

The N-terminal domain of the flagellar pocket collar protein BILBO1 from T. brucei was cloned, overexpressed, purified and crystallized. Anomalous X-ray diffraction data were collected to 1.7 Å resolution.
Keywords: BILBO1, flagellar pocket, Trypanosoma brucei
Abstract
Trypanosoma brucei is a unicellular parasite that causes sleeping sickness in sub-Saharan Africa. It has a unique flagellar pocket (FP) at the base of the single flagellum. The FP is the sole site for endocytosis and exocytosis activity and plays crucial roles in the defence of the cell against the host immune response. In the neck region of the FP is an electron-dense material termed the flagellar pocket collar (FPC). T. brucei BILBO1 (TbBILBO1) was the first cytoskeletal protein to be characterized in the FPC. This protein is highly conserved among trypanosomatids and is essential for FP biogenesis. Structural information is needed to better understand the molecular mechanism of TbBILBO1 function in the cell. Here, the expression, purification and preliminary crystallographic analysis of the N-terminal domain of TbBILBO1 are reported. The protein was overexpressed in Escherichia coli strain BL21 (DE3), purified by multi-step chromatography and crystallized using the vapour-diffusion method. The crystal diffracted to 1.69 Å resolution and belonged to space group P21, with unit-cell parameters a = 29.69, b = 50.80, c = 37.22 Å, β = 94.61°. There was one molecule in the asymmetric unit.
1. Introduction
Trypanosoma brucei is a protist parasite that causes trypanosomiasis in sub-Saharan Africa, colloquially known as sleeping sickness (in humans) and nagana (in animals). The disease is transmitted by the tsetse fly from the genus Glossina and affected approximately 10 000 people in 2009 (Malvy & Chappuis, 2011 ▶; Simarro et al., 2011 ▶). Currently, there is no vaccine available to treat the disease and the available drugs can have lethal side effects (Absalon et al., 2008 ▶).
T. brucei is a unicellular and serpentine-shaped organism that has a single flagellum emerging from near the posterior end of the cell. The flagellum is important for parasite motility and attachment to the salivary-gland epithelium of the tsetse fly (Tetley & Vickerman, 1985 ▶). In the area where the flagellum emerges from the cytoplasm, there is a specialized organelle termed the flagellar pocket (FP). The FP is the invagination of the plasma membrane and has an important role as the only site for endocytosis and exocytosis of the cell (Overath & Engstler, 2004 ▶). This organelle is also essential for periodically changing its major variant surface glycoproteins so that the parasite is able to evade immune responses of the host cells (O’Beirne et al., 1998 ▶).
At the neck region of the FP is a dense cytoskeletal barrier element called the flagellar pocket collar (FPC). The diameter of this region is slightly larger than the flagellum. The FPC presumably serves to anchor the membrane and the axoneme and to position the flagellum in the cytoskeleton arrangement (Field & Carrington, 2009 ▶; Lacomble et al., 2009 ▶). T. brucei BILBO1 (TbBILBO1) was the first identified component of the FPC; its depletion by RNAi was shown to prevent FP and FPC biogenesis (Bonhivers et al., 2008 ▶). TbBILBO1 is only found in kinetoplastids, without apparent orthologues in humans or other organisms (Bonhivers et al., 2008 ▶). Homology-based predictions suggested that TbBILBO1 consists of three main domains: the N-terminal domain (NTD; residues 1–110), two putative EF-hands (EFh; residues 183–213 and 221–249) and a C-terminal coiled-coil domain (residues 263–578).
Currently, there is no high-resolution structure available for this protein. The availability of the structure would be important to assist in analysis of the function of TbBILBO1 in the cell. Compared with other domains of TbBILBO1, no homologous structures of the NTD can be detected based on primary-sequence comparisons, implying that it might have a novel fold with essential functions. Here, we describe the cloning, overexpression, purification, crystallization and preliminary crystallographic analysis of the TbBILBO1-NTD.
2. Materials and methods
2.1. Cloning
The full-length TbBILBO1 (TbBILBO1-FL; accession code XP_829277) open reading frame was amplified by PCR from genomic DNA and ligated into the custom vector HM15b, which provides both an N-terminal 6×His tag (cleavable by thrombin) and a maltose-binding protein (MBP) tag. DNA encoding TbBILBO1-NTD-EFh (residues 1–249) was amplified from the construct of the HM15b-TbBILBO1-FL by PCR using the primers 5′-GCAGCTGCATATGGCGTTTCTCGTACAAGTAG-3′ and 5′-CGACGGATCCTTAGATTTGTTCTTCTTCCCAAAAAGC-3′, which contain an NdeI and a BamHI cutting site, respectively. After double digestion with NdeI/BamHI, the DNA product was ligated into the custom expression vector MalpET. This vector provides an N-terminal MBP tag plus a 10×His tag [cleavable by Tobacco etch virus (TEV) protease]. The DNA sequence was subsequently confirmed by sequencing.
2.2. Overexpression and protein purification
For protein expression, the plasmid was transformed into Escherichia coli BL21 (DE3) cells. The cells were grown at 37°C in Luria–Bertani (LB) medium containing 30 µg ml−1 kanamycin to an OD600 of ∼0.6–0.8 and were then subjected to cold shock on ice for 30 min. Recombinant protein expression was induced by the addition of isopropyl β-d-1-thiogalactopyranoside to a final concentration of 0.5 mM and protein production was continued overnight at 16°C.
The cells were lysed in an EmulsiFlex-C3 homogenizer (Avestin) in lysis buffer [20 mM Tris–HCl pH 8.0, 300 mM NaCl, 20 mM imidazole, 5%(v/v) glycerol] and centrifuged (16 000g, 40 min, 4°C) to remove cell debris. The supernatant was filtered (0.45 µm pore size) and loaded onto an Ni-HiTrap column (GE Healthcare) pre-equilibrated with the same lysis buffer. The column was washed with five column volumes (CV) of lysis buffer and bound protein was eluted by a linear gradient concentration of imidazole (20–600 mM, 10 CV) in lysis buffer. The N-terminal MBP-His10 tag was removed by the addition of 2%(w/w) TEV protease, which leaves two vector-derived amino acids, Gly-His, at the N-terminus of the target protein. The sample was dialyzed against lysis buffer (including 20 mM imidazole) to remove excess imidazole (overnight, 4°C). The sample was loaded again onto an Ni-HiTrap column to remove the MBP-His10 tag. The protein in the flowthrough fraction was cut by thrombin to separate the NTD from the EFh (there is a thrombin cleavage site after Arg175). The sample was then loaded onto an anion-exchange HiTrap QFF column (GE Healthcare) and bound proteins were eluted using running buffer [20 mM Tris–HCl pH 8.0, 50 mM NaCl, 5%(v/v) glycerol] with a linear gradient of NaCl concentration (50 mM–1 M, 25 CV). The NTD fraction was further purified on a Superdex-200 16/60 column (GE Healthcare) pre-equilibrated with a buffer consisting of 20 mM Tris–HCl pH 8.0, 100 mM NaCl, 5%(v/v) glycerol. The sample was concentrated to a final concentration of 20 mg ml−1 by centrifugation in an Ultra-15 Centrifugal Filter Unit (Amicon) with a 3 kDa molecular-weight cutoff.
SeMet-substituted TbBILBO1-NTD-EFh was generated following the protocols reported by Doublié (1997 ▶). Briefly, cells were grown in M9 minimal medium supplemented with all amino acids (2 mg ml−1) except for methionine. Prior to induction, l-SeMet (80 mg l−1), threonine (100 mg l−1), lysine (100 mg l−1), phenylalanine (100 mg l−1), leucine (50 mg l−1), isoleucine (50 mg l−1) and valine (50 mg l−1) were added. The protein was purified as described above using the same buffer with the addition of 15 mM β-mercaptoethanol for purification on the Ni-HiTrap column and 10 mM dithiothreitol (DTT) for size-exclusion chromatography.
2.3. Crystallization and data collection
An initial crystallization screen was performed at both 4 and 22°C by the sitting-drop vapour-diffusion method using the commercial screens Crystal Screen HT (Hampton Research), JBScreen Classics 1–4 (Jena Bioscience), Morpheus and PACT Premier (Molecular Dimension) in MRC 2-well crystallization plates (Hampton Research) carried out by a Crystal Phoenix robot (Art Robbins Instruments). Crystals were observed after 9 d at 22°C in a condition from JBScreen Classics 1–4 consisting of 0.1 M sodium acetate, 0.1 M MES–NaOH pH 6.5, 30%(w/v) polyethylene glycol monomethyl ether 2000 (PEG MME 2000). The crystals could only be reproduced by micro-seeding. Given that no homologous structures are available for molecular replacement, we tried directly to crystallize SeMet-substituted protein for de novo phase determination. For this, original native crystals were crushed and suspended in 50 µl of original mother liquor to prepare a seed stock. Crystallization was performed manually by the hanging-drop vapour-diffusion method. The drops consisted of 1 µl TbBILBO1-NTD (SeMet), 0.8 µl mother liquor and 0.2 µl seed stock. The drops were equilibrated against 200 µl of the same mother liquor with varied concentrations of PEG MME 2000. Plate-shaped crystals were obtained from a condition consisting of 0.1 M sodium acetate, 0.1 M MES–NaOH pH 6.5, 35%(w/v) PEG MME 2000, 5 mM DTT after 2 d. Single crystals of suitable dimensions were transferred into the same mother-liquor solution supplemented with 10%(v/v) glycerol as a cryoprotectant, mounted on a CryoLoop (Hampton Research) and flash-cooled in liquid nitrogen for X-ray diffraction studies.
A single anomalous dispersion (SAD) data set at the absorption edge of Se (λ = 0.9792 Å) was collected on the ID14-4 beamline at the European Synchrotron Radiation Facility (ESRF), Grenoble, France (McCarthy et al., 2009 ▶). 300 frames of diffraction images were collected on a Q315r CCD detector (ADSC) with an oscillation angle of 1.4° and a crystal-to-detector distance of 241.7 mm. The diffraction images were integrated and scaled using iMosflm (Battye et al., 2011 ▶) and SCALA (Evans, 2006 ▶), respectively (Table 1 ▶).
Table 1. Data-collection and processing statistics.
Values in parentheses are for the highest resolution shell
| Wavelength (Å) | 0.9792 |
| Space group | P21 |
| Unit-cell parameters (Å, °) | a = 29.69, b = 50.80, c = 37.22, β = 94.61° |
| Matthews coefficient (Å3 Da−1) | 2.15 |
| Solvent content (%) | 42.85 |
| No. of molecules per asymmetric unit | 1 |
| Light source | ID14-4, ESRF |
| Detector | Q315r CCD (ADSC) |
| Temperature (°C) | −173 |
| Resolution range (Å) | 37.10–1.69 (1.78–1.69) |
| No. of observed reflections | 98166 (13228) |
| No. of unique reflections | 12398 (1744) |
| Completeness (%) | 99.7 (97.9) |
| Average I/σ(I) | 12.7 (2.1) |
| Multiplicity | 7.9 (7.6) |
| R merge † | 0.099 (0.972) |
| Wilson B factor (Å2) | 33.5 |
R
merge = 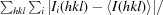
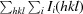 , where Ii(hkl) is the ith observation of reflection hkl and 〈I
i(hkl)〉 is the weighted average intensity for all observations of reflection hkl.
, where Ii(hkl) is the ith observation of reflection hkl and 〈I
i(hkl)〉 is the weighted average intensity for all observations of reflection hkl.
3. Results and discussion
TbBILBO1-NTD-EFh was cloned and successfully overexpressed in E. coli BL21 (DE3). Initial affinity purification was carried out on an Ni-HiTrap column. After elution by imidazole, TEV protease was added to the eluted fraction to cut off the MBP-His10 tag. Subsequently, thrombin was used to separate the NTD and the EFh domains by cleavage after residue Arg175. Owing to the different isoelectric points (pIs) of the NTD (pI = 7.94) and the EFh (pI = 4.7), the protein sample was then applied onto a HiTrap QFF anion-exchange column to separate the NTD from the EFh. After size-based fractionation of the NTD on a Superdex 200 16/60 column, two major bands of protein were observed by SDS–PAGE with molecular weights of 18 and 13 kDa (Fig. 1 ▶ b, lane 1). Both bands were confirmed to be the NTD based on our Western blot analysis using a polyclonal antibody against TbBILBO1 residues 1–110 (data not shown). The protein was concentrated to approximately 20 mg ml−1 and used for crystallization.
Figure 1.

Protein constructs and SDS–PAGE of TbBILBO1-NTD. (a) Schematic depicting the construct used for protein expression. Amino-acid numbers are indicated above the schematic. (b) 15% SDS–PAGE of protein used for crystallization (lane 1) and washed crystals (lane 2). Lane M, molecular-weight marker (labelled in kDa).
An initial crystallization hit was observed from the JBScreen Classics 1–4 screen. An attempt to reproduce crystals manually in this condition did not succeed. Given that no obvious homologous structures could be found for TbBILBO1-NTD, we produced SeMet-substituted crystals for phase determination by the SAD method. Crystallization of SeMet-substituted protein by micro-seeding yielded plate-shaped crystals with dimensions of 0.2 × 0.1 × 0.02 mm after 2 d of incubation (Fig. 2 ▶). When the crystals were subjected to SDS–PAGE, only a single band at approximately 13 kDa was observed, which corresponded to the lower band of the protein sample used for crystallization (Fig. 1 ▶ b, lane 2).
Figure 2.

A typical crystal of the SeMet-substituted TbBILBO1-NTD.
X-ray diffraction data were collected on the ID14-4 beamline at the ESRF in Grenoble, France. An X-ray fluorescence scan of a SeMet-substituted TbBILBO1-NTD crystal was initially carried out both to confirm the presence of SeMet and to select the appropriate wavelength for anomalous data collection (Fig. 3 ▶). The crystal diffracted to a maximum resolution of approximately 1.7 Å (Fig. 4 ▶). The crystal belonged to space group P21, with unit-cell parameters a = 29.69, b = 50.80, c = 37.22 Å, β = 94.61°. The Matthews coefficient of 2.15 Å3 Da−1 and the solvent content of 42.85% (assuming a molecular weight of 13 kDa) calculated using the Matthews Probability Calculator (http://www.ruppweb.org/mattprob/) suggests that there is likely to be one molecule in the asymmetric unit (Matthews, 1968 ▶; Kantardjieff & Rupp, 2003 ▶). Data-collection and processing statistics are shown in Table 1 ▶. All three selenium sites (Met1, Met63, Met107) were successfully located by AutoSol in the PHENIX software suite (Adams et al., 2010 ▶), and were used to phase the data to an initial figure of merit of 0.54. Structure refinement is in progress and a full analysis of the new structural insights will be published elsewhere.
Figure 3.

X-ray fluorescence scan profile of a SeMet-substituted TbBILBO1-NTD crystal.
Figure 4.

X-ray diffraction image of the TbBILBO1-NTD crystal obtained on beamline ID14-4 of the ESRF.
Acknowledgments
This research received institutional support from the Max F. Perutz Laboratories and grant P24383-B21 from the Austrian Science Fund (FWF) to GD. KV was supported by an Österreichischer Austauschdienst (OeAD) graduate scholarship during 2009–2012. We appreciate B. Morriswood for critical reading of the manuscript.
References
- Absalon, S., Blisnick, T., Bonhivers, M., Kohl, L., Cayet, N., Toutirais, G., Buisson, J., Robinson, D. & Bastin, P. (2008). J. Cell Sci. 121, 3704–3716. [DOI] [PubMed]
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Battye, T. G. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. W. (2011). Acta Cryst. D67, 271–281. [DOI] [PMC free article] [PubMed]
- Bonhivers, M., Nowacki, S., Landrein, N. & Robinson, D. R. (2008). PLoS Biol. 6, e105. [DOI] [PMC free article] [PubMed]
- Doublié, S. (1997). Methods Enzymol. 276, 523–530. [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Field, M. C. & Carrington, M. (2009). Nature Rev. Microbiol. 7, 775–786. [DOI] [PubMed]
- Kantardjieff, K. A. & Rupp, B. (2003). Protein Sci. 12, 1865–1871. [DOI] [PMC free article] [PubMed]
- Lacomble, S., Vaughan, S., Gadelha, C., Morphew, M. K., Shaw, M. K., McIntosh, J. R. & Gull, K. (2009). J. Cell Sci. 122, 1081–1090. [DOI] [PMC free article] [PubMed]
- Malvy, D. & Chappuis, F. (2011). Clin. Microbiol. Infect. 17, 986–995. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCarthy, A. A., Brockhauser, S., Nurizzo, D., Theveneau, P., Mairs, T., Spruce, D., Guijarro, M., Lesourd, M., Ravelli, R. B. G. & McSweeney, S. (2009). J. Synchrotron Rad. 16, 803–812. [DOI] [PMC free article] [PubMed]
- O’Beirne, C., Lowry, C. M. & Voorheis, H. P. (1998). Mol. Biochem. Parasitol. 91, 165–193. [DOI] [PubMed]
- Overath, P. & Engstler, M. (2004). Mol. Microbiol. 53, 735–744. [DOI] [PubMed]
- Simarro, P. P., Diarra, A., Ruiz Postigo, J. A., Franco, J. R. & Jannin, J. G. (2011). PLoS Negl. Trop. Dis. 5, e1007. [DOI] [PMC free article] [PubMed]
- Tetley, L. & Vickerman, K. (1985). J. Cell Sci. 74, 1–19. [DOI] [PubMed]