

Cloning, expression, purification, crystallization and preliminary X-ray diffraction analysis of N-acetylmannosamine kinase from methicillin-resistant S. aureus, a novel antibiotic target within sialic acid catabolism, are reported.
Keywords: N-acetylmannosamine kinase, NanK, sialic acid catabolism, Staphylococcus aureus, MRSA
Abstract
N-Acetylmannosamine kinase (EC 2.7.1.60) is involved in the catabolism of sialic acid for many bacterial pathogens implicated in human disease such as Escherichia coli, Staphylococcus aureus, Vibrio cholerae and V. vulnificus. Interestingly, some human commensals and bacterial pathogens can scavenge sialic acids from their surrounding environment and degrade them as a source of carbon, nitrogen and energy. This process requires a cluster of genes known as the ‘Nan-Nag cluster’, which have proven to be essential for S. aureus growth on sialic acids, suggesting that the pathway is a viable antimicrobial drug target. The enzyme N-acetylmannosamine kinase is involved in the catabolism of sialic acid, transferring a phosphate group from adenosine-5′-triphosphate to the C6 position of N-acetylmannosamine to generate N-acetylmannosamine-6-phosphate. The gene was cloned into an appropriate expression vector; recombinant protein was expressed in E. coli BL21 (DE3) cells and purified via anion-exchange chromatography, hydrophobic interaction chromatography and size-exclusion chromatography. Purified N-acetylmannosamine kinase was screened for crystallization. The best crystal diffracted to a resolution of beyond 2.6 Å in space group P2. Understanding the structural nature of this enzyme from methicillin-resistant S. aureus will provide insights necessary for the development of future antimicrobials.
1. Introduction
Sialic acids (more commonly known as N-acetylneuraminic acids) comprise a large family of nine-carbon amino sugars located on the surface of eukaryotic cells that line mucous-rich niches such as the human respiratory tract and gut (Vimr et al., 2004 ▶). In such environments, some bacterial pathogens can scavenge and catabolize sialic acid into carbon, nitrogen and energy precursors, which can then be used in a diverse range of cellular processes (Almagro-Moreno & Boyd, 2009a ▶; Vimr et al., 2004 ▶). This process requires a cluster of genes, known as the ‘Nan-Nag cluster’ (Almagro-Moreno & Boyd, 2009a ▶), that have proven necessary for colonization and persistence of Escherichia coli (Chang et al., 2004 ▶), Vibrio cholerae (Almagro-Moreno & Boyd, 2009b ▶) and V. vulnificus (Jeong et al., 2009 ▶) in mouse models. More significantly, utilization of sialic acid as a carbon source for growth has been proven for Staphylococcus aureus (Olson et al., 2013 ▶), making the pathway a viable target for antibiotic drug design. S. aureus is a Gram-positive bacterial pathogen that is responsible for a diverse spectrum of clinical infections (Furuya & Lowy, 2006 ▶). Notorious for its ability to evolve resistance mechanisms against novel antibiotics, antibiotic-resistant staphylococcal strains such as methicillin-resistant S. aureus have reached epidemic proportions worldwide (Chambers & DeLeo, 2007 ▶; Grundmann et al., 2006 ▶). Thus, it is necessary to characterize novel drug targets and develop new antibiotics against this pathogen. The enzymes involved in the catabolism of sialic acid are targets that have yet to be exploited.
The sialic acid catabolic pathway is depicted in Fig. 1 ▶(a). Following the retro-aldol cleavage of N-acetylneuraminic acid by N-acetylneuraminate lyase to form N-acetyl-d-mannosamine and pyruvate (Barbosa et al., 2000 ▶; Izard et al., 1994 ▶; North et al., 2013 ▶), N-acetylmannosamine kinase transfers a phosphate group from adenosine-5′-triphosphate (ATP) to the C6 position of N-acetyl-d-mannosamine to generate N-acetylmannosamine-6-phosphate (Fig. 1 ▶ b). N-Acetylmannosamine kinase (EC 2.7.1.60) is a phosphotransferase belonging to the ROK (repressor, open reading frame, kinase) superfamily. ROK kinases usually contain a conserved N-terminal ATP-binding motif denoted by the sequence DXGXT and a zinc-binding motif denoted by the sequence CXCGXXGC (Holmes et al., 1993 ▶; Larion et al., 2007 ▶).
Figure 1.

(a) Sialic acid catabolism. The enzymes involved in the uptake and subsequent catabolism of sialic acid are in bold. Lyase, N-acetylneuraminate lyase; kinase (blue), N-acetylmannosamine kinase; epimerase, N-acetylmannosamine-6-phosphate 2-epimerase; deacetylase, N-acetylglucosamine-6-phosphate deacetylase; deaminase, glucosamine-6-phosphate deaminase. (b) The detailed reaction catalyzed by N-acetylmannosamine kinase.
Crystal structures have been solved for the N-acetylmannosamine kinase domain of the bifunctional UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase from Homo sapiens (PDB entries 2yhw, 2yhy and 2yi1; Martinez et al., 2012 ▶). However, only one bacterial crystal structure of N-acetylmannosamine kinase has been determined (PDB entry 2aa4, 17% amino-acid identity; New York SGX Research Center for Structural Genomics, unpublished work), belonging to the bacterial pathogen E. coli. There are, however, a number of other bacterial structures available for sequence-related kinases belonging to the ROK superfamily; for example, PDB entry 2gup (from Streptococcus pneumoniae, 22% amino-acid identity; Midwest Center for Structural Genomics, unpublished work).
Here, we describe for the first time the cloning, expression, purification, crystallization and preliminary X-ray diffraction analysis of N-acetylmannosamine kinase present in methicillin-resistant S. aureus USA300.
2. Materials and methods
2.1. Cloning of N-acetylmannosamine kinase
The nanK gene encoding N-acetylmannosamine kinase from S. aureus USA300 (accession No. YP_493030) was synthesized commercially by GenScript and supplied in a cloning vector designated pUC57-Kan. The primer pair NanK-F (5′-CAT ATG TAT TAC ATC GCA ATC GAT ATT GG-3′) and NanK-R (5′-AAG CTT TCA TTG CAA ACA GCC-3′) was used to incorporate NdeI and HindIII restriction sites into the 5′ and 3′ ends of the nanK gene by the polymerase chain reaction (PCR). The resulting PCR product was subcloned into pCR2.1-TOPO using a TOPO TA cloning kit (Invitrogen), creating pCR2.1-TOPO/NanK. The nanK gene was further digested with NdeI and HindIII (New England Biolabs) restriction enzymes and subcloned into the pET30ΔSE expression vector (Suzuki et al., 2014 ▶), purified using an agarose gel DNA extraction kit (Roche) and ligated with T4 DNA ligase at 293 K for 50 min to create pET30ΔSE/NanK. The nucleotide sequence and corresponding amino-acid sequence of the expressed protein product are shown in Fig. 2 ▶.
Figure 2.

The nucleotide sequence (in red) and corresponding amino-acid sequence (in blue) of N-acetylmannosamine kinase from S. aureus.
2.2. Expression and purification
The recombinant expression vector was transformed into XL1-Blue competent cells, the plasmid was DNA purified and verified by sequencing and further transformed into E. coli BL21 (DE3) cells for protein overexpression. A single colony was transferred into 10 ml Luria broth containing 30 µg ml−1 kanamycin and cultured at 310 K and 180 rev min−1. This culture was transferred into 2 l Luria broth containing 30 µg ml−1 kanamycin and grown at 310 K and 180 rev min−1 until an OD600 of 0.6 was reached. After approximately 5 h, an OD600 of 0.6 was reached; expression of N-acetylmannosamine kinase was induced by the addition of isopropyl β-d-1-thiogalactopyranoside to a final concentration of 1 mM. The culture was further incubated overnight at 299 K and 180 rev min−1. Cells were harvested by centrifugation using a Thermo Sorvall RC-6-Plus centrifuge for 10 min at 8000 rev min−1 and 277 K. Cells were resuspended in buffer consisting of 20 mM Tris–HCl pH 8.0 and lysed by sonication using a Hielscher UP200S Ultrasonic Processor at 70% amplitude in cycles of 0.5 s on, 0.5 s off for 10 min. Cell debris was pelleted by centrifugation at 10 000 rev min−1 for 10 min at 277 K.
Purification of N-acetylmannosamine kinase was conducted via a three-step procedure: anion-exchange chromatography, hydrophobic interaction chromatography and size-exclusion chromatography. For anion-exchange chromatography the soluble fraction was applied onto a 20 ml Q Sepharose column (GE Healthcare) pre-equilibrated with 20 mM Tris–HCl pH 8.0. The column was washed with this buffer until a steady baseline absorbance was observed. N-Acetylmannosamine kinase was then eluted using an increasing concentration gradient to 20 mM Tris–HCl pH 8.0, 1 M NaCl. For hydrophobic interaction chromatography, ammonium sulfate was added to the eluate of the anion-exchange step to a concentration of 1 M and applied onto a 20 ml Phenyl Sepharose column (GE Healthcare) pre-equilibrated with 20 mM Tris–HCl pH 8.0, 1 M ammonium sulfate. The column was washed in this buffer until a steady baseline absorbance was observed. N-Acetylmannosamine kinase was eluted using a decreasing concentration gradient of ammonium sulfate to 20 mM Tris–HCl pH 8.0. Size-exclusion chromatography was conducted using a HiLoad 16/60 Superdex 200 column (GE Healthcare) with 20 mM Tris–HCl pH 8.0. All purification steps were carried out at 277 K. N-Acetylmannosamine kinase was concentrated using a 10 kDa molecular-weight cutoff centrifugal concentrator (Millipore) and flash-cooled for storage at 193 K. Protein concentration was determined after each purification step using the Bradford assay (Bradford, 1976 ▶).
2.3. Sequence analysis of bacterial N-acetylmannosamine kinase
A multiple protein sequence alignment was performed between N-acetylmannosamine kinase from S. aureus USA300 (YP_493030), two additional Gram-positive bacterial species [Clostridium botulinum (YP_004395330) and S. mitis (WP_004261832)] and three Gram-negative bacterial species [E. coli (2AA4_A), Haemophilus influenzae (YP_247864) and Yersinia pestis KIM10+ (NP_668781)], all of which are human bacterial pathogens. In addition, three mammalian sequences [H. sapiens (Q9Y223), Mus musculus (Q91WG8), Rattus norvegicus (O35826)] coding for the N-acetylmannosamine kinase domain of the eukaryotic bifunctional UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase were added to the alignment (extracted from http://www.ncbi.nlm.nih.gov, protein accession numbers in parentheses). This alignment was performed using multiple sequence alignment by ClustalW (Larkin et al., 2007 ▶; http://www.genome.jp/tools/clustalw/), with manual editing. Prediction of secondary-structure conservation from the sequence alignment was performed using the PSIPRED server (Buchan et al., 2013 ▶; Jones, 1999 ▶; http://bioinf.cs.ucl.ac.uk/psipred/).
2.4. Crystallization of N-acetylmannosamine kinase
Crystallization studies were initially conducted using a 10 mg ml−1 preparation of N-acetylmannosamine kinase from S. aureus in 20 mM Tris–HCl pH 8.0. Initial protein crystallization trials were performed at the Collaborative Crystallization Centre (C3; http://www.csiro.au/c3/). Using The PACT Suite and The JCSG+ Suite (Newman et al., 2005 ▶, 2008 ▶), crystal screens were performed using the sitting-drop vapour-diffusion method at 281 and 293 K, with droplets consisting of 150 nl protein solution and 150 nl reservoir solution. Various crystals and morphologies were produced in both The PACT Suite and The JCSG+ Suite. Condition C8 [0.2 M ammonium chloride, 20%(w/v) polyethylene glycol 6000, 0.1 M HEPES pH 7.0] from The PACT Suite produced small, delicate and needle-like crystals after 3 d at 293 K (Fig. 3 ▶). The crystals produced in this condition at C3 were suitable for diffraction.
Figure 3.

Crystallization of N-acetylmannosamine kinase from S. aureus. Crystals were obtained from condition C8 of The PACT Suite. Scale bar indicates 0.3 mm.
2.5. Data collection and processing
High-quality X-ray diffraction data (Fig. 4 ▶) were collected on the MX2 beamline at the Australian Synchrotron (Victoria, Australia). For data collection, crystals from condition C8 of The PACT Suite screen were briefly soaked in cryoprotectant solution containing 85% reservoir solution [0.2 M ammonium chloride, 20%(w/v) polyethylene glycol 6000, 0.1 M HEPES pH 7.0] and 15% of a 1:1 ethylene glycol:glycerol mixture, mounted onto a Cryo-Loop (Hampton Research) and flash-cooled in liquid nitrogen. Crystals were mounted onto the beamline in a stream of nitrogen gas at 110 K. The detector was positioned 350 mm from the crystal and data were collected in 0.5° increments for one 180° pass, with 90% attenuation and an exposure time of 1 s.
Figure 4.

X-ray diffraction of N-acetylmannosamine kinase from S. aureus. Diffraction to 2.6 Å resolution is marked with a ring.
The data were indexed and integrated using XDS (Kabsch, 2010 ▶). Scaling and data reduction were then performed using AIMLESS (Evans, 2006 ▶, 2011 ▶) from the CCP4 program suite (Winn et al., 2011 ▶). The resulting intensity data were analyzed using phenix.xtriage (Adams et al., 2010 ▶). Molecular-replacement methods for phase determination were attempted with Phaser using either PDB entry 2aa4 (17% amino-acid identity) or PDB entry 2gup (22% amino-acid identity) as the search model (McCoy et al., 2007 ▶). A summary of relevant data-collection statistics is given in Table 1 ▶.
Table 1. X-ray data-collection statistics for N-acetylmannosamine kinase from S. aureus .
Values in parentheses are for the highest resolution shell. The Matthews coefficient and solvent content are based on eight monomers, with a combined molecular mass of 253 464 Da, in the asymmetric unit.
| Wavelength (Å) | 0.9537 |
| No. of images | 360 |
| Oscillation range (°) | 0.5 |
| Space group | P2 |
| Unit-cell parameters (Å, °) | a = 83.7, b = 117.9, c = 130.6,α = 90, β = 103.9, γ = 90 |
| Resolution (Å) | 46.9–2.60 (2.66–2.60) |
| Observed reflections | 198489 (12927) |
| Unique reflections | 69158 (4557) |
| Completeness (%) | 91.2 (93.5) |
| R merge † | 0.081 (0.469) |
| R r.i.m ‡ | 0.115 (0.663) |
| R p.i.m. § | 0.081 (0.468) |
| Mean I/σ(I) | 9.0 (1.8) |
| Multiplicity | 2.9 (2.8) |
| Wilson B value (Å2) | 34.2 |
| Molecules per asymmetric unit¶ | 8 |
| V M ¶ (Å3 Da−1) | 2.47 |
| Solvent content¶ (%) | 50.2 |
R
merge = 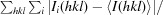
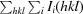 .
.
R
r.i.m. = 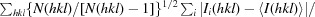 .
.
R
p.i.m. = 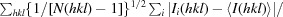 , where Ii(hkl) is the ith intensity measurement of reflection hkl, 〈I(hkl)〉 is its average and N(hkl) is the redundancy of a given reflection.
, where Ii(hkl) is the ith intensity measurement of reflection hkl, 〈I(hkl)〉 is its average and N(hkl) is the redundancy of a given reflection.
The most probable values, assuming eight molecules in the asymmetric unit, are given.
3. Results and discussion
3.1. Expression and purification of N-acetylmannosamine kinase
Purification of N-acetylmannosamine kinase from S. aureus was carried out using anion-exchange chromatography, hydrophobic interaction chromatography and size-exclusion chromatography. From 2 l of bacterial cell culture, approximately 250 mg protein was obtained from anion-exchange chromatography, 120 mg from hydrophobic interaction chromatography and 80 mg from size-exclusion chromatography. The purity of the sample following size-exclusion chromatography was at least 95%, as estimated by SDS–PAGE (Fig. 5 ▶).
Figure 5.
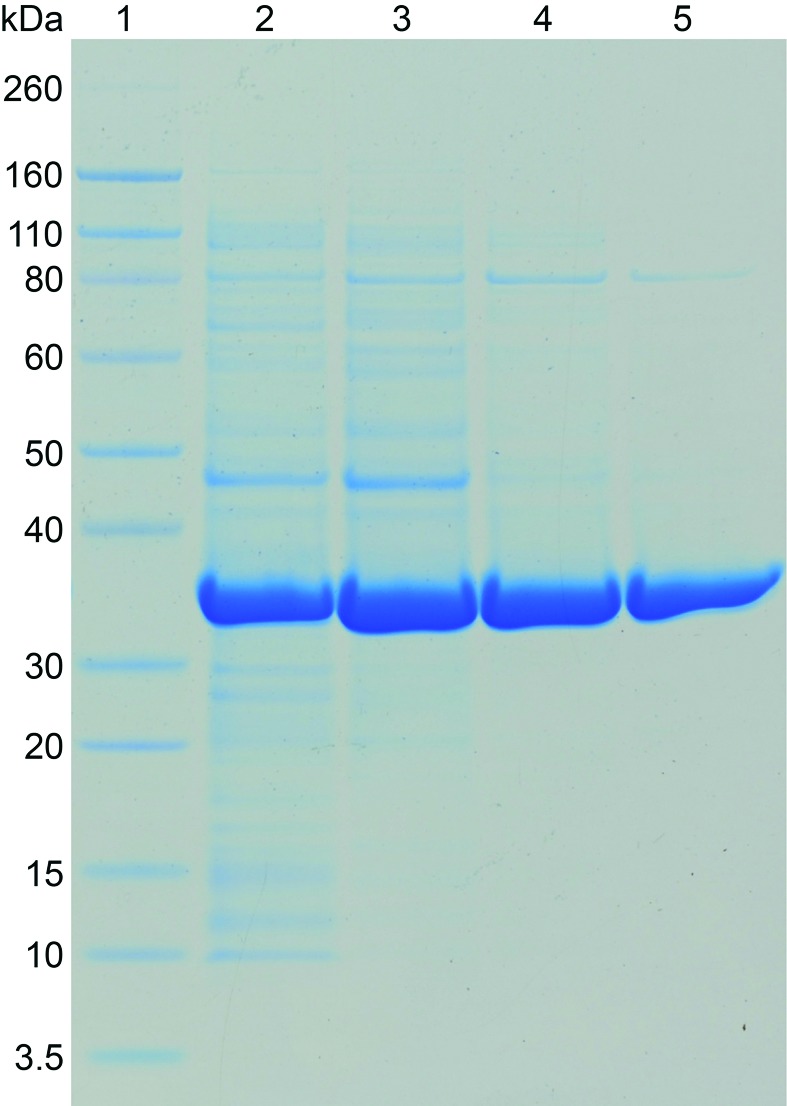
Purification of recombinant N-acetylmannosamine kinase from S. aureus. Lane 1, molecular-weight markers (labelled in kDa); lane 2, crude cell lysate; lane 3, pooled fractions from anion-exchange chromatography; lane 4, pooled fractions from hydrophobic interaction chromatography; lane 5, pooled fractions from size-exclusion chromatography.
3.2. Sequence analysis of bacterial N-acetylmannosamine kinase
A multiple protein sequence alignment was performed between N-acetylmannosamine kinase from S. aureus, two additional Gram-positive bacterial species (C. botulinum and S. mitis), three Gram-negative bacterial species (E. coli, H. influenzae and Y. pestis) and three mammalian species (H. sapiens, M. musculus and R. norvegicus). Mammalian sequences include only the N-acetylmannosamine kinase domain of the bifunctional UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase (amino-acid residues 406–720; Martinez et al., 2012 ▶). These were included in the alignment as efforts to understand how bacterial N-acetylmannsoamine kinase differs from its mammalian equivalents are vital for the design of viable and selective drugs. The sequence alignment (Fig. 6 ▶) allows the identification of amino-acid residues that are potentially involved in ligand binding and catalysis, or those that may be important for protein stability. Secondary-structure elements that are conserved are indicated above the alignment. As mentioned previously, ROK kinases contain a conserved N-terminal ATP-binding motif denoted by the sequence DXGXT and a zinc-binding motif denoted by the sequence CXCGXXGC (Holmes et al., 1993 ▶; Larion et al., 2007 ▶). Highlighted with a red box in Fig. 6 ▶ is an N-terminal ATP-binding motif that is present between residues 7 and 11 (amino-acid numbering corresponds to that of S. aureus) of all species aligned. A purple box is used in Fig. 6 ▶ to highlight the zinc-binding motif. For S. aureus and S. mitis, a zinc-binding motif is not obvious throughout the entire sequence. Zinc is proposed to play a key structural role for the active site within many ROK kinases (Larion et al., 2007 ▶). The lack of a zinc-binding motif suggests that the geometry and stability of the active site may be affected in some other way. This difference could be explored as a target for inhibitory molecules.
Figure 6.

Sequence alignment of N-acetylmannosamine kinase from three Gram-positive species of bacteria, three Gram-negative species of bacteria and three mammalian species. Highly conserved residues are highlighted in blue. Conserved amino-acid residues are numbered according to S. aureus. The ATP-binding motif is highlighted using a red box. The zinc-binding motif is highlighted using a purple box. Conserved secondary-structure elements are indicated above the alignment, where β-strands are depicted as orange arrows and α-helices are depicted as green wavy lines. Amino-acid positions are numbered down the right-hand side for all sequences. Mammalian amino-acid positions are numbered according to the position of the N-acetylmannosamine kinase domain within the bifunctional UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase.
Despite the lack of literature published on bacterial N-acetylmannosamine kinase, much is known about its bifunctional eukaryotic homologue UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase. It has been proposed that this bifunctional enzyme is involved in mammalian biosynthesis of sialic acid, as opposed to sialic acid catabolism (Hinderlich et al., 1997 ▶). The crystal structure of the H. sapiens N-acetylmannosamine kinase domain of UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase has been solved in complex with its substrate N-acetyl-d-mannosamine (PDB entry 2yhw; Martinez et al., 2012 ▶). Based on this structure, it has been determined that N-acetyl-d-mannosamine is hydrogen bonded to eight amino acids (Gly476, Arg477, Thr489, Asn516, Asp517, Glu566, His569 and Glu588). Interestingly, these residues appear to be only partially conserved within all of the aligned sequences. In particular, residues Gly476, Asn516, Asp517, Glu566 and Glu588 (which correspond to residues Gly68, Asn107, Asp108, Glu157 and Glu172 of S. aureus, respectively) are conserved within S. aureus, C. botulinum, H. sapiens, M. musculus and R. norvegicus (Fig. 6 ▶). More importantly, Asn516 and Asp517 (corresponding to residues Asn107 and Asp108 of S. aureus, respectively) are conserved throughout all of the species aligned (Fig. 6 ▶). Because of its proximity to the hydroxyl moiety at position C6 of N-acetyl-d-mannosamine, Asp517 of the H. sapiens N-acetylmannosamine kinase domain (corresponding to residue Asp108 of S. aureus) was proposed to be a key catalytic residue; mutation at this position resulted in attenuation of kinase activity, demonstrating a definite catalytic role (Martinez et al., 2012 ▶). Future work conducted on N-acetylmannosamine kinase from S. aureus should mimic these mutagenesis studies, with the aim of obtaining experimental data supporting the importance of Asp108 and other substrate-binding residues in their hypothesized roles.
Residues Arg477 and Thr489 (which correspond to residues Val69 and Gly81 of S. aureus, respectively) are conserved within mammalian sequences, but are non-conserved between the mammalian and bacterial sequences analyzed. In the H. sapiens N-acetylmannosamine kinase domain crystal structure with bound N-acetyl-d-mannosamine, Arg477 forms a hydrogen bond to the carbonyl group and the C3 hydroxyl group of N-acetyl-d-mannosamine (Martinez et al., 2012 ▶). In the absence of a ligand-bound bacterial N-acetylmannosamine kinase structure, it is unclear what the consequence of a change in this position would be on function. However, we speculate that the binding mode may be altered and/or the enzyme has altered specificity. In the substrate-bound crystal structure of the H. sapiens N-acetylmannosamine kinase domain, Thr489 forms a hydrogen bond with the carbonyl group of N-acetyl-d-mannosamine via its backbone (Martinez et al., 2012 ▶). Thus, it is likely that the side-chain functionality is not important for its interaction with N-acetyl-d-mannosamine and can therefore be freely exchanged without affecting the enzyme activity. Interestingly, residue His569 (corresponding to residue Tyr160 of S. aureus) is conserved between all mammalian and bacterial sequences analyzed, except for those of S. aureus and S. mitis. In H. sapiens this residue functions to coordinate both zinc and the C1 hydroxyl group of N-acetyl-d-mannosamine (Martinez et al., 2012 ▶). As mentioned previously, S. aureus and S. mitis appear to lack a zinc-binding motif; therefore, it is not surprising that these two organisms have a non-conserved residue in this position.
3.3. Crystallization, data collection and processing of N-acetylmannosamine kinase
X-ray diffraction data to a resolution of 2.6 Å were collected from a single crystal that grew in crystallization condition C8 of The PACT Suite. Analysis of the intensity data using POINTLESS (Evans, 2006 ▶) suggested that the crystal belonged to the monoclinic space group P2. The Matthews coefficient (Matthews, 1968 ▶) was estimated to be 2.47 Å3 Da−1 with a corresponding solvent content of 50.2%, assuming eight monomers in the asymmetric unit. However, it is also plausible that there are seven or nine molecules in the asymmetric unit: seven molecules give a Matthews coefficient of 2.82 Å3 Da−1 and a corresponding solvent content of 56.4%, whereas nine molecules give a Matthews coefficient of 2.19 Å3 Da−1 and a corresponding solvent content of 43.9%. As a result of low sequence identity to other structurally known kinases (22% to PDB entry 2gup and 17% to PDB entry 2aa4) the crystal structure of N-acetylmannosamine kinase from S. aureus has not yet been determined by molecular replacement. Thus, selenomethionine labelling to use single-wavelength or multi-wavelength anomalous dispersion (SAD and MAD, respectively) methods is in progress as a phase-solution strategy.
Acknowledgments
We acknowledge the support and assistance of the friendly staff at the CSIRO Collaborative Crystallization Centre at CSIRO Material Science and Engineering, Parkville, Melbourne and the MX beamline scientists at the Australian Synchrotron, Victoria, Australia. Parts of this research were undertaken at the MX2 beamline of the Australian Synchrotron. Travel to the Australian Synchrotron was supported by the New Zealand Synchrotron Group. RCJD acknowledges the following for funding support, in part: (i) the Ministry of Business, Innovation and Employment (contract UOCX1208); (ii) the New Zealand Royal Society Marsden Fund (contract UOC1013); and (iii) the US Army Research Laboratory and US Army Research Office under contract/grant number W911NF-11-1-0481. HS acknowledges FY 2012 Researcher Exchange Program between the Japan Society for the Promotion of Science and the Royal Society of New Zealand for salary support. We especially thank Jackie Healy for her mountainous technical support.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Almagro-Moreno, S. & Boyd, E. F. (2009a). BMC Evol. Biol. 9, 118. [DOI] [PMC free article] [PubMed]
- Almagro-Moreno, S. & Boyd, E. F. (2009b). Infect. Immun. 77, 3807–3816. [DOI] [PMC free article] [PubMed]
- Barbosa, J. A., Smith, B. J., DeGori, R., Ooi, H. C., Marcuccio, S. M., Campi, E. M., Jackson, W. R., Brossmer, R., Sommer, M. & Lawrence, M. C. (2000). J. Mol. Biol. 303, 405–421. [DOI] [PubMed]
- Bradford, M. M. (1976). Anal. Biochem. 72, 248–254. [DOI] [PubMed]
- Buchan, D. W. A., Minneci, F., Nugent, T. C. O., Bryson, K. & Jones, D. T. (2013). Nucleic Acids Res. 41, 340–348. [DOI] [PMC free article] [PubMed]
- Chambers, H. F. & DeLeo, F. R. (2007). Nature Rev. Microbiol. 7, 629–641. [DOI] [PMC free article] [PubMed]
- Chang, D. E., Smalley, D. J., Tucker, D. L., Leatham, M. P., Norris, W. E., Stevenson, S. J., Anderson, A. B., Grissom, J. E., Laux, D. C., Cohen, P. S. & Conway, T. (2004). Proc. Natl Acad. Sci. USA, 101, 7427–7432. [DOI] [PMC free article] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Evans, P. R. (2011). Acta Cryst. D67, 282–292. [DOI] [PMC free article] [PubMed]
- Furuya, E. Y. & Lowy, F. D. (2006). Nature Rev. Microbiol. 4, 36–45. [DOI] [PubMed]
- Grundmann, H., Aires-de-Sousa, M., Boyce, J. & Tiemersma, E. (2006). Lancet, 368, 874–885. [DOI] [PubMed]
- Hinderlich, S., Stäsche, R., Zeitler, R. & Reutter, W. (1997). J. Biol. Chem. 272, 24313–24318. [DOI] [PubMed]
- Holmes, K. C., Sander, C. & Valencia, A. (1993). Trends Cell Biol. 3, 53–59. [DOI] [PubMed]
- Izard, T., Lawrence, M. C., Malby, R. L., Lilley, G. G. & Colman, P. M. (1994). Structure, 2, 361–369. [DOI] [PubMed]
- Jeong, H. G., Oh, M. H., Kim, B. S., Lee, M. Y., Han, H. J. & Choi, S. H. (2009). Infect. Immun. 77, 3209–3217. [DOI] [PMC free article] [PubMed]
- Jones, D. T. (1999). J. Mol. Biol. 292, 195–202. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Larion, M., Moore, L. B., Thompson, S. M. & Miller, B. G. (2007). Biochemistry, 46, 13564–13572. [DOI] [PubMed]
- Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J. & Higgins, D. G. (2007). Bioinformatics, 23, 2947–2948. [DOI] [PubMed]
- Martinez, J., Nguyen, L. D., Hinderlich, S., Zimmer, R., Tauberger, E., Reutter, W., Saenger, W., Fan, H. & Moniot, S. (2012). J. Biol. Chem. 287, 13656–13665. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Newman, J., Egan, D., Walter, T. S., Meged, R., Berry, I., Ben Jelloul, M., Sussman, J. L., Stuart, D. I. & Perrakis, A. (2005). Acta Cryst. D61, 1426–1431. [DOI] [PubMed]
- Newman, J., Pham, T. M. & Peat, T. S. (2008). Acta Cryst. F64, 991–996. [DOI] [PMC free article] [PubMed]
- North, R. A., Kessans, S. A., Atkinson, S. C., Suzuki, H., Watson, A. J. A., Burgess, B. R., Angley, L. M., Hudson, A. O., Varsani, A., Griffin, M. D. W., Fairbanks, A. J. & Dobson, R. C. J. (2013). Acta Cryst. F69, 306–312. [DOI] [PMC free article] [PubMed]
- Olson, M. E., King, J. M., Yahr, T. L. & Horswill, A. R. (2013). J. Bacteriol. 195, 1779–1788. [DOI] [PMC free article] [PubMed]
- Suzuki, H., Tabata, K., Morita, E., Kawasaki, M., Kato, R., Dobson, R. C. J., Yoshimori, T. & Wakatsuki, S. (2014). Structure, 22, 47–58. [DOI] [PubMed]
- Vimr, E. R., Kalivoda, K. A., Deszo, E. L. & Steenbergen, S. M. (2004). Microbiol. Mol. Biol. Rev. 68, 132–153. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.