

Cloning, expression, purification, crystallization and preliminary X-ray diffraction analysis to 1.84 Å resolution of N-acetylmannosamine-6-phosphate 2-epimerase from methicillin-resistant S. aureus, an enzyme involved in the catabolism of sialic acid and a novel antimicrobial drug target, are reported.
Keywords: N-acetylmannosamine-6-phosphate 2-epimerase, NanE, sialic acid catabolism, Staphylococcus aureus, MRSA
Abstract
Sialic acids are one of the most important carbohydrate classes in biology. Some bacterial pathogens can scavenge sialic acids from their surrounding environment and degrade them as a source of carbon, nitrogen and energy. This sequestration and subsequent catabolism of sialic acid require a cluster of genes known as the ‘Nan-Nag’ cluster. The enzymes coded by these genes are important for pathogen colonization and persistence. Importantly, the Nan-Nag genes have proven to be essential for Staphylococcus aureus growth on sialic acids, suggesting that the pathway is a viable antibiotic drug target. The enzyme N-acetylmannosamine-6-phosphate 2-epimerase is involved in the catabolism of sialic acid; specifically, the enzyme converts N-acetylmannosamine-6-phosphate into N-acetylglucosamine-6-phosphate. The gene was cloned into an appropriate expression vector, and recombinant protein was expressed in Escherichia coli BL21 (DE3) cells and purified via a three-step procedure. Purified N-acetylmannosamine-6-phosphate 2-epimerase was screened for crystallization. The best crystal diffracted to a resolution of beyond 1.84 Å in space group P21212. Understanding the structural nature of this enzyme from methicillin-resistant S. aureus will provide us with the insights necessary for the development of future antibiotics.
1. Introduction
Staphylococcus aureus is a Gram-positive bacterial pathogen that is the causative agent of a diverse range of clinical infections, particularly among those with compromised immune systems (Furuya & Lowy, 2006 ▶; Olson et al., 2013 ▶). S. aureus is an extraordinarily adaptable pathogen that is notorious for its ability to evolve resistance mechanisms against novel antibiotics (Furuya & Lowy, 2006 ▶). As a result, the overall burden of antibiotic-resistant staphylococcal strains, particularly methicillin-resistant S. aureus, has reached epidemic proportions worldwide (Chambers & DeLeo, 2007 ▶; Grundmann et al., 2006 ▶). The ever-increasing prevalence of this superbug and the continual progression of antibiotic resistance highlight the urgent need to characterize novel drug targets and develop new drugs against this pathogen. The enzymes involved in the catabolism of sialic acids are promising targets that have yet to be exploited.
Sialic acids (e.g. N-acetylneuraminic acid) constitute a large family of amino sugars that are found at the terminus of glycan molecules that are attached to the surface of eukaryotic cells (Vimr et al., 2004 ▶). Interestingly, bacterial pathogens capable of colonizing heavily sialylated niches can scavenge sialic acid from their surrounding environment and degrade it as a source of carbon, nitrogen and energy (Almagro-Moreno & Boyd, 2009a ▶; Vimr et al., 2004 ▶). This sequestration and subsequent catabolism of sialic acid require a cluster of genes known as the ‘Nan-Nag cluster’ (Almagro-Moreno & Boyd, 2009a ▶). The Nan-Nag cluster of genes encodes enzymes that scavenge sialic acid from the host, transport it into the bacterial cell and degrade it via a series of reactions into fructose-6-phosphate. This pathway has been documented in several bacterial pathogens and has proven to be necessary for colonization and persistence in mouse models for Escherichia coli (Chang et al., 2004 ▶), Vibrio cholerae (Almagro-Moreno & Boyd, 2009b ▶) and V. vulnificus (Jeong et al., 2009 ▶). More significantly, the utilization of sialic acid as a carbon source for growth has been proven for S. aureus (Olson et al., 2013 ▶), making the pathway a viable target for drug design against this pathogen.
The sialic acid catabolic pathway is depicted in Fig. 1 ▶(a). The first committed step in the catabolism of sialic acid is the retro-aldol cleavage of N-acetylneuraminic acid by the enzyme N-acetylneuraminate lyase, removing a pyruvate group to yield N-acetyl-d-mannosamine (Barbosa et al., 2000 ▶; Izard et al., 1994 ▶; North et al., 2013 ▶). N-Acetylmannosamine kinase then transfers a phosphate from adenosine-5′-triphosphate (ATP) to the C6 position of N-acetyl-d-mannosamine, generating N-acetylmannosamine-6-phosphate. Subsequently, N-acetylmannosamine-6-phosphate 2-epimerase converts N-acetylmannosamine-6-phosphate into N-acetylglucosamine-6-phosphate (detailed in Fig. 1 ▶ b). Next, N-acetylglucosamine-6-phosphate deacetylase removes the acetyl group from N-acetylglucosamine-6-phosphate and yields glucosamine-6-phosphate. Glucosamine-6-phosphate deaminase then catalyses the isomerization and deamination of glucosamine-6-phosphate into fructose-6-phosphate (Almagro-Moreno & Boyd, 2009a ▶).
Figure 1.

(a) Sialic acid catabolism. The enzymes involved in the uptake and subsequent catabolism of sialic acid are in bold. Lyase, N-acetylneuraminate lyase; kinase, N-acetylmannosamine kinase; epimerase (red), N-acetylmannosamine-6-phosphate 2-epimerase; deacetylase, N-acetylglucosamine-6-phosphate deacetylase; deaminase, glucosamine-6-phosphate deaminase. (b) The detailed reaction catalyzed by N-acetylmannosamine-6-phosphate 2-epimerase.
Crystal structures of N-acetylmannosamine-6-phosphate 2-epimerase from methicillin-resistant S. aureus (PDB entry 1y0e; Midwest Center for Structural Genomics, unpublished work), Salmonella enterica (PDB entry 3igs; Center for Structural Genomics of Infectious Diseases, unpublished work) and Streptococcus pyogenes (PDB entry 1yxy; Midwest Center for Structural Genomics, unpublished work) have been solved and published in the PDB. However, these structures have not been published in the literature. Here, we present for the first time the cloning, expression, purification and preliminary X-ray diffraction analysis of N-acetylmannosamine-6-phosphate 2-epimerase from methicillin-resistant S. aureus USA300. The amino-acid sequence of PDB entry 1y0e differs from the construct presented in this article by only two residues. Firstly, PDB entry 1y0e contains an extra alanine residue at position 1, which precedes the methionine located at position 1 of the construct presented. Secondly, a valine residue located at position 220 of PDB entry 1y0e corresponds to an isoleucine residue located at position 219 of the construct presented.
2. Materials and methods
2.1. Sequence analysis of bacterial N-acetylmannosamine-6-phosphate 2-epimerase
A multiple protein sequence alignment was performed between N-acetylmannosamine-6-phosphate 2-epimerase from methicillin-resistant S. aureus USA300 (YP_493032), four additional Gram-positive bacterial species [Clostridium botulinum (WP_003363467), Gemella haemolysans (WP_003144293), Lactobacillus plantarum subsp. plantarum P-8 (YP_007988511) and S. pneumoniae (WP_000448318)] and six Gram-negative bacterial species [E. coli (WP_001261214), Haemophilus influenzae (WP_005691497), S. enterica subsp. enterica serovar Typhimurium strain LT2 (NP_462247), V. cholerae (WP_002023168), V. vulnificus (WP_017421644) and Yersinia pestis KIM10+ (NP_668779)] (extracted from http://www.ncbi.nlm.hih.gov, protein accession numbers in parentheses). This alignment was performed using multiple sequence alignment by ClustalW (Larkin et al., 2007 ▶; http://www.genome.jp/tools/clustalw/) with manual editing.
2.2. Cloning of N-acetylmannosamine-6-phosphate 2-epimerase
The nanE gene encoding N-acetylmannosamine-6-phosphate 2-epimerase from methicillin-resistant S. aureus USA300 (accession No. YP_493032) was synthesized commercially by GenScript and supplied in a cloning vector designated pUC57-Kan. NdeI and HindIII restriction sites were incorporated into the 5′ and 3′ ends of the nanE gene by the polymerase chain reaction (PCR) using the primer pair NanE-F (5′-CAT ATG TTA CCA CAT GGA TTA ATA GTA TCT TGT CAG GC-3′) and NanE-R (5′-AAG CTT TTA ATC TTC CAT AAT TTG AAC AAA ACG TTT CG-3′). The resulting PCR product was subcloned into pCR2.1-TOPO using a TOPO TA cloning kit (Invitrogen) to create pCR2.1-TOPO/NanE. The nanE gene was further subcloned into the pET30ΔSE expression vector (Suzuki et al., 2014 ▶) by digestion with NdeI and HindIII restriction enzymes (New England Biolabs), purified using an agarose gel DNA extraction kit (Roche) and ligated with T4 DNA ligase at 293 K for 50 min to create pET30ΔSE/NanE.
2.3. Expression and purification
The recombinant expression vector was transformed into XL1-Blue competent cells and the plasmid DNA was successfully purified. Following sequence verification, plasmid DNA was transformed into E. coli BL21 (DE3) cells for protein overexpression. A single colony was transferred into 10 ml Luria broth containing 30 µg ml−1 kanamycin and cultured at 310 K and 180 rev min−1. This culture was transferred into 2 l Luria broth containing 30 µg ml−1 kanamycin and grown for approximately 5 h at 310 K and 180 rev min−1 until an OD600 of 0.6 was reached. Expression of N-acetylmannosamine-6-phosphate 2-epimerase was induced by the addition of isopropyl β-d-1-thiogalactopyranoside to a final concentration of 1 mM, followed by further incubation overnight at 299 K and 180 rev min−1. Cells were harvested by centrifugation using a Thermo Sorvall RC-6-Plus centrifuge for 10 min at 8000 rev min−1 and 277 K. The cells were then resuspended in buffer consisting of 20 mM Tris–HCl pH 8.0 and lysed by sonication using a Hielscher UP200S Ultrasonic Processor at 70% amplitude in cycles of 0.5 s on, 0.5 s off for 10 min. Cell debris was pelleted by centrifugation at 10 000 rev min−1 for 10 min at 277 K.
The cell lysate was first purified by anion-exchange chromatography. The soluble fraction was applied onto a 20 ml Q Sepharose column (GE Healthcare), which was pre-equilibrated with 20 mM Tris–HCl pH 8.0. The column was washed in this buffer until a steady baseline absorbance was observed. N-Acetylmannosamine-6-phosphate 2-epimerase was eluted via an increasing concentration gradient to 20 mM Tris–HCl pH 8.0, 1 M NaCl. Hydrophobic interaction chromatography was implemented as the second purification step; 1 M ammonium sulfate was added to the eluate of the anion-exchange step and applied onto a Phenyl Sepharose column (GE Healthcare), which was pre-equilibrated with 20 mM Tris–HCl pH 8.0, 1 M ammonium sulfate. The column was washed in this buffer until a steady baseline absorbance was observed. N-Acetylmannosamine-6-phosphate 2-epimerase was eluted using a decreasing concentration gradient of ammonium sulfate to 20 mM Tris–HCl pH 8.0. Size-exclusion chromatography was employed as the final purification step using a HiLoad 16/60 Superdex 200 column (GE Healthcare) with 20 mM Tris–HCl pH 8.0. All purification steps were carried out at 277 K. N-Acetylmannosamine-6-phosphate 2-epimerase was concentrated using a 10 kDa molecular-weight cutoff Centricon (Millipore). Protein concentration was determined after each purification step using the Bradford assay (Bradford, 1976 ▶).
2.4. Crystallization of N-acetylmannosamine-6-phosphate 2-epimerase
Crystallization studies were initially conducted using a 10 mg ml−1 preparation of N-acetylmannosamine-6-phosphate 2-epimerase from methicillin-resistant S. aureus in 20 mM Tris–HCl pH 8.0. Initial protein crystallization trials were performed at the Collaborative Crystallization Centre (C3; http://www.csiro.au/c3/) using The PACT Suite and The JCSG+ Suite crystal screens (Newman et al., 2005 ▶, 2008 ▶) at 281 and 293 K. Crystal screens were performed using the sitting-drop vapour-diffusion method with droplets consisting of 150 nl protein solution and 150 nl reservoir solution. The JCSG+ Suite produced crystals in several of the screen conditions. Condition E1 (1 M trisodium citrate, 0.1 M sodium cacodylate pH 6.5) produced crystals in a cluster after 1 d at 281 K (Fig. 2 ▶). These crystals were suitable for the collection of high-resolution X-ray diffraction data.
Figure 2.

Crystallization of N-acetylmannosamine-6-phosphate 2-epimerase from methicillin-resistant S. aureus. Crystals from condition E1 of The JCSG+ Suite. The scale bar indicates 0.3 mm.
2.5. Data collection and processing
High-quality X-ray diffraction data (Fig. 3 ▶) were collected on the MX2 beamline at the Australian Synchrotron, Victoria, Australia. Crystals from The JCSG+ Suite condition E1 were briefly soaked in cryoprotectant solution containing 85% reservoir solution (1 M trisodium citrate, 0.1 M sodium cacodylate pH 6.5) and 15% of a 1:1 ethylene glycol:glycerol mixture, mounted onto a Cryo-Loop (Hampton Research) and flash-cooled in liquid nitrogen. Crystals were mounted onto the beamline in a stream of nitrogen gas at 110 K. The ADSC Quantum 315r detector was positioned 280 mm from the crystal and data were collected in 0.5° increments for one 180° pass, with 90% attenuation and an exposure time of 1 s.
Figure 3.

X-ray diffraction of N-acetylmannosamine-6-phosphate 2-epimerase from methicillin-resistant S. aureus. Diffraction to 1.84 Å resolution is marked with a ring.
The data were indexed and integrated using xdsme (Kabsch, 2010 ▶). Scaling and data reduction were then performed using AIMLESS (Evans, 2006 ▶, 2011 ▶) from the CCP4 program suite (Winn et al., 2011 ▶). The resulting intensity data were analyzed using phenix.xtriage (Adams et al., 2010 ▶). Molecular replacement was performed using the program Phaser (McCoy et al., 2007 ▶) via CCP4. The monomer of S. aureus N-acetylmannosamine-6-phosphate 2-epimerase (PDB entry 1y0e) was used as the search model. The resulting structure is currently being rebuilt using Coot (Emsley et al., 2010 ▶) and refined using both REFMAC (Murshudov et al., 1997 ▶, 2011 ▶) and phenix.refine (Afonine et al., 2012 ▶). All relevant data-collection and processing parameters are given in Table 1 ▶. Images will be made available via the TARDIS server (Androulakis et al., 2008 ▶) when the structure is published.
Table 1. X-ray data-collection statistics for N-acetylmannosamine-6-phosphate 2-epimerase from methicillin-resistant S. aureus .
Values in parentheses are for the highest resolution shell. The Matthews coefficient and solvent content are based on two monomers, with a combined molecular weight of 49 090 Da, in the asymmetric unit.
| Wavelength (Å) | 0.9537 |
| No. of images | 360 |
| Oscillation range (°) | 0.5 |
| Space group | P21212 |
| Unit-cell parameters (Å, °) | a = 77.1, b = 173.7, c = 44.4, α = β = γ = 90 |
| Resolution (Å) | 46.3–1.84 (1.88–1.84) |
| Observed reflections | 189024 (9200) |
| Unique reflections | 52704 (3125) |
| Completeness (%) | 99.8 (98.5) |
| R merge † | 0.082 (0.549) |
| R r.i.m. ‡ | 0.112 (0.738) |
| R p.i.m. § | 0.076 (0.490) |
| Mean I/σ(I) | 10.8 (1.8) |
| Multiplicity | 3.6 (2.9) |
| Wilson B value (Å2) | 9.6 |
| Molecules in asymmetric unit | 2 |
| V M (Å3 Da−1) | 3.04 |
| Solvent content (%) | 59.5 |
R
merge = 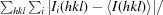
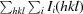 .
.
R
r.i.m. = 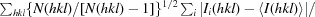 .
.
R
p.i.m. = 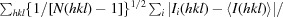 , where Ii(hkl) is the ith intensity measurement of reflection hkl, 〈I(hkl)〉 is its average and N(hkl) is the redundancy of a given reflection.
, where Ii(hkl) is the ith intensity measurement of reflection hkl, 〈I(hkl)〉 is its average and N(hkl) is the redundancy of a given reflection.
3. Results and discussion
3.1. Sequence analysis of bacterial N-acetylmannosamine-6-phosphate 2-epimerase
A multiple protein sequence alignment was performed between N-acetylmannosamine-6-phosphate 2-epimerase from methicillin-resistant S. aureus, four additional Gram-positive bacterial species (C. botulinum, G. haemolysans, L. plantarum and S. pneumoniae) and six Gram-negative bacterial species (E. coli, H. influenzae, S. enterica, V. cholerae, V. vulnificus and Y. pestis). This was performed to investigate the homology of N-acetylmannosamine-6-phosphate 2-epimerase from methicillin-resistant S. aureus with additional Gram-positive and Gram-negative bacterial species. Furthermore, such sequence analysis may allow the identification of amino-acid residues that are potentially involved in catalysis. Owing to a lack of literature published on N-acetylmannosamine-6-phosphate 2-epimerase, it is difficult to predict which residues are involved in catalysis. However, the crystal structure of N-acetylmannosamine-6-phosphate 2-epimerase from S. enterica with a bound ligand, N-acetylglucosmine-6-phosphate, has been solved (PDB entry 3igs). Here, N-acetylglucosamine-6-phosphate is predicted to form hydrogen bonds to Arg83 and Lys74 (amino-acid numbering corresponds to that of S. enterica). As illustrated in Fig. 4 ▶, Lys74 is conserved between all of the Gram-positive and Gram-negative bacterial species analyzed. In contrast, Arg83 is only highly conserved between S. enterica, H. influenzae, V. cholerae and Y. pestis. S. aureus and the remaining organisms have either a phenylalanine or a tyrosine residue in this position. The strict conservation of Lys74 indicates that this residue is likely to be important for the function of N-acetylmannosamine-6-phosphate 2-epimerase. Ligand-bound crystal structures of N-acetylmannosamine-6-phosphate 2-epimerase from methicillin-resistant S. aureus will further confirm this.
Figure 4.

Sequence alignment of N-acetylmannosamine-6-phosphate 2-epimerase from five Gram-positive and six Gram-negative species of bacteria. Highly conserved residues are highlighted in red. Conserved amino-acid residues are numbered according to S. enterica.
3.2. Expression and purification of N-acetylmannosamine-6-phosphate 2-epimerase
Purification of methicillin-resistant S. aureus N-acetylmannosamine-6-phosphate 2-epimerase was carried out using anion-exchange chromatography, hydrophobic interaction chromatography and size-exclusion chromatography. From 2 l of bacterial cell culture, approximately 370 mg protein was obtained after anion-exchange chromatography, 200 mg after hydrophobic interaction chromatography and 140 mg after size-exclusion chromatography. The purity of the sample following size-exclusion chromatography was at least 95%, as estimated by SDS–PAGE (Fig. 5 ▶).
Figure 5.
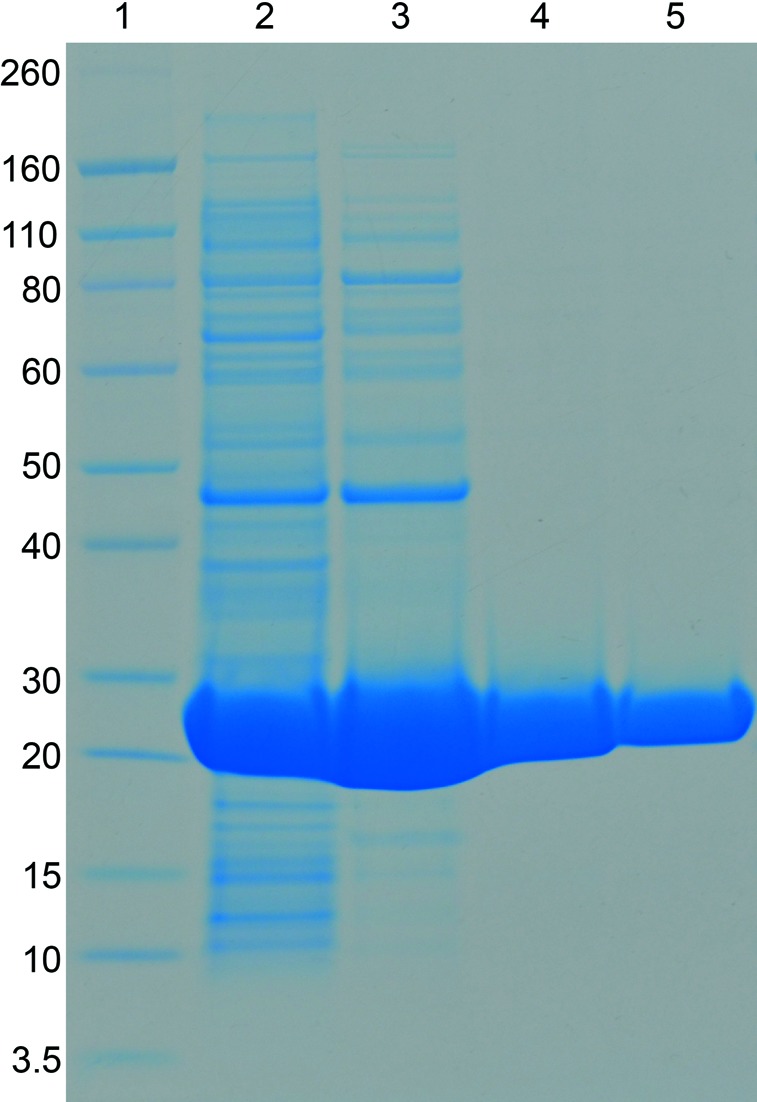
Purification of recombinant N-acetylmannosamine-6-phosphate 2-epimerase from methicillin-resistant S. aureus. Lane 1, molecular-weight markers (labelled in kDa); lane 2, crude cell lysate; lane 3, pooled fractions from the anion-exchange chromatography step; lane 4, pooled fractions from the hydrophobic interaction chromatography step; lane 5, pooled fractions from the size-exclusion chromatography step.
3.3. Crystallization, data collection and processing of N-acetylmannosamine-6-phosphate 2-epimerase
X-ray diffraction data were collected to a resolution of 1.84 Å from a crystal that grew in condition E1 of The JCSG+ Suite in a screen perfomed at C3. The crystal grew as a cluster, which was broken apart prior to mounting it into the nylon loop. The crystal was very delicate and crushed easily, so great care was required. Analysis of the intensity data using POINTLESS (Evans, 2006 ▶) suggested that the crystals belong to the orthorhombic space group P21212. The presence of two screw axes was verified by the presence of systematic absences in only two of the axes. The Matthews coefficient (Matthews, 1968 ▶) was estimated to be 3.04 Å3 Da−1 with a corresponding solvent content of 59.5%; this assumes two monomers in the asymmetric unit that form a dimer. Molecular replacement was conducted using the program Phaser with data scaled in P21212. The monomer of S. aureus N-acetylmannosamine-6-phosphate 2-epimerase (PDB entry 1y0e) was used as the search model, producing a final solution with a translation-function Z-score of 38.1 and a log-likelihood gain of 8222. The construct presented in this article differs by only two amino-acid residues from that of PDB entry 1y0e, yet our structure has a unique cell and symmetry; PDB entry 1y0e has space group P212121 and the unit-cell parameters are a = 53.0, b = 76.3, c = 183.66 Å, α = β = γ = 90.0°. Structure determination is currently under way.
In summary, the cloning, expression, purification, crystallization and preliminary X-ray diffraction analysis of N-acetylmannosamine-6-phosphate 2-epimerase from methicillin-resistant S. aureus USA300 are presented for the first time.
Acknowledgments
We acknowledge the support and assistance of the friendly staff at the CSIRO Collaborative Crystallization Centre at CSIRO Material Science and Engineering, Parkville, Melbourne and the MX beamline scientists at the Australian Synchrotron, Victoria, Australia. Parts of this research were undertaken at the MX2 beamline of the Australian Synchrotron. Travel to the Australian Synchrotron was supported by the New Zealand Synchrotron Group. RCJD acknowledges the following for funding support, in part: (i) the Ministry of Business, Innovation and Employment (contract UOCX1208); (ii) the New Zealand Royal Society Marsden Fund (contract UOC1013); and (iii) the US Army Research Laboratory and US Army Research Office under contract/grant No. W911NF-11-1-0481. We especially thank Jackie Healy for her steely technical support.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Afonine, P. V., Grosse-Kunstleve, R. W., Echols, N., Headd, J. J., Moriarty, N. W., Mustyakimov, M., Terwilliger, T. C., Urzhumtsev, A., Zwart, P. H. & Adams, P. D. (2012). Acta Cryst. D68, 352–367. [DOI] [PMC free article] [PubMed]
- Almagro-Moreno, S. & Boyd, E. F. (2009a). BMC Evol. Biol. 9, 118. [DOI] [PMC free article] [PubMed]
- Almagro-Moreno, S. & Boyd, E. F. (2009b). Infect. Immun. 77, 3807–3816. [DOI] [PMC free article] [PubMed]
- Androulakis, S. et al. (2008). Acta Cryst. D64, 810–814.
- Barbosa, J. A., Smith, B. J., DeGori, R., Ooi, H. C., Marcuccio, S. M., Campi, E. M., Jackson, W. R., Brossmer, R., Sommer, M. & Lawrence, M. C. (2000). J. Mol. Biol. 303, 405–421. [DOI] [PubMed]
- Bradford, M. M. (1976). Anal. Biochem. 72, 248–254. [DOI] [PubMed]
- Chambers, H. F. & DeLeo, F. R. (2007). Nature Rev. Microbiol. 7, 629–641. [DOI] [PMC free article] [PubMed]
- Chang, D. E., Smalley, D. J., Tucker, D. L., Leatham, M. P., Norris, W. E., Stevenson, S. J., Anderson, A. B., Grissom, J. E., Laux, D. C., Cohen, P. S. & Conway, T. (2004). Proc. Natl Acad. Sci. USA, 101, 7427–7432. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Evans, P. R. (2011). Acta Cryst. D67, 282–292. [DOI] [PMC free article] [PubMed]
- Furuya, E. Y. & Lowy, F. D. (2006). Nature Rev. Microbiol. 4, 36–45. [DOI] [PubMed]
- Grundmann, H., Aires-de-Sousa, M., Boyce, J. & Tiemersma, E. (2006). Lancet, 368, 874–885. [DOI] [PubMed]
- Izard, T., Lawrence, M. C., Malby, R. L., Lilley, G. G. & Colman, P. M. (1994). Structure, 2, 361–369. [DOI] [PubMed]
- Jeong, H. G., Oh, M. H., Kim, B. S., Lee, M. Y., Han, H. J. & Choi, S. H. (2009). Infect. Immun. 77, 3209–3217. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J. & Higgins, D. G. (2007). Bioinformatics, 23, 2947–2948. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Newman, J., Egan, D., Walter, T. S., Meged, R., Berry, I., Ben Jelloul, M., Sussman, J. L., Stuart, D. I. & Perrakis, A. (2005). Acta Cryst. D61, 1426–1431. [DOI] [PubMed]
- Newman, J., Pham, T. M. & Peat, T. S. (2008). Acta Cryst. F64, 991–996. [DOI] [PMC free article] [PubMed]
- North, R. A., Kessans, S. A., Atkinson, S. C., Suzuki, H., Watson, A. J. A., Burgess, B. R., Angley, L. M., Hudson, A. O., Varsani, A., Griffin, M. D. W., Fairbanks, A. J. & Dobson, R. C. J. (2013). Acta Cryst. F69, 306–312. [DOI] [PMC free article] [PubMed]
- Olson, M. E., King, J. M., Yahr, T. L. & Horswill, A. R. (2013). J. Bacteriol. 195, 1779–1788. [DOI] [PMC free article] [PubMed]
- Suzuki, H., Tabata, K., Morita, E., Kawasaki, M., Kato, R., Dobson, R. C. J., Yoshimori, T. & Wakatsuki, S. (2014). Structure, 22, 47–58. [DOI] [PubMed]
- Vimr, E. R., Kalivoda, K. A., Deszo, E. L. & Steenbergen, S. M. (2004). Microbiol. Mol. Biol. Rev. 68, 132–153. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.