407 share a sequence identity of 67%, a number of substitutions are present between amino-acid residues within the NADPH binding pocket that impact on the enzyme kinetic activity rates between the orthologues (Fig. 4 ▶). Of the 21 amino-acid residues from MSMEG_2407 interacting with NADPH, there is a difference of four residues in Rv2971. Residues Gly30, Gly196, Leu200 and Ile236 in MSMEG_2407 are substituted with residues Ala29, Cys195, Val199 and Val235 (Fig. 4 ▶). The most significant difference is the substitution of a glycine (Gly196) for cysteine (Cys195). Cys195 is present in two alternate conformations in the structure of Rv2971, both facing into the NADPH binding pocket. One of the conformations forms van der Waals interactions with the chloride ion present (Fig. 4 ▶ a), and indicates the potential position of the residue in the presence of NADPH, owing to the position of a hydroxyl group of NADPH bound in MSMEG_2407 (Fig. 4 ▶ b). The loss of glycine residues involved in NADPH contacts indicates a more constrained binding site, in particular the substitution of Gly30 for Ala29 at the nicotinamide binding site, and may provide a more complete holoenzyme crystal structure of AKR5H1, which appeared disordered in the MSMEG_2407 holoenzyme crystal structure (Scoble et al., 2010 ▶).
Figure 4.

Structural comparison between Rv2971 and MSMEG_2407. Ball-and-stick representation of the NADPH binding pockets of Rv2971 and (a) MSMEG_2407 in its apo form (PDB entry 2wzt) and (b) in its holoenzyme form in the presence of NADPH (PDB entry 2wzm). Visualized is the structural comparison between amino-acid residues in contact with NADPH in the MSMEG_2407 holoenzyme structure with residues of identical position in Rv2971. Residues of Rv2971, the MSMEG_2407 apo form and the MSMEG_2407 holoenzyme are represented in green, wheat and blue, respectively. Bound malonate ions and a single chloride ion are represented as cyan lines and magenta spheres, respectively. The bound NADPH molecule in the MSMEG_2407 holoenzyme is represented in pink. Missing residues are indicated in parentheses. (c) Sequence alignment between Rv2971 and MSMEG_2407. The alignment was prepared with the program ClustalW2 and visualized using ESPript v.2.2. The secondary-structure elements correspond to the structure of Rv2971. Strict sequence-identical residues are denoted with a red background, while similar residues are visualized in red text with white background. Sequence similarities in groups are denoted by blue boxes. MSMEG_2407 residues interacting with NADPH by hydrogen bonding or van der Waals interactions are indicated by green arrows.
4. Concluding remarks
While valuable structural insight into the architecture of Rv2971 has been obtained, the successful determination of further crystal structures of Rv2971, both in its holoenzyme form and in complex with the INH–NADP adduct, will aid in further characterization of its function, as well as in gaining a more in-depth characterization of INH resistance.
Supplementary Material
PDB reference: Rv2971, 4otk
Acknowledgments
We thank the staff of the Australian Synchrotron and Monash Macromolecular Crystallization Facility for assistance with crystallization and X-ray data collection. This work was supported by the Australian Research Council (ARC) Centre of Excellence in Structural and Functional Microbial Genomics and the National Health and Medical Research Council of Australia. JR is an NHMRC Australia Fellow and TB is a Pfizer Australian Research Fellow.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Argyrou, A., Jin, L., Siconilfi-Baez, L., Angeletti, R. H. & Blanchard, J. S. (2006). Biochemistry, 45, 13947–13953. [DOI] [PMC free article] [PubMed]
- Baker, N. A., Sept, D., Joseph, S., Holst, M. J. & McCammon, J. A. (2001). Proc. Natl Acad. Sci. USA, 98, 10037–10041. [DOI] [PMC free article] [PubMed]
- Banner, D. W., Bloomer, A. C., Petsko, G. A., Phillips, D. C., Pogson, C. I., Wilson, I. A., Corran, P. H., Furth, A. J., Milman, J. D., Offord, R. E., Priddle, J. D. & Waley, S. G. (1975). Nature (London), 255, 609–614. [DOI] [PubMed]
- Battye, T. G. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. W. (2011). Acta Cryst. D67, 271–281. [DOI] [PMC free article] [PubMed]
- Brünger, A. T. (1992). Nature (London), 355, 472–475. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Dessen, A., Quémard, A., Blanchard, J. S., Jacobs, W. R. Jr & Sacchettini, J. C. (1995). Science, 267, 1638–1641. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Engh, R. A. & Huber, R. (1991). Acta Cryst. A47, 392–400.
- Grant, A. W., Steel, G., Waugh, H. & Ellis, E. M. (2003). FEMS Microbiol. Lett. 218, 93–99. [DOI] [PubMed]
- Grimshaw, C. E. (1992). Biochemistry, 31, 10139–10145. [DOI] [PubMed]
- Lovell, S. C., Davis, I. W., Arendall, W. B., de Bakker, P. I., Word, J. M., Prisant, M. G., Richardson, J. S. & Richardson, D. C. (2003). Proteins, 50, 437–450. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Painter, J. & Merritt, E. A. (2006). Acta Cryst. D62, 439–450. [DOI] [PubMed]
- Penning, T. M. & Drury, J. E. (2007). Arch. Biochem. Biophys. 464, 241–250. [DOI] [PMC free article] [PubMed]
- Rozwarski, D. A., Grant, G. A., Barton, D. H. R., Jacobs, W. R. Jr & Sacchettini, J. C. (1998). Science, 279, 98–102. [DOI] [PubMed]
- Sassetti, C. M., Boyd, D. H. & Rubin, E. J. (2003). Mol. Microbiol. 48, 77–84. [DOI] [PubMed]
- Scoble, J., McAlister, A. D., Fulton, Z., Troy, S., Byres, E., Vivian, J. P., Brammananth, R., Wilce, M. C. J., Le Nours, J., Zaker-Tabrizi, L., Coppel, R. L., Crellin, P. K., Rossjohn, J. & Beddoe, T. (2010). J. Mol. Biol. 398, 26–39. [DOI] [PubMed]
- Timmins, G. S. & Deretic, V. (2006). Mol. Microbiol. 62, 1220–1227. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- World Health Organization (2013). Global Tuberculosis Report 2013 Geneva: World Health Organization. http://www.who.int/tb/publications/global_report/en/.
- Zhu, H. (2011). DSSP and Stride Plugin for PyMOL Biotechnology Center (BIOTEC), TU Dresden, Germany. http://www.biotec.tu-dresden.de/~hongboz/dssp_pymol/dssp_pymol.html.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Rv2971, 4otk