

Abstract
The advance in technological tools for massively parallel, high-throughput sequencing of DNA has enabled the comprehensive characterization of somatic mutations in large number of tumor samples. Here, we review recent cancer genomic studies that have assembled emerging views of the landscapes of somatic mutations through deep sequencing analyses of the coding exomes and whole genomes in various cancer types. We discuss the comparative genomics of different cancers, including mutation rates, spectrums, and roles of environmental insults that influence these processes. We highlight the developing statistical approaches used to identify significantly mutated genes, and discuss the emerging biological and clinical insights from such analyses as well as the challenges ahead translating these genomic data into clinical impacts.
Introduction
While it is recognized that cancer is a collection of complex pathological entities with diverse biological capabilities1, much of our current understanding of cancer genetics is grounded on the principle that cancer arises from a clone that has accumulated the requisite somatically-acquired genetic aberrations, leading to malignant transformation2. Progression of such a transformed clone to disseminated disease has previously been thought to be a linear process driven by serially acquired new mutations, including base pair substitutions, small insertions and deletions (indels) of bases, chromosomal rearrangements, and gains and losses in gene copy number3. However, recent insights have emerged from comprehensive genomic characterization using next-generation sequencing (NGS) technologies, which has allowed for the sequencing of the coding portion of the genome through hybrid-capture whole-exome sequencing (WES) or nearly all base pairs in a tumor-normal pair by whole-genome sequencing (WGS) (Box 1) (reviewed in4,5), revealing unanticipated complexities in the patterns of somatic alterations in cancers.
Prior to the more widespread use of NGS technologies, studies using PCR-amplification and Sanger-based capillary sequencing methodology were limited by cost and throughput, dictating either one of two study designs: a limited number of genes sequenced in a large cohort or all coding genes sequenced in a small number of samples. Mutations in many of the known cancer genes, so-called ‘mountains’ (genes altered in a high percentage of tumors), were discovered by focused sanger sequencing or cytogenetic analyses, some of which express mutated proteins that have become successful drug targets2,4. The first efforts to interrogate the complete protein coding sequence of the cancer genome of colon, breast and glioblastoma necessitated the generation of up to 208,311 primer pairs6,7 for Sanger-based sequencing. These pioneering studies led to the identification of a highly recurrent mutation in a novel oncogene, isocitrate dehydrogenase 1 (IDH1)6, involved in both cell metabolism8,9 and DNA methylation10, reinforcing the promise of unbiased genomic sequencing in the identification of novel genetic driving events in human cancers.
The advancement in NGS technology has allowed the cancer research community to employ systematic sequencing to identify additional ‘mountains’, which include the discovery of frequent mutations in epigenetic regulators and pre-mRNA splicing machinery in many cancers (Figure 1 and described in detail below), and so-called ‘hills’ (genes altered less frequently in cancer). Furthermore, these efforts have uncovered various mutational mechanisms underlying tumorigenesis and progression, such as chromothripsis11,12, chromoplexy13,14 and kategeis15-17. Following the launch of the Cancer Genome Project (CGP) in the United Kingdom in 2000 and The Cancer Genome Atlas (TCGA) in the United States in 2006, the International Cancer Genome Consortium (ICGC) was created in 2007 to coordinate the generation of comprehensive catalogues of genome alterations from 52 different cancers18. To date, disease working groups from TCGA and ICGC have released comprehensive genomic analyses for a number of cancer types19-33. In addition, a major ongoing effort is to molecularly characterize rare malignancies and cancers that are common in diverse geographical regions in different populations, such as gastric cancer34,35, viral hepatitis (B or C) associated hepatocellular carcinoma (HCC)27,36-38, and parasite-induced cholangiocarcinoma (CCA)39.
Figure 1. Frequent mutations of epigenetic regulators and pre-mRNA splicing machinery in cancers.

Representative genes involved in (A) chromatin remodelling, histone and DNA methylation (Me), acetylation (Ac), and ubiquitination (Ub), and (B) pre-mRNA splicing that were identified to be recurrently mutated in various types of cancers by whole-exome and whole-genome sequencing are displayed. Predicted consequences of mutations, either gain of function (GOF) or loss of function (LOF), are also shown.
The general approach to sequencing and bioinformatics analysis, as well as the potential of large-scale cancer genomics by NGS, has been discussed in depth in recent review articles2,4,5,40,41. Here, we attempt to keep pace with the explosion of NGS studies by summarizing a selection of findings from recently published genomic studies (2008 to July 2013), with a focus on analysis of genes targeted by point mutations (base pair substitutions) and indels. We review WES and WGS studies that have provided the clearest landscape of somatic mutations in major adult tumor types (majority of which involve deep sequencing analyses of the coding exomes and whole genomes of 20 or more samples per study), TCGA and ICGC publications, as well as a few examples of rare malignancies and cancers more prevalent in diverse geographical regions (a selection of studies is summarized in Table 1). We discuss our current understanding of the mutational landscapes of these different tumor types and underscore the biological insights into the etiology of cancer gained through integrative and pathway analysis with various genomic platforms, while highlighting the challenges of identifying driver mutations and functional validation of significantly mutated genes (SMGs). While structural rearrangement can also be identified through NGS, the analytics for defining such structural alterations are much less matured, thus we will not be covering this important aspect of genomic alterations in detail in this review.
Table 1.
Overview of example cancer genomics studies for different cancer types.
| Cancer type | Mutation analysis: # of samples | Highlighted or novel mutated genes | References |
|---|---|---|---|
| Glioblastoma | PCR amplify and sequenced 20,661 genes: 22 | IDH1 • † | 6 |
| WES: 48 (6 matched normal DNA) *pediatric GBM | H3F3A•†, ATRX/DAXX#, TP53 | 118 | |
| ccRCC | WES: 7 | PBRM1# | 47 |
| WGS: 1 | BAP1# | 53 | |
| WES: 106 WGS: 14 |
TCEB1•#, KEAP1#, TET2, mTOR† | 55 | |
| WES: 417 WGS: 22 |
TCEB1•#, NFE2L2†, ARID1A#, mTOR† (Confirmed PBRM1#, SETD2#, KDM5C#, PTEN#, BAP1#, TP53, PIK3CA•†) | 19 | |
| HNSCC | WES: 32 | NOTCH1# | 56 |
| WES: 74 | NOTCH1# | 57 | |
| HGS-OvCa | WES: 316 | TP53 (universal) | 25 |
| Melanoma | WGS: 25 | PREX2a (see Box 4) | 61 |
| WES: 121 | RAC1•†, PPP6C•, STK19•, ARID2# | 43 | |
| WES: 147 (99 matched normal DNA) | RAC1•†, PPP6C•#, ARID2# | 62 | |
| Lung AC | WES: 159 WGS/WES: 23 WGS : 1 |
U2AF1•†, RBM10#, ARD1A# | 64 |
| Lung SCC | WES: 178 | HLA-A#, NFE2L2†, KEAP1(#) | 26 |
| Small-cell lung cancer | WES: 53 | SOX family (+22 SMGs identified) | 65 |
| Prostate | WES: 112 | SPOP, FOXA1, MED12• | 68 |
| WES: 50 | CHD1#, MLL2, FOXA1† | 69 | |
| Colorectal | WES: 224 | ARID1A#, SOX9#, FAM123B# | 22 |
| WES: 72 WGS: 2 |
TET family, ERBB3†, ATM# | 70 | |
| Gastric | WES: 22 | ARID1A# | 34 |
| WES: 15 | FAT4#, ARID1A#, MLL3, MLL | 35 | |
| Breast | WES: 54 WES/WGS: 4 WGS:15 |
Confirmed TP53, PIK3CA•†, RB1#, PTEN# | 75 |
| WES: 31 WGS: 46 |
MAP3K1#, CDKN1B#, TBX3, RUNX1, LDLRAP1, STNM2, MYH9, AGTR2, STMN2, SF3B1• and CBFB# | 73 | |
| WES: 100 | AKT2†, ARID1B#, CASP8#, CDKN1B#, MAP3K1#, MAP3K13#, NCOR1#, SMARCD1#, TBX3 | 31 | |
| WES: 86 WES/WGS: 17 WGS: 5 |
CBFB# | 72 | |
| WES: 510 | GATA3•, PIK3CA•†, MAP3K1#, MAP2K4# (BOX3) | 23 | |
| PDAC | WES: 142 | EPC1, ARID2, ATM, ZIM2, MAP2K4, NALCN, SLC16A4, MAGEA6, axon guidance pathway genes (SLIT/ROBO signaling) | 21 |
| HCC | WES: 10 | ARID2# | 37 |
| WES: 24 | ARID1A#, RPS6KA3#, NFE2L2†, IRF2# | 36 | |
| WGS: 27 | ARID1A#, ARID1B#, ARID2#, MLL#, MLL3# | 27 | |
| WES: 10 | ARID1A# | 38 | |
| CCA | WES: 8 | TP53, KRAS†, SMAD4#, MLL3#, ROBO2#, RNF43#, PEG3#, GNAS•† | 39 |
| AML | WGS: 24 | SMC3#, SMC1A#, STAG2#, RAD21# | 88 |
| WGS: 50 WES: 150 |
Confirmed 23 SMGs; FLT3•†, NPM1, DNMT3A•#, IDH1/2•†, TET2#, RUNX1*#, TP53#, NRAS*†, CEBPA#, WT1#, PTPN11†, KIT*†, U2AF1, KRAS•†, SMC1A#, SMC3#, PHF6#, STAG2#, RAD21#, FAM5C, EZH2•#, HNRNPK | 33 | |
| MDS | WES: 29 | SF3B1•, SRSF2•, U2AF1•, ZRSR2, SF3A1, PRPF40B, U2AF2, SF1 | 89 |
| WES: 9 | SF3B1• | 28 | |
| CLL | WES: 5 | NOTCH1 • † | 94 |
| WGS: 4 | NOTCH1•†, MYD88•†, XPO1•†, KLHL6 | 29 | |
| WES: 88 WGS: 3 |
SF3B1• | 92 | |
| WES: 105 | SF3B1• | 30 | |
| DLBCL | WES: 6 | MLL2#, CREBBP#, EP300# | 100,101 |
| WES or WGS: 13 | MLL2#, MEF2B • † | 98 | |
| WES: 55 | MEF2B•†, MLL2#, BTG1, GNA13, ACTB, P2RY8, PCLO, TNFRSF14#, BCL2# | 99 | |
| MM | WES: 16 WGS: 23 WES/WGS: 1 |
DIS3#, FAM46C, LRRK2, BRAF•†, IRF4†, 11 NFκB pathway genes | 103 |
Summarized information of whole-exome (WES) and whole-genome sequencing (WGS) studies of selected publications with highlighted novel mutated genes indicated. ccRCC = clear cell renal cell carcinoma, HNSCC = head-and-neck squamous cell carcinoma, HGS-OvCa = high grade serous ovarian carcinoma, HCC = hepatocellular carcinoma, AML = acute myeloid leukemia, MDS = myelodysplastic syndromes, CLL = chronic lymphocytic leukemia, DLBCL = diffuse large B cell lymphoma, MM = multiple myeloma, MSI = microsatelite instable, MSS = microsatelite stable. Predicted or reported consequences of mutations are shown: •hotspot identified, †activating, or likely activating #inactivating, or likely inactivating.
hotspot identified
activating, or likely activating
inactivating, or likely inactivating
Features of somatic mutation genomic studies
Mutation rates and spectrum across cancer types
A meta-analysis of 2,957 whole exomes and 126 whole genomes from 27 cancer types performed at the Broad Institute, recently illustrated the mutational heterogeneity in diverse cancer types42. First, the variation in mutation frequency can partly be explained by cancer type. For example, the mutation rates for pediatric and haematological cancers possess the lowest mutation rates (~1 mutations/Mb for chronic lymphocytic leukemia (CLL))30, compared to cancers where environmental mutagens are known to increase the mutation burden, such as melanoma and lung cancer (~15 mutations/Mb for melanoma)43. In addition, mutation rates can vary tremendously within a cancer type, often due to the degree of exposure to an environmental mutagen, or dependent on which genes are mutated (e.g. tumors possessing mutations in mismatch repair genes). Second, the mutation spectrums also vary across cancer types. For example, clustering analysis on all possible mutations (considering context of flanking residues) demonstrated natural groupings of mutation spectrum and cancer types consistent with known signatures of carcinogenesis mechanisms: lung tumors possess a high fraction of G>T transversions, attributable to exposure of polycyclic aromatic hydrocarbons from tobacco smoke; melanomas possess a high fraction of C>T transitions in dipyrimidines caused by UV-induced DNA damage and misrepair; gastrointestinal tumors (oesophageal, colorectal and gastric) possess a high frequency of transition mutations at CpG dinucleotides that may be a reflection of elevated methylation levels in these tumors22; cervical, bladder, some head-and-neck and breast cancers possess frequent mutations at Cs in the context of TpC, characteristic mutations caused by the APOBEC family of cytidine deaminases15-17; and leukemic samples (acute myeloid leukaemia (AML) and CLL) possess A to T mutations in the TpA context42. Finally, analysis of WGS confirmed mutational heterogeneity across the genome that is heavily influenced by two factors: gene expression level and DNA replication time. In light of this known mutational heterogeneity, correcting for such factors has been shown to be important in determining SMGs42,43.
Approaches to identify cancer-associated genes
Identifying which mutations are likely to be ‘drivers’ in pathogenesis and elucidating how mutated genes affect the biology of a given tumor, are fundamental challenges in cancer genomics. Statistical tools to identify SMGs that possess a higher mutation rate than the expected (calculated/estimated) background mutation rate (BMR), indicating positive selection during tumorigenesis have been developed by a number of groups (Box 2). Following the recent surge of WES and WGS studies, a more comprehensive census of human cancer genes has emerged. However, this is just the first step in the translation of these findings to clinical benefits for patients. Additionally, many of the published studies have collected data on other genomic dimensions (e.g. DNA copy number, DNA methylation, mRNA and miRNA expression profiles), which can be used for integrative analysis (Box 3) and for elucidating mechanisms of disease pathogenesis; we highlight a few informative examples of such integration in the sections on specific cancers.
Landscape of somatic mutations in solid tumors
Glioblastoma
The Sanger-sequencing of 20,661 protein coding genes from 22 glioblastoma samples mentioned above revealed a recurrent heterozygous IDH1 mutation targeting amino acid R132 in 12% of samples, which correlated with improved patient survival6. In parallel, glioblastoma was the first cancer to undergo comprehensive genomic characterization by TCGA Research Network, which used a targeted approach consisting of Sanger-based capillary sequencing of 601 selected genes in 91 glioblastoma tumor–normal pairs to identify somatic mutations, which revealed frequent mutations in the phosphatidylinositol 3-kinase (PI3K) regulatory subunit, PIK3R124. The targeted sequencing was complemented by analyses of DNA copy number, mRNA expression and DNA methylation from 206 glioblastomas. Integrative analysis demonstrated statistically significant deregulation of the RTK–RAS–PI3K, p53, and RB signaling pathways in glioblastoma24. This study set the framework for comprehensive multi-dimensional genomic characterization in large-scale studies (Box 3). Investigation of RNA sequencing (RNA-seq) data from glioblastomas also revealed translocations involving fibroblast growth factor receptor (FGFR) genes to the transforming acidic coiled-coil (TACC) coding domains of TACC1 or TACC3, in approximately 3% (3/97) of cases44. Glioblastoma cells expressing the fusion demonstrated sensitivity to FGFR kinase inhibition in preclinical assays44. The examples above demonstrate the power of unbiased genomic sequencing in the identification of clinically relevant genetic changes.
Clear-cell renal cancer
Somatic or germline inactivating mutations in the tumor suppressor gene, von Hippel–Lindau (VHL), a master regulator of HIF transcription factors and the hypoxia response, are found in most clear-cell renal-cell carcinoma (ccRCC) cases (somatic mutations reported in >55% of sporadic ccRCC)45,46. However, studies from model organisms have pointed to the requirement for additional genetic alterations to drive tumor development. Indeed, focused Sanger sequencing revealed mutations in neurofibromin 2 (NF2) and in genes involved in histone methylation and demethylation: SETD2, JARID1c (also known as KDM5C) and UTX (also known as KMD6A)46 (Box 3 and Figure 1a). A follow-up WES analysis led to the identification of the second most frequently mutated gene in ccRCC, polybromo 1 (PBRM1)47, which possessed truncating mutations in 41% (92/227) of cases. PBRM1 encodes for the chromatin-targeting subunit of the PBAF SWI/SNF chromatin remodeling complex, which regulates transcription and has essential roles in maintenance of stem pluripotency (reviewed in48) (Figure 1a). The importance of the PBAF complex was initially suggested from studies that found inactivating mutations in its core component, SMARCB1 (SNF5), in rhabdoid tumors49,50. PBRM1 mutations were found in the context of loss of heterozygosity, and functional data supported its role as a tumor suppressor, suggesting PBRM1 represents the second major genetic event that cooperates with VHL in ccRCC47. A second WES study identified frequent mutations in the two-hit tumor suppressor BRCA1-associated protein-1 (BAP1) in ccRCC, previously found to be frequently mutated in uveal melanoma51 and pleural mesothelioma52, that functions as a deubiquitinating enzyme and regulator of histone H2A lysine 119 ubiquitination53 (Figure 1a). Interestingly, mutations in BAP1 and PBRM1 were anti-correlated, and tumors possessing BAP1 mutations were associated with high tumor grade53,54. Recent efforts from a larger WES/WGS study55 and TCGA19 confirmed PBRM1, SETD2, KDM5C and BAP1 to be significantly mutated in a cohort of 417 ccRCC tumors, as well as a novel hotspot mutation in a component the VHL E3 ligase complex, TCEB119,55. Importantly, TCGA was able to show widespread DNA hypomethylation in SETD2-mutant tumors and transcriptional network analysis suggest mutations in the chromatin remodeling complex (PBRM1, ARID1A, and SMARCA4) are linked to RAS signaling, immune function, DNA repair, β-catenin and TGF-β signaling. RNA-seq analysis also revealed recurrent SFPQ–TEF3 fusions found by in 5/416 samples, all five lacking a VHL mutation19.
Head-and-neck squamous cell carcinomas (HNSCCs)
A first global view of the somatic mutations in HNSCC was published in two studies in 2011, based on WES characterization of 32 and 74 HNSCC tumor–normal pairs, respectively56,57. Epidemiologically, tobacco use, alcohol consumption, and infection with human papilloma virus (HPV) are known major risk factors for HNSCC. These studies reported that tumors isolated from patients with a history of tobacco use possessed higher mutation rates than those from non-smokers, that HPV-associated tumors possessed far fewer mutated genes, and that mutations in TP53 and HPV infection were mutually exclusive56,57. These observations demonstrate how environmental causes of cancer leave footprints in the genome, and these may offer hints to the molecular mechanism of pathogenesis.
In terms of genes that are frequently mutated in HNSCC, Agarwal et al.56 identified 6 recurrently mutated genes based on the frequency of mutations, and Stransky et al.57 defined 39 SMGs using the MutSig algorithm (Box 2; Supplementary Figure 1). Stransky et al. also looked for enrichment in functional gene sets among the list of SMGs and discovered that the highest scoring set was involved in epidermal development, particularly in squamous cell differentiation, pointing to disruption of the stratified squamous differentiation program as a candidate route to HNSCC.
Both studies identified previously unrecognized NOTCH1 mutations in HNSCC in approximately 10-15% of samples. Although oncogenic activating NOTCH1 mutations have been observed in a number of hematological malignancies29, the mutations in NOTCH1 identified in HNSCC possessed characteristics indicative of loss of function (LOF) mutations, hence suggesting a tumor-suppressive role in this cancer. Phenotypes observed in mice are consistent with the idea that Notch1 can function as a tumor suppressor58. γ-secretase inhibitors target NOTCH downstream signaling, but the clinical development of an inhibitor in humans has been halted, partly due to an increased association with skin cancer risk59. This example highlights the increasingly recognized context-dependent nature of cancer gene functions and that different mutations of the same gene likely confer different cancer-relevant biological activities (Box 4), adding to the complexity of applying genomic information in therapeutic decisions.
Ovarian cancer
Integrated analysis of high-grade serous ovarian adenocarcinoma (HGS-OvCa) by TCGA included mRNA, miRNA, promoter methylation and DNA copy number analyses from 489 tumors as well as WES data on 316 of these samples25. Although 9 SMGs were identified using two algorithms (MutSig and MuSiC), the landscape of somatic mutations in this cancer type was dominated by near universal presence of TP53 mutations (found in 96% of samples). However, there was also evidence for substantial complexity, with multiple infrequently SMGs identified (Supplementary Figure 1). The HGS-OvCa genomes had many somatic copy-number aberrations (SCNAs), with 113 statistically significant recurrent SCNAs as defined by the statistical approach, GISTIC. This level of SCNAs was greater than that for other reported TCGA studies (for glioblastoma, colon, breast and lung cancers)22-26.
Melanoma
The discovery of the BRAF V600E mutation in over 50% of melanomas in 200260 and the subsequent development of an inhibitor to treat patients with BRAF-mutant metastatic disease is the proof-of-concept for genomics-informed personalized therapy. It is known that an additional 20% of melanomas are characterized by recurrent NRAS hotspot mutations; however, the driver mutations in the remaining melanoma cases remain poorly understood.
The search for additional driver mutations in melanoma has been complicated by the fact that melanoma has the highest basal mutation rate of any cancer sequenced to date43,61,62, which can be almost entirely attributable to the abundance of UV-induced C>T transitions in dipyrimidines. As a result, identifying SMGs is highly susceptible to discovery cohort bias. To overcome this challenge, statistically powered discovery cohorts of many patients and modified analytics that take into account this high basal mutation rate have been needed to make sense of the mutation data for this tumor type, as demonstrated by two recent studies that analyzed WES data of cohorts of >100 tumor–normal pairs43,62 (Supplementary Figure 1). For example, an algorithm called InVEx43 was developed to identify SMGs (Box 2). This method led to the identification of several novel SMGs, many of which harbored hotspot mutations, a pattern of mutation that signifies strong biological selection. One of these was a hotspot mutation in the Rho GTPase, RAC1, identified in both studies at a frequency of 4–9%43,62. This example illustrates that different numbers of samples and analytical algorithms are required for different tumor types based on the status of the cancer genome.
Lung cancer
Lung cancer is classified into two major histological types: small-cell lung cancer (SCLC) and non-small-cell lung cancer (NSCLC). NSCLC is further subdivided into squamous cell carcinoma (SCC), adenocarcinoma (AC) and large-cell carcinoma subtypes. Significant progress in personalized treatment for this disease has been made in recent years with the demonstration that epidermal growth factor receptor (EGFR)-activating mutations or gene fusion events involving the receptor tyrosine kinase gene ALK identify NSCLC patients who are responsive to inhibitors of these tyrosine kinases (reviewed in63).
Recently, WES and WGS data have provided a more detailed view of the somatic mutational landscape of various lung cancer subtypes: a combination of WES and WGS on 183 lung adenocarcinoma tumor–normal DNA pairs64, an integrated analysis of 178 lung SCCs26, and WES of 53 SCLC samples65. These studies showed that lung cancer possesses the second highest reported mutation rate after melanoma (mean mutation rate as high as 12.9 mutations/Mb for smokers), with strong mutation signatures associated with tobacco smoke exposure (i.e. G>T transversions and C>T transitions in the setting of CpG dinucleotides)26,64-66. In one study, the InVEx algorithm was used to address the challenge of this high mutation rate and 25 SMGs were identified64 (Supplementary Figure 1). The authors found that mutations in U2 small nuclear RNA auxiliary factor 1 (U2AF1) (Figure 1b) and TP53 correlated negatively with progression-free survival. Together with frequent mutations in RNA binding motif protein 10 (RBM10), the mutations in U2AF1 point towards a role for RNA splicing deregulation in lung cancer as seen in a number of hematological malignancies (Figure 1b and described in detail below).
A modified version of MutSig was used26, which took into account gene expression level, GC content, local gene density, and local relative replication time, to identify 10 SMGs in the lung SCC TCGA study (Box 2; Supplementary Figure 1). These included previously unreported LOF mutations in HLA-A and widespread TP53 mutations in nearly all samples analyzed26. Integrative analysis identified pathways frequently deregulated in lung SCC, which included confirmation of the oxidative stress response due to frequent mutations and SCNAs in the CUL3 and KEAP1 components of an E3 ubiquitin ligase and its target substrate, NFE2L2, in 34% of samples26 (Box 3). Finally, Rudin et al. took into account gene expression levels in their significance analysis to identify 22 SMGs (Supplementary Figure 1), including frequent mutations and SCNAs in SOX family members not previously recognized in SCLC65. This series of analyses nicely illustrates the genomically distinct nature of these three subtypes of lung cancer despite their common organ site involvement, heralding the need for genomic classification to complement traditional histopathological diagnosis.
Prostate cancer
Frequent chromosomal rearrangements of ETS transcription factor genes with androgen-responsive promoters, commonly through TMPRSS2–ERG fusions, were discovered as driver events in up to 50% of prostate cancers in 200567. Recurrent somatic point mutations were thought to play a less important role in prostate tumorigenesis, until two WES studies shed light on novel SMGs in this disease. An analysis68 of WES data from 112 treatment-naїve prostate adenocarcinoma–normal pairs identified 12 SMGs (Supplementary Figure 1); notably, SPOP, a substrate-binding subunit of a Cullin-based E3 ubiquitin ligase complex, was mutated in ~13% of samples. A WES analysis69 on 50 lethal, heavily pre-treated castration-resistant prostate cancers (CRPCs) and 11 treatment-naїve, high-grade localized prostate cancers identified 9 SMGs. Integrating WES and copy-number data found CHD1, an ATP-dependent chromatin-remodeling enzyme, to be frequently altered (Figure 1a). Interestingly, both SPOP-mutant and CHD1-deleted tumors lacked ETS-family gene rearrangements, thus demonstrating how molecular characterization of sizeable tumor cohorts can help identify novel genetic events defining a molecularly distinct subset of a tumor type.
Colorectal and gastric cancer
A molecular characterization of colorectal carcinoma (CRC) by TCGA analyzed WES, DNA copy number, promoter methylation, mRNA and miRNA expression from >200 samples22. Interestingly, the somatic mutation rates varied considerably among samples. Approximately 16% of tumors were designated as hypermutated (having >12 mutations/Mb). These were characterized by high levels of microsatellite instability (MSI), frequent epigenetic silencing of the DNA mismatch-repair pathway gene, MLH1, mutations in other mismatch repair genes or a DNA polymerase catalytic subunit, POLE, providing molecular insights into the underlying causes for the elevated mutation rate. Interestingly, the hypermutated samples possessed few SCNAs and harbored frequent concurrent BRAF V600E mutations. After removal of non-expressed genes, MutSig and prior biological knowledge were used to identify 15 mutated genes in the hypermutated cancers, and 17 in the non-hypermutated samples, thought to be important for CRC (Supplementary Figure 1). The two signature genes of CRC, TP53 and the WNT signaling pathway antagonist APC, were found to be more frequently mutated in the non-hypermutated tumors. Across the hypermutated and non-hypermutated groups it was found that there was near universal deregulation (92–97%) of the WNT signaling pathway and the PI3K pathways (~50%). However, deregulation of the transforming growth factor-β (TGFβ) and RTK–RAS signaling pathway was observed more frequently in the hypermutated subtype.
In another study, WES analysis70 of 15 MSI and 57 microsatellite stable (MSS) colorectal cancer samples identified 23 SMGs in the MSS cohort. Interestingly, RNA-seq analysis discovered recurrent gene fusions predicted to produce functional proteins involving R-spondin family members, RSPO2 and RSPO3, in 3% (2/68) and 8% (5/68) of samples, respectively. The RSPO fusions were found to be mutually exclusive with APC mutations, and exogenous expression of plasmids encoding fusions were shown to activate WNT signaling in a human colon cancer cell line. In a separate WGS study71, a recurrent fusion of VTI1A and TCF7L2 (encodes a WNT signaling effector, TCF4 transcription factor) was found in 3/97 of colorectal cancer samples, and a colorectal carcinoma cell line harboring the fusion was shown to be dependent on its expression for anchorage-independent growth. These multiple findings of frequent deregulation of WNT signaling is consistent with other evidence of APC as an initiating event in CRC and points to various components of WNT signaling as drivers of this disease, particularly in the hypermutated subtype.
Gastric cancer has high prevalence in East Asia and WES analysis on >15 gastric tumors was performed in two studies34,35. Both studies identified cell adhesion/junction organization and chromatin modification as the most enriched pathways affected by SMGs (Supplementary Figure 1). Interestingly, a member of the SWI/SNF chromatin remodeling family, ARID1A, was found to be mutated more frequently and had decreased expression in MSI (83%) and Epstein–Barr virus (EBV)-infected MSS (73%) gastric cancers, compared to non-EBV-infected MSS cancers (11%)34. Alterations in ARID1A were also predictive of improved disease-free survival, suggesting deregulation of the SWI/SNF complex represents a unique mechanism of carcinogenesis associated with a distinct clinical behavior.
Breast cancer
Clinically, breast cancer is categorized into three basic groups: estrogen receptor (ER) and progesterone receptor (PR) positive; ERBB2 (also known as HER2) amplified; and triple-negative breast cancer (TNBC), which lacks ER, PR and ERBB2 overexpression. Recently, a number of large-scale WES and WGS studies of breast cancer have developed new algorithms to reconstruct the clonal evolution of the tumors (Box 5), shedding light on the mutational processes responsible for the generation of somatic mutations in breast cancer in addition to identifying SMGs that correlated with well-established clinically relevant subtypes16,23,31,72-75.
For example, WES/WGS analysis of 65 TNBC cases75 identified 6 SMGs, confirming TP53 as the most frequently mutated gene in this subtype. Clonal frequency analysis provided evidence that somatic mutations in TP53, PIK3CA and PTEN are clonally dominant in most tumors in which they are found, consistent with a founder mutation status role in most, but not all TNBCs.
The largest sequencing studies to date for breast cancer include WES of 79 ER+ and 21 ER-breast tumors31, WES of 103 and WGS of 22 breast tumors from diverse subtypes72, and the TCGA WES analysis of 510 breast tumors from 507 patients23. Although these studies used diverse approaches to understand the mutational landscape of breast cancer, they shared remarkable overlap in the SMGs that were identified (Supplementary Figure 1)23,31,72, including nearly all genes previously associated with breast cancer, as well as novel SMGs in the transcription factors TBX331 and CBFB72, and a recurrent, potentially druggable MAGI3–AKT3 fusion in 3.4% (8/235) of samples72.
Finally in a study of the responsiveness of ER+ breast cancers (of both the luminal A and luminal B expression subtypes) to estrogen deprivation, WES and WGS were performed on pretreatment tumor biopsies from patients who were subsequently treated with the neoadjuvant aromatase inhibitors73. The authors identified 18 SMGs by MuSiC, and found that GATA3 mutations correlated with response to aromatase inhibitor treatment. Furthermore, integrative analysis of mutations, mRNA expression and clinical attributes suggested that for patients with MAP3K1-mutant luminal A tumors, neoadjuvant aromatase inhibitors would prove a favorable option, but not for patients with TP53-mutant luminal B tumors73.
Pancreatic cancer
Recently, the ICGC published a WES and copy-number analysis from a prospectively accrued cohort of 142 early (stage I and II) sporadic pancreatic ductal adenocarcinoma (PDAC) samples21. MuSiC identified 16 SMGs (Supplementary Figure 1) and pathway analysis of these genes using GeneGO ascertained known cancer pathways (such as the G1/S checkpoint, apoptosis, and TGFβ signaling) as mechanisms important for PDAC development, as well as a novel pathway involved in axon guidance. Importantly, expression levels of two axon guidance genes, ROBO2 and ROBO3, were associated with patient survival21. Forward genetics (in the form of a Sleeping Beauty transposon mutagenesis screen in a mouse model of PDAC) and functional genomics (in the form of a shRNA screen in pancreatic cell lines) were also leveraged to explore the functional relevance of SMGs identified by sequencing. Thus, large-scale genomic analysis coupled with forward genetics and functional genomics can provide insights into pathways not previously linked to cancer.
Liver cancer
Hepatocellular carcinoma (HCC) has a strong association with chronic liver disease such as viral hepatitis (B or C) and aflatoxin B exposure, and is more prevalent outside of North America and Europe. Four recent genomic studies27,36-38 using WES and WGS data from discovery tumor cohorts varying from 4 to 27 samples have provided insights into the genetic drivers in HCC arising from various etiologies. All the studies confirmed previously known mutations in TP53 in HCC, but they also shed light on the importance of deregulation by somatic mutations of genes involved in chromatin remodeling, the WNT–β-catenin pathway, cell cycle control, the PI3K pathway, and oxidative and endoplasmic reticulum stress pathway27,36-38 (Supplementary Figure 1). Although most of the mutations were not associated with a specific type of chronic liver disease, in one study, mutations in interferon regulatory factor 2 (IRF2) were exclusively found in hepatitis B virus (HBV)-related tumors36. WGS has also revealed that the number of HBV integration sites in HCC tumors was associated with poor survival, and identified recurrent integration events in the TERT76, MLL4 and CCNE1 loci, which resulted in concurrent increase in gene expression27,77, reaffirming the powerful analytical capacity of NGS to investigate the potential role of pathogens in human cancers5.
One form of fatal hepatobiliary cancer that is highly prevalent in certain parts of Southeast Asia, is cholangiocarcinoma (CCA) associated with ingestion of the Opisthorchis viverrini parasite present in raw or undercooked fish. Interestingly, the biliary tree contains stem cell compartments for the liver, pancreas and bile duct, and analysis of WES from 8 Opisthorchis viverrini-related CCAs identified a mutational landscape that appeared more similar to PDAC than HCC (Supplementary Figure 1)39. This reinforces the importance of genomic classification in diagnosis, as we begin to understand cancers on a molecular level in addition to their organ-site and pathological features.
Landscape of somatic mutations in hematological malignancies
Myeloid malignancies
Genomic characterization of hematological malignancies has been at the forefront in the field of cancer genetics, and the most active in terms of clinical translation. Identification of gene fusions as the predominant drivers of certain leukemias has led to the development and clinical success of targeted therapies, including imatinib in chronic myeloid leukemia (CML) and acute lymphocytic leukemia (ALL) cases with the BCR–ABL1 fusion and all-trans-retinoic acid (ATRA) in acute promyelocytic leukemia (APL) cases with PML–RARA fusion. However, substantial numbers of leukemias do not possess such gene fusions, and the search for other genetic drivers led to the identification of somatic mutations in genes such as FLT3, RAS, CEBPA, KIT, JAK2, RUNX1, TET2, ASXL1, EZH2, and TP53 prior to the era of NGS-based studies78-82. The first WGS of a patient with French-American-British (FAB) classification M1 acute myeloid leukemia (AML) not only confirmed mutations in previously known genes, but led to the subsequent identification of new somatic mutations in genes involved in DNA methylation, such as DNA methyltransferase 3A (DNMT3A), IDH1, and IDH2 (Figure 1a)83-86.
Although there is some inter-patient heterogeneity, frequency of somatic mutations identified in myeloid luekemias is generally lower than that of solid tumors. The recent TCGA WES and WGS analysis of 200 patients of de novo acute myeloid leukemia (AML) revealed that an average of 13 coding mutations per sample (range: 0-51), and the mutation rate was even lower in cases that harbor known fusion variants, such as MLL-X or PML-RARA fusion33. In this study, MuSiC identified 23 SMGs that included known mutations in AML, as well as mutations in the mRNA splicing machinery (U2AF1) (Figure 1b) or cohesin complex (SMC1A, SMC3. STAG2, and RAD21) that were recently identified in myeloid leukemias (Supplementary Figure 2)87,88.
WES analyses of myelodysplastic syndromes (MDS) have shown that the mutation rate is similar to that of AML (median 9 mutations per sample), and share a similar spectrum of mutated genes (Supplementary Figure 2)28,89. However, some genes and pathways are overrepresented in MDS compared to AML and vice versa. For example, mutations in spliceosome complex genes are more abundant in MDS than AML and approximately 40% of MDS cases are found to have mutations in one of the spliceosome complex genes (SF3B1, SRSF2, U2AF1, ZRSR2, SF3A1, PRPF40B, U2AF2, and SF1) in a mutually exclusive manner, suggesting that deregulation in pre-mRNA splicing plays a crucial role in MDS pathogenesis (Figure 1b)89,90. Recently, sequencing of longitudinal samples from MDS patients identified hotspot mutations in SET binding protein 1 (SETBP1) that was acquired during leukemic evolution to AML91. This data has set the framework for elucidating the genomic basis of transformation from MDS to AML.
Lymphoid malignancies
The pattern of driver mutations identified in lymphoid malignancies differs from that of myeloid malignancies, although there is some overlap of mutated genes found in CLL29,30,92-94, acute lymphoblastic leukemia (ALL)95-97, diffuse large-B-cell lymphoma (DLBCL)98-101, mantle-cell lymphoma (MCL)102, and multiple myeloma (MM)103,104 (Supplementary Figure 2). Examples of findings for CLL include three independent WES and WGS studies29,30,92 that revealed known mutations in TP53105,106 and ATM107, and previously unknown SMGs in NOTCH1, myeloid differentiation primary response gene 88 (MYD88), and the splicing factor SF3B1 (Figure 1b)29,30,92. In addition, WES and WGS analysis from 91 CLL cases92 defined five core molecular pathways crucial for disease pathogenesis: DNA damage repair and cell cycle control, NOTCH signaling, inflammatory pathways, WNT signaling, and RNA splicing and processing pathways.
Some shared features have been reported in other lymphoid malignancies. For example, RNA-seq has identified hotspot mutations in NOTCH1 in some cases of MCL, suggesting a common role of NOTCH signaling deregulation in B cell malignancies102. MYD88 mutations were also observed in DLBCL99 and Waldenstrom macroglobulinemia108. The landscape of somatic mutations has also been characterized in multiple myeloma in two recent WES/WGS studies, confirming known deregulation of RAS, NF-κB, and histone methyltransferase activity, while revealing previously unknown mutations in genes involved in RNA processing and protein homeostasis103,104. The pattern of somatic mutations in DLBCL is more complex99, but a significant number of cases harbor mutations in regulators of histone and chromatin modification including MLL2, CREBBP, EP300 and activating mutation in EZH2 (Figure 1a)98,100,101. Of interest, WGS of pediatric early T cell precursor ALL revealed a similar somatic mutation landscape to that of myeloid leukemia, suggesting that therapies effective for patients with myeloid leukemia might also be effective in this aggressive form of pediatric leukemia97.
NGS also has the potential to reveal major actionable genetic alterations in rare cancers, such as the discovery of NOTCH2 mutations in 25% of splenic marginal zone lymphoma (SMZL)109, mutations in signal transducer and activator of transcription 3 (STAT3) in 40% of large granular lymphocytic leukemia (LGL)110, and BRAF V600E mutation present in 100% of hairy-cell leukemia (HCL) samples tested to date111. The BRAF inhibitor, vemurafenib, has already shown efficacy in the case of an individual with refractory HCL, and Phase II clinical trials are ongoing to validate these findings (clinicaltrials.gov identifier: NCT01711632)112 [link to http://clinicaltrials.gov/show/NCT01711632] These examples show how systematic integration of data from diverse tumor types has the potential to transform the diagnostic and treatment paradigm for a rare disease.
Genomically defined cancer subtypes
Cancer genomics has shown that histopathologically distinct cancer subtypes of the same organ site often have divergent underlying genomic alterations. In addition to the examples described above, TP53 is mutated in 96% of HGS-OvCa samples, but clear-cell and endometrioid ovarian cancer tumors have lower rates of TP53 mutations and instead possess frequent recurrent mutations in PIK3CA113 and ARID1A114,115, which were identified using NGS technology. Most cutaneous melanomas are driven by BRAF or NRAS mutations; by contrast ocular melanomas, have frequent hotspot mutations in the G-proteins GNA11, GNAQ, and LOF mutations in deubiquitinating enzyme, BAP151,116,117. Recent glioblastoma studies support the notion that pediatric and adult cancers need to be characterized separately at the molecular level, as pediatric and adult glioblastoma tumors possess distinct genetic driving events, which includes mutations in ATRX, DAXX, and the replication-independent histone variant, H3F3A, which were much more prevalent in the pediatric setting (Figure 1a and Supplementary Figure 1)118.
Genomically defined cancer subtypes have been shown to carry diagnostic and/or prognostic significance. One example of this is the V617F mutation in Janus kinase 2 (JAK2) in the diagnosis of the myeloproliferative neoplasm, polycythemia vera (PV), the incidence of which is estimated at 95% and is currently incorporated as one of the diagnostic criteria for PV119. Based on recent findings, the BRAF V600E mutation might be used as a diagnostic tool for HCL, a disease for which morphological diagnosis has been a challenge, but further validation is required111. Classically, genetic information has been actively incorporated into the diagnosis and prognostication of AML and MDS120,121. Chromosomal alterations remain the strongest prognostic factor in both AML and MDS, but recent efforts incorporating somatic mutations have shown promise in creating more sophisticated prognostic models122-124. For instance, in intermediate-risk AML identified by the conventional prognostic model, by incorporating information of additional genetic alterations, which include the internal tandem duplication in fms-related tyrosine kinase 3 gene (FLT3-ITD) and mutations in NPM1, CEBPA, and MLL genes, physicians are able to identify patients that will benefit from stem cell transplant during the first complete remission125. Such prognostication studies help not only to identify “biological drivers” but also to identify “clinically relevant drivers”.
Conclusions and future directions
A number of important future directions have emerged from the genomic studies described above. First, the characterization of rare cancers and clinically important genetic subtypes, such as NSCLCs that lack genetic aberrations in EGFR, KRAS and ALK or melanomas that are wild-type for BRAF and NRAS will undoubtedly provide valuable information into the genetic etiology of these cancers. Second, the analysis of somatic mutations has focused almost entirely on the protein coding regions of the genome. However, projects such as the Encyclopedia of DNA elements (ENCODE) are elucidating the functional elements encoded in the roughly 80% of the genome that is non-coding, including promoter and enhancer regions, providing a significant opportunity to understand the landscape of somatic mutation across the entire genome126. In this regard, two groups have recently shown recurrent mutations in the TERT promoter in approximately 70% of melanomas127,128, which highlights the discovery potential in examining somatic mutations in non-coding regions in cancer. Third, we need to begin to systematically explore the interaction of host genome variation with the somatic genome of the tumor in ultimately influencing outcomes. Fourth, recent studies have brought forward the important issue of tumor heterogeneity and the clinical implications for targeted therapy129 (Box 5). In this respect, in addition to extending genomic study design to include multi-region and longitudinal sampling of tumors, linking of the complex genomic data to dynamic clinical history of that specific patient will be most informative in guiding the analyses and extracting the most clinically relevant insights from the cancer genomes. Such approaches will undoubtedly be essential for the study of drug resistance mechanisms of cancer, which remains an important area of future focus. Lastly, we must not underestimate the challenge and necessity of deep biological investigation in the search for efficient and effective translation of new genomic discoveries into clinically impactful endpoints.
Supplementary Material
Box 1: WES and WGS: complementarity and pitfalls.
Briefly, to identify mutated cancer-relevant genes, the first step is to generate high quality NGS data covering the genomic regions of interest (e.g. WGS or WES or targeted panel of selected genes) from both the germline and tumor DNAs. Next, the sequencing raw reads from the sequencers need to be mapped back to the reference human genome so they can be properly “stitched” together to reconstruct the linear DNA sequence corresponding to each chromosome. Once aligned, sophisticated analytical algorithms are used to call sequence variants in the tumor DNA compared to the human reference genome, and to determine whether a variant is “somatic” by comparison with the germline DNA. Deep coverage (most WES and WGS studies aim for an average of 100- to 150-fold and 30- to 60- fold coverage, respectively) is necessary due to the heterogeneous nature of most tumors4,5,40,41. Tumors can be comprised of many subclones129, possess gross numerical and structural chromosome abnormalities resulting in large variation in actual DNA content (ploidy)130, and often reside in a community of “genomically normal” non-cancerous cells, all of which reduce the sensitivity to detect mutations. This can be further exacerbated in genomic regions of high GC content, which typically results in low to absent coverage. Therefore, it is important that the analytics to call a somatic mutation consider whether the genomic locus is adequately covered.
Box 2: Determining significantly mutated genes and driver mutations.
Significantly mutated genes (SMGs)
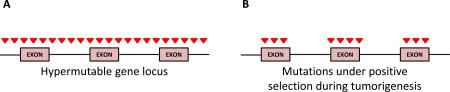
A number of statistical tools to identify SMGs that possess a higher mutation rate than the calculated BMR have been developed7,131-135, which include MutSig24,26,136, MuSiC137, and InVEx algorithms43. Recent analyses have illustrated how challenging this task is in the context of tumors possessing a high and heterogeneous BMR26,43,64. Controlling for type I errors (false positives) is difficult as mutations in a gene can occur in excess of the calculated BMR if resident in a hypermutable region of the genome (a vs b). Studies have shown mutation rates correlate across the genome with levels of heterochromatin-associated histone H3K9me3 modification, gene expression due to transcription-coupled repair, the size of genomic footprint of a gene, and replication time of DNA during the cell cycle42,103,138-141. Statistical tests assuming a uniform BMR across the genome may identify genes that are significantly mutated due to an elevated locus-specific BMR and not because of positive selection of the mutated gene in the tumor. SMG lists often include genes not expressed in the tumor (e.g olfactory receptors) and extremely large genes (e.g. genes involved in muscle development)42,65. Ongoing refinements in algorithms that take into consideration such factors to increase the accuracy of BMR calculations42,43 are improving our ability to identify mutated genes undergoing selection. For example, InVEx leverages sequencing data from intronic and untranslated regions (UTRs) to infer locus (gene)-specific mutation rates. Based on the assumption that mutations in intronic and UTR sequences are more likely under neutral selective pressure, whereas coding mutations will undergo selection during tumor progression, InVEx employs a permutation-based framework to determine whether the observed coding mutations experience positive selection against the inferred gene-specific BMR43. Recently, the algorithm MutSigCV42 has been developed, which corrects for the mutation variation by taking into account patient-specific mutation frequency and spectrum, as well as incorporating expression level and replication time.
WES and WGS analysis of a handful of samples has demonstrated the ability to discover new ‘mountains in cancer’, such as the case of PBRM1 in ccRCC47. However, given driver mutations can be present at low frequency in many cancer genes, the ICGC calculated that 500 tumor samples are needed to detect, with 80% power, genes mutated in 3% of samples assuming a typical BMR of 1.5 mutations/Mb4,18. As illustrated above, sequencing more samples will not be the only solution, and applying evolving algorithms are equally important.
Distinguishing driver from passenger mutations
A major challenge in interpreting and translating genomic discoveries is determining whether SMGs prioritized based on statistical significance truly play functional roles in processes important for tumorigenesis, and which mutations are modulating what gene functions. There are at least three reasons why a gene is found to be significantly mutated in a sequencing study. First, the identified mutations are technical artifacts. However, in most cases these genes will be eliminated from the analysis by manual inspection and verification studies. Second, the mutations in a gene are found to occur in excess of the calculated basal mutation rate (BMR) because they reside in a hypermutable region as illustrated above. Third, a gene is found to be a SMG because it possesses driver mutations that are selected during tumorigenesis, and warrants further functional investigation (Box 4). Additional characteristics can provide supportive evidence of driver mutations. Hotspot mutations and clustered mutations are strong indicators of positive selection, especially when located in functional domains or affecting amino acid residues shown to be important by 3D protein structures. The same mutations may be found as drivers in other cancers. Evolutionary conservation of a mutated residue may indicate importance of the mutational event. Algorithms that score mutational impact by amino acid conservation can infer likely functional mutations in cancer genes142-144. Similarly, the tool Cancer-Specific High-Throughput Annotation of Somatic Mutations (CHASM) that separates candidate driver and passenger mutations based on machine-learning of approximately 50 characteristics associated with cancer-causing mutations can help identify driver mutations145.
Box 3: Integrative and pathway analysis.
Integration of multidimensional genomic data has been crucial to our understanding of biologically and clinically relevant subtypes in cancer. An example of insights from integrative analyses is the definition of four transcriptomic subtypes in glioblastoma annotated as proneural, neural, classical and mesenchymal based on gene expression profiles146. Integration with copy number and mutation data showed that the classical subtype is enriched for EGFR alterations, whereas the mesenchymal subtype contains cases with NF1 loss, and the proneural subtype has specific alterations in PDGFRA and IDH1146. Similarly, analysis of promoter methylation alterations in 272 glioblastoma tumors identified a subset of patient samples with characteristic promoter DNA methylation classified as glioma CpG island methylator phenotype (G-CIMP) which is associated with IDH1 mutations10. Consistent with previous studies, patients possessing G-CIMP tumors tended to be diagnosed earlier in life, and showed better overall survival. In recent ccRCC studies, integration of mutation and expression data provided insight into potential mechanisms of novel SMGs in tumorigenesis. For example, NF2 mutations were found in tumors possessing a non-hypoxic signature and were mutually exclusive with VHL mutations, suggesting mutations in NF2 is a key driver event in this ccRCC subgroup46. Conversely, significant co-occurrence of mutations in epigenetic regulators, SETD2, JARID1C and PBRM1 were found in tumor possessing VHL mutations or hypoxic expression signature, supporting the notion that mutations in this class of genes may act as the second hit to widespread inactivation of VHL in ccRCC46,47. The authors of the TCGA lung squamous cell carcinoma (SQCC) study observed many of the genes involved in oxidative stress and squamous cell differentiation pathways frequently altered by mutations, SCNAs, and identified genomic alterations associated with reported lung SQCC gene signatures (classical, primitive, basal, and secretory)26. Similarly, authors in the TCGA breast cancer analysis were able to show expression subtype-associated enrichment for SMGs. Such associations include GATA3, high frequency of PIK3CA, and likely inactivating MAP3K1 and MAP2K4 mutations in the luminal A expression subtype, enrichment of TP53 and PIK3CA mutations in the HER2 expression subtype, and a high frequency of TP53 mutations in the basal-like expression tumors.
The recent unbiased genomic characterization of a number of different cancers has provided a view into the landscape of somatic mutation that has been valuable in establishing correlations with other genetic aberrations, and has provided insights into driving events in genetic subtypes of cancers that were not well understood. Understanding the landscape can provide both mechanistic and biological insight into the role of SMGs in a given cancer. Mutual exclusivity of genetic alterations is often seen for somatic alterations that possess redundant biological effects in a given pathway. One example is the mutually exclusive mutations in the CUL3–KEAP1 E3 ligase complex and its target substrate NFE2L2 in the oxidative response pathway in lung squamous carcinomas26,147. Based on this principle, the method Mutual Exclusivity Modules in cancer (MeMO) can be used to identify candidate driver networks across a set of patient samples148.
The utility of testing whether known pathways are enriched in SMG lists has been demonstrated in many studies, including the WES analysis of HNSCC that observed deregulation of squamous cell differentiation via somatic mutations57, and the recent ICGC pancreatic study that observed enrichment of mutations in genes in the axon guidance pathway21. The reverse is also possible: identifying SMG networks using computational strategies, such as HotNet, may help discover additional pathways that otherwise would not have been identified using more traditional approaches149.
Box 4: Functional validation of SMGs.
Functional validation of SMGs and candidate driver mutations requires equal consideration in the selection and interpretation of biological assays employed to determine cancer relevance. In the case of newly discovered SMGs, it may not be entirely clear what stage in the tumorigenic process the gene may play a role, or how easily a gene's function can be measured by standard assays. The context and relevant cell lines chosen to perform experiments are also crucial. The example of NOTCH1, which acts as a tumor suppressor with loss-of-function mutations in some tumor types (a) and as an oncogene with activating mutation in another tumor type (b), highlights the challenges in functional validation of SMGs and candidate driver mutations identified by cancer genomics. In addition, the genetic context of the biological system used to test the function of a SMG is significant. For example, if functional assays are performed in primary cells that have been immortalized by engineering specific genetic alterations, or cancer lines possessing inactivation of a tumor suppressor or activation of an oncogene that a candidate SMG may function through, a phenotype may not be observed due to prior deregulation. Similarly, SMGs may require co-operation with additional cancer genes to affect tumorigenesis or bypass oncogene-induced senescence. An example is the lineage-specific melanoma oncogene, MITF, which, when overexpressed alone, did not affect the proliferation or colony-formation of immortalized melanocytes in two-dimensional culture but did so with co-expression of BRAF V600E mutant, which co-occur at a high frequency in melanoma samples150. Biological assays may also provide insight into the function of mutations that would not have been accurately predicted by computational approaches. For example, PREX2A, a gene found to possess widespread distribution of missense and truncating mutations discovered from WGS of 25 metastatic melanoma–normal pairs indicative of a tumor-suppressive function61. However, missense mutations were found to produce mild phenotypes in in vivo tumorigenic assays, whereas, truncating mutations were shown to possess oncogenic activity that could have only been elucidated through functional experiments. A similar observation was made in the case of a p53-inducible phosphatase, PPM1D, where truncating variants were shown to possess gain-of-function effects in functional studies151. The examples highlighted above serve to remind us the need for improved methods to functionally validate candidate cancer genes, such as the development of non-germline mouse models that offer increased speed and reduced costs in developing more high throughput in vivo models152.
Incorporation of forward genetics has been shown by researchers to aid in the determination of the biological relevance of SMGs as demonstrated by the recent ICGC pancreatic study21. Cross-species comparative oncogenomics is another approach that can be employed to identify driving events. In contrast to humans, mouse cancer genomes from genetically engineered mouse models have far fewer genetic alterations in addition to the engineered cancer associated alleles4. The presence of homologous, orthologous or paralogous somatic mutations for a given gene in samples obtained from both human and GEM models may indicate strong selective pressure providing evolutionary evidence that such a mutation is a driving event. This approach has been successfully used by a number of groups to identify novel cancer genes targeted by SCNAs in different cancers153-155. More recently, WGS of mouse APL tumors was performed to identify cooperating somatic mutations in a mouse model expressing a PML-RARA fusion oncogene known to initiate APL in mice156. The authors identified a deletion in a demethylase gene, Kdm6a, and a recurrent mutation in Jak1 V657F. The human JAK1 V658F mutation has been previously reported in APL, demonstrating analysis of WGS from GEM model tumors can be used as an unbiased approach for discovering functionally relevant mutations.

Box 5: Understanding the role of SMGs in intra-tumoral heterogeneity and clonal evolution.
Several pioneering studies have leveraged the sensitivity of NGS to systematically explore the heterogeneity inherent within the same tumor or the same patient, and how such information can be used to infer the temporal order of acquisition of genomic alterations (reviewed in detail129). Approaches used to measure tumor heterogeneity include mathematical algorithms74,130, single cell sequencing157, longitudinal158(a) and multi-region sampling159,160(b), whereby biopsies are taken from different portions of the tumor or from metastatic lesions from the same patient. With such data, one can reconstruct the life history of a tumor by mapping the distribution of clonal and subclonal mutations and SCNAs (c). These analyses can inform whether SMGs are found in the dominant clone, subclones, or in a corresponding metastatic tumor, which may infer its prognostic importance and biological role in tumor progression. For example, WGS analysis from 17 NSCLC tumor–normal pairs validated the presence of EGFR and KRAS mutations in founder clones supporting their roles in cancer initiation161. Studies employing longitudinal sampling involving biopsies taken at different time points during the course of a disease (i.e. pre- and post-treated samples) have been useful in identifying driver clones that play major roles in disease progression, or surviving clones in response to therapeutic regimens by obtaining biopsies of cancer cells from patients before and following treatment158. Recently, geographical sampling and multi-region sequencing of primary renal carcinomas and associated distant metastatic sites demonstrated that 63–69% of all somatic mutations were not detected across every tumor region, and gene expression signatures indicative of both good and poor prognosis were detected in different regions of the same tumor159. The insights gained from the study-design and mathematical tools employed in these studies represent a major breakthrough in the analysis of genomic data and should be considered and incorporated in all future genomic analyses where possible.

Online summary.
The emergence of next-generation sequencing (NGS) technology has made important contributions to our understanding of cancer genomes.
We present a current view of the mutational landscapes of diverse cancer types
There are various challenges to identifying driver mutations and functionally validating significantly mutated genes (SMGs).
Whole-exome and whole-genome sequencing studies, and integrative analysis with other genomic platforms have provided biological insights into the etiology of cancer.
Such studies are enabling classifications of cancers based on genetic alterations rather than tissues of origin.
Acknowledgements
We sincerely apologize for omission of any pertinent work related to this review. We thank Denise Spring and members of the Chin lab for their helpful comments and feedback.
GLOSSARY TERMS
- Next generation sequencing (NGS)
All post-Sanger sequencing methods, most commonly referring to massively parallel sequencing technology.
- Hybrid-capture sequencing
A target enrichment approach wherein custom oligonucleotides (bait set) are designed and optimized to hybridize to specific regions of the genome so specific fragments of DNA can be enriched by hybridization for NGS sequencing.
- Whole-exome sequencing (WES)
Sequence by NGS all protein-coding exomes after capture utilizing hybridization to a whole-exome bait set designed to enrich DNAs in all protein coding portion of the genome. The most common implementation targets miRNA genes in addition. The size of the captured DNA is approximately 40 Mb.
- Whole-genome sequencing (WGS)
Sequencing of the entire genome, usually via a random fragment (shotgun) and to sufficient coverage to ensure adequate representation of all alleles. A variation specifically utilizing low-coverage WGS is sometimes leveraged to assess rearrangements in the genome.
- Significantly mutated genes (SMGs)
A gene that possesses a somatic mutation rate above the calculated background mutation rate (BMR) as determined by a given statistical calculation.
- Driver mutations
Somatic mutations in a gene that confer a selective advantage to cancer cells as reflected in statistical evidence of positive selection. This is not a definition based on functional activity.
- Passenger mutations
Neutral mutations in a gene that do not provide a selective advantage for cancer cells as reflected in lack of statistical evidence for positive or negative selection. This is not a definition based on functional activity.
- Hotspot mutations
Recurrent mutations resulting in the same amino acid change in a gene observed in cancer, signifying strong positive selection.
- Cancer gene
A gene is considered a cancer gene if it harbors a cancer driving genetic aberration (note: cancer genes may possess both driver and passenger somatic alterations) as defined by criteria that can include statistical evidence of selection, recurrence pattern or by functional activity.
- Two-hit tumor suppressor
The Knudson two-hit hypothesis was proposed to explain the early onset of cancer in hereditary syndromes whereby inheritance of one germline copy of a mutated gene in all cells substantially increases the likelihood any cell undergoing mutation of the other allele, thus giving rise to earlier onset disease compared to sporadic forms of the disease. It specifically relates to the necessity to inactivate both alleles of a recessive cancer gene.
- Epstein-Barr Virus (EBV)
Member of the Herpes virus family associated with the development of particular forms of cancer.
- Background mutation rate (BMR)
The rate of mutation in a tumor sample as a consequence of exposure to environmental mutagens (e.g. UV exposure) and/or random generation and misrepair processes.
- CpG island methylator phenotype
A classification of cancers by their degree of methylation at CpG rich promoter regions, first characterized in human colorectal cancers, and often associated with distinct epidemiology, histological and molecular distinct features.
- Triple negative breast cancer
One of the subtypes of breast cancer that is defined by the absence of staining for estrogen receptor (ER), progesterone receptor (PR) and HER2 (ERBB2) by immunohistochemistry.
- Neoadjuvant aromatase inhibitors
Aromatase inhibitors used to treat estrogen receptor positive breast cancer patients prior to surgical resection. This approach is applied in cases where tumor size needs to be reduced for breast conserving surgery. Neoadjuvant aromatase inhibitor treatment is not considered a standard of care at this point and is conducted under clinical trials.
- Sleeping Beauty Transposon System
A genetically engineered insertional mutagenesis system involving synthetic DNA transposons, which can be applied to various model systems to ascertain gene function.
- Aflatoxin B
Aflatoxin B is one of the mycotoxins that are produced by Aspergillus Flavus. High-level exposure to aflatoxins is known to cause acute liver necrosis or cirrhosis resulting in the development of hepatocellular carcinoma (HCC).
- French-American-British (FAB) classification M1
FAB classification of acute leukemias was first proposed in 1976 by French-American-British cooperative group. It classified acute myeloid leukemia (AML) into 8 different categories (M0-M7) and acute lymphoblastic leukemia (ALL) into 3 different categories (L1-L3) based on their morphological findings. The classification was updated in 1989.
- RNAsequencing (RNAseq)
Whole-transcriptome shotgun sequencing of cDNA to determine the sequence of RNA used for expression analysis and the identification of gene-gene fusions.
- Microsatellite instability (MSI):
Is a hypermutable phenotype caused from germline, somatic, or epigenetic inactivation in DNA Mismatch Repair (MMR) activity
- Chromothripsis
Greek for chromosome “shattering”, whereby up to hundreds of genomic rearrangements take place in a single cellular crisis event that develop from errors in mitosis that occur in approximately 2-3% of cancers.
- Chromoplexy
Greek for chromosome “weave” or “braid”. Analysis of prostate cancer genomes revealed copy-neutral rearrangements consisting of between 4-12 distinct breakpoint junctions, which tend to occur at transcriptionally active portions of chromatin, forming a closed chain called chromoplexy.
- Kategeis
Greek for “shower” or “thunderstorm”. A phenomenon, identified in breast cancers, of localized hypermutations almost exclusively involving C base pair substitutions at TpC dinucleotides. This mutation pattern has been linked to the APOBEC family of cytidine deaminases.
- Actionable genetic alterations
A genetic alteration with sufficient scientific evidence supporting its use to inform a treatment decision
Biography
Lynda Chin, M.D., is a Professor and Chair of the Department of Genomic Medicine and the Scientific Director of the Institute of Applied Cancer Science at The University of Texas MD Anderson Cancer Center in Houston. Dr. Chin is a Principal Investigator of The Cancer Genome Atlas (TCGA) Genome Data Analysis Center at the Broad Institute and co-Principal Investigator of the TCGA Genome Characterization Center at Harvard Medical School. She is also a member of the Scientific Steering Committee of the International Cancer Genome Consortium (ICGC). Dr. Chin received her M.D. from the Albert Einstein College of Medicine in New York and conducted her clinical and research training at Albert Einstein College of Medicine and Columbia Presbyterian Medical Center. Her research program focuses on mining and translating complex multi-dimensional genomic data through comparative oncogenomics — the comparison of mouse and human cancers — and the integration with functional genomics to identify novel cancer targets and diagnostic biomarkers.
Andrew Futreal, Ph.D., is a Professor in the Department of Genomic Medicine at The University of Texas, MD Anderson Cancer Center in Houston and Honorary Faculty member at the Wellcome Trust Sanger Insitute. He received his Ph.D. in Pathology from the University of North Carolina at Chapel Hill. He was a faculty member at Duke University until 2000, when he moved to the Wellcome Trust Sanger Institute to become a Co-Founder/Director of the Cancer Genome Project and Head of Cancer Genetics and Genomics. His work has focused on identifying somatic mutations in human cancers and discoverying susceptibility genes for breast and ovarian cancers, which includes his pioneering work on BRCA1 and BRCA2. Dr. Futreal has characterized a number of somatic alterations driving cancer, such as the discovery of somatic BRAF mutations in melanoma and other cancers; ERBB2 intragenic mutations in lung cancer; FBXW7 and PTEN mutations in T-cell leukemia and frequent mutations in genes encoding epigenetic regulators in clear cell renal cell carcinoma.
Ian Watson, Ph.D., is a Postdoctoral Fellow in the laboratory of Dr. Lynda Chin at the Department of Genomic Medicine at The University of Texas, MD Anderson Cancer Center in Houston. He currently holds a Canadian Institute of Health Research (CIHR) Postdoctoral Fellowship. He trained at the Dana Farber Cancer Institute in Boston from 2010-2011 and was a visiting scientist at the Broad Institute prior to moving to MD Anderson Cancer Center. His current area of research focuses on characterizing the genetic driving events in melanoma through integration of computational approaches and functional genomics.
Koichi Takahashi, M.D., received his M.D. from Niigata University, Niigata, and completed his internship and residency in medicine at both Toranomon Hospital, Tokyo, and at Beth Israel Medical Center, New York. He is currently a Clinical Fellow in Hematology and Oncology at The University of Texas, MD Anderson Cancer Center in Houston and is a research scholar at Department of Hematology and Oncology at Kyoto University, Kyoto, Japan. He is currently training in the laboratory of Dr. Lynda Chin. His research focus involves uncovering genomic mechanisms of disease progression and transformation in hematologic malignancies.
Footnotes
Competing interests statement
The authors have no competing interests to declare.
Weblinks:
International Cancer Genome Consortium (ICGC) Data Portal: http://dcc.icgc.org/web/
The Cancer Genome Atlas (TCGA) data portal: https://tcga-data.nci.nih.gov/tcga/
Broad Institute Cancer Portals http://www.broadinstitute.org/scientific-community/science/programs/cancer/cancer-portals-overview
Broad Institute Genome Data Analysis Center (GDAC) https://confluence.broadinstitute.org/display/GDAC/Home
Wellcome Trust Sanger Institute Scientific Cancer Genome Project http://www.sanger.ac.uk/research/projects/cancergenome/
Catalogue of Somatic Mutations in Cancer (COSMIC) http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/
Memorial Sloan-Kettering Cancer Center cBioPortal: http://www.cbioportal.org/public-portal/
Institute for Systems Biology and MD Anderson Cancer Center web portal http://www.cancerregulome.org/cancerstudies.html
UCSC Cancer Genomics Browser https://genome-cancer.ucsc.edu/
UCSC Encyclopedia of DNA Elements http://genome.ucsc.edu/ENCODE/
ClinicalTrials.gov: http://clinicaltrials.gov NCT01711632
Subject categories:
Biological sciences / Cancer / Cancer genomics [URI /631/67/69]
Biological sciences / Genetics / Genomics / Genome evolution [URI /631/208/212/2304]
Biological sciences / Genetics / Functional genomics / Mutagenesis [URI /631/208/191/1908]
Biological sciences / Genetics / Sequencing / Next-generation sequencing [URI /631/208/514/2254]
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Stratton MR. Exploring the genomes of cancer cells: progress and promise. Science. 2011;331:1553–8. doi: 10.1126/science.1204040. [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends in genetics : TIG. 1993;9:138–41. doi: 10.1016/0168-9525(93)90209-z. [DOI] [PubMed] [Google Scholar]
- 4.Chin L, Hahn WC, Getz G, Meyerson M. Making sense of cancer genomic data. Genes Dev. 2011;25:534–55. doi: 10.1101/gad.2017311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nature reviews. Genetics. 2010;11:685–96. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- 6.Parsons DW, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sjoblom T, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 8.Dang L, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–44. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ward PS, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer cell. 2010;17:225–34. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noushmehr H, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–22. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crasta K, et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482:53–8. doi: 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stephens PJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baca SC, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153:666–77. doi: 10.1016/j.cell.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berger MF, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–20. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burns MB, et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013;494:366–70. doi: 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nik-Zainal S, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–93. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor BJ, et al. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. eLife. 2013;2:e00534. doi: 10.7554/eLife.00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.International Cancer Genome, C. et al. International network of cancer genome projects. Nature. 2010;464:993–8. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Creighton CJ, et al. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013 doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kandoth C, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Biankin AV, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cancer Genome Atlas Research, N. et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–25. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujimoto A, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760–4. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- 28.Papaemmanuil E, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365:1384–95. doi: 10.1056/NEJMoa1103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puente XS, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–5. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quesada V, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2012;44:47–52. doi: 10.1038/ng.1032. [DOI] [PubMed] [Google Scholar]
- 31.Stephens PJ, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–4. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Totoki Y, et al. High-resolution characterization of a hepatocellular carcinoma genome. Nat Genet. 2011;43:464–9. doi: 10.1038/ng.804. [DOI] [PubMed] [Google Scholar]
- 33.Cancer Genome Atlas N. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N Engl J Med. 2013 doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang K, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011;43:1219–23. doi: 10.1038/ng.982. [DOI] [PubMed] [Google Scholar]
- 35.Zang ZJ, et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet. 2012;44:570–4. doi: 10.1038/ng.2246. [DOI] [PubMed] [Google Scholar]
- 36.Guichard C, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694–8. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li M, et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat Genet. 2011;43:828–9. doi: 10.1038/ng.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang J, et al. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat Genet. 2012;44:1117–21. doi: 10.1038/ng.2391. [DOI] [PubMed] [Google Scholar]
- 39.Ong CK, et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat Genet. 2012;44:690–3. doi: 10.1038/ng.2273. [DOI] [PubMed] [Google Scholar]
- 40.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 41.Vogelstein B, et al. Cancer genome landscapes. Science. 2013;339:1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lawrence MS, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013 doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hodis E, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–63. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh D, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337:1231–5. doi: 10.1126/science.1220834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Banks RE, et al. Genetic and epigenetic analysis of von Hippel-Lindau (VHL) gene alterations and relationship with clinical variables in sporadic renal cancer. Cancer research. 2006;66:2000–11. doi: 10.1158/0008-5472.CAN-05-3074. [DOI] [PubMed] [Google Scholar]
- 46.Dalgliesh GL, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–3. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Varela I, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539–42. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nature reviews. Cancer. 2011;11:481–92. doi: 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- 49.Versteege I, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–6. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- 50.Wong AK, et al. BRG1, a component of the SWI-SNF complex, is mutated in multiple human tumor cell lines. Cancer research. 2000;60:6171–7. [PubMed] [Google Scholar]
- 51.Harbour JW, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410–3. doi: 10.1126/science.1194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Testa JR, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nature genetics. 2011;43:1022–5. doi: 10.1038/ng.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pena-Llopis S, et al. BAP1 loss defines a new class of renal cell carcinoma. Nature genetics. 2012;44:751–9. doi: 10.1038/ng.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hakimi AA, et al. Adverse Outcomes in Clear Cell Renal Cell Carcinoma with Mutations of 3p21 Epigenetic Regulators BAP1 and SETD2: A Report by MSKCC and the KIRC TCGA Research Network. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:3259–67. doi: 10.1158/1078-0432.CCR-12-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sato Y, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nature genetics. 2013 doi: 10.1038/ng.2699. [DOI] [PubMed] [Google Scholar]
- 56.Agrawal N, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–7. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stransky N, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–60. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nicolas M, et al. Notch1 functions as a tumor suppressor in mouse skin. Nature genetics. 2003;33:416–21. doi: 10.1038/ng1099. [DOI] [PubMed] [Google Scholar]
- 59.Extance A. Alzheimer's failure raises questions about disease-modifying strategies. Nature reviews. Drug discovery. 2010;9:749–51. doi: 10.1038/nrd3288. [DOI] [PubMed] [Google Scholar]
- 60.Davies H, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 61.Berger MF, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485:502–6. doi: 10.1038/nature11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krauthammer M, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44:1006–14. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mok TS. Personalized medicine in lung cancer: what we need to know. Nat Rev Clin Oncol. 2011;8:661–8. doi: 10.1038/nrclinonc.2011.126. [DOI] [PubMed] [Google Scholar]
- 64.Imielinski M, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–20. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rudin CM, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012;44:1111–6. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pleasance ED, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–90. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tomlins SA, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–8. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 68.Barbieri CE, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–9. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grasso CS, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Seshagiri S, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488:660–4. doi: 10.1038/nature11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bass AJ, et al. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion. Nature genetics. 2011;43:964–8. doi: 10.1038/ng.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Banerji S, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486:405–9. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ellis MJ, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486:353–60. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nik-Zainal S, et al. The life history of 21 breast cancers. Cell. 2012;149:994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shah SP, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–9. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Paterlini-Brechot P, et al. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene. 2003;22:3911–6. doi: 10.1038/sj.onc.1206492. [DOI] [PubMed] [Google Scholar]
- 77.Sung WK, et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nature genetics. 2012;44:765–9. doi: 10.1038/ng.2295. [DOI] [PubMed] [Google Scholar]
- 78.Grimwade D, Hills RK. Independent prognostic factors for AML outcome. Hematology Am Soc Hematol Educ Program. 2009:385–95. doi: 10.1182/asheducation-2009.1.385. [DOI] [PubMed] [Google Scholar]
- 79.Abdel-Wahab O, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114:144–7. doi: 10.1182/blood-2009-03-210039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gelsi-Boyer V, et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 2009;145:788–800. doi: 10.1111/j.1365-2141.2009.07697.x. [DOI] [PubMed] [Google Scholar]
- 81.Makishima H, et al. Novel homo- and hemizygous mutations in EZH2 in myeloid malignancies. Leukemia. 2010;24:1799–804. doi: 10.1038/leu.2010.167. [DOI] [PubMed] [Google Scholar]
- 82.Ernst T, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42:722–6. doi: 10.1038/ng.621. [DOI] [PubMed] [Google Scholar]
- 83.Ley TJ, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ley TJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–33. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yan XJ, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet. 2011;43:309–15. doi: 10.1038/ng.788. [DOI] [PubMed] [Google Scholar]
- 86.Mardis ER, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–66. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Graubert TA, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nature genetics. 2012;44:53–7. doi: 10.1038/ng.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Welch JS, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–78. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yoshida K, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–9. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 90.Walter MJ, et al. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia. 2013;27:1275–82. doi: 10.1038/leu.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Makishima H, et al. Somatic SETBP1 mutations in myeloid malignancies. Nature genetics. 2013 doi: 10.1038/ng.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang L, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365:2497–506. doi: 10.1056/NEJMoa1109016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Quesada V, Ramsay AJ, Lopez-Otin C. Chronic lymphocytic leukemia with SF3B1 mutation. N Engl J Med. 2012;366:2530. doi: 10.1056/NEJMc1204033. [DOI] [PubMed] [Google Scholar]
- 94.Fabbri G, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. 2011;208:1389–401. doi: 10.1084/jem.20110921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mullighan CG, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–64. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 96.Mullighan CG, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471:235–9. doi: 10.1038/nature09727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang J, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–63. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Morin RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lohr JG, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. 2012;109:3879–84. doi: 10.1073/pnas.1121343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pasqualucci L, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–95. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pasqualucci L, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43:830–7. doi: 10.1038/ng.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kridel R, et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood. 2012;119:1963–71. doi: 10.1182/blood-2011-11-391474. [DOI] [PubMed] [Google Scholar]
- 103.Chapman MA, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–72. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Walker BA, et al. Intraclonal heterogeneity and distinct molecular mechanisms characterize the development of t(4;14) and t(11;14) myeloma. Blood. 2012;120:1077–86. doi: 10.1182/blood-2012-03-412981. [DOI] [PubMed] [Google Scholar]
- 105.Zenz T, et al. TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol. 2010;28:4473–9. doi: 10.1200/JCO.2009.27.8762. [DOI] [PubMed] [Google Scholar]
- 106.Trbusek M, et al. Missense mutations located in structural p53 DNA-binding motifs are associated with extremely poor survival in chronic lymphocytic leukemia. J Clin Oncol. 2011;29:2703–8. doi: 10.1200/JCO.2011.34.7872. [DOI] [PubMed] [Google Scholar]
- 107.Austen B, et al. Mutations in the ATM gene lead to impaired overall and treatment-free survival that is independent of IGVH mutation status in patients with B-CLL. Blood. 2005;106:3175–82. doi: 10.1182/blood-2004-11-4516. [DOI] [PubMed] [Google Scholar]
- 108.Treon SP, et al. MYD88 L265P somatic mutation in Waldenstrom's macroglobulinemia. N Engl J Med. 2012;367:826–33. doi: 10.1056/NEJMoa1200710. [DOI] [PubMed] [Google Scholar]
- 109.Kiel MJ, et al. Whole-genome sequencing identifies recurrent somatic NOTCH2 mutations in splenic marginal zone lymphoma. J Exp Med. 2012;209:1553–65. doi: 10.1084/jem.20120910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Koskela HL, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366:1905–13. doi: 10.1056/NEJMoa1114885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tiacci E, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011;364:2305–15. doi: 10.1056/NEJMoa1014209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dietrich S, et al. BRAF inhibition in refractory hairy-cell leukemia. N Engl J Med. 2012;366:2038–40. doi: 10.1056/NEJMc1202124. [DOI] [PubMed] [Google Scholar]
- 113.Kuo KT, et al. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am J Pathol. 2009;174:1597–601. doi: 10.2353/ajpath.2009.081000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jones S, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–31. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wiegand KC, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363:1532–43. doi: 10.1056/NEJMoa1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Van Raamsdonk CD, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Van Raamsdonk CD, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363:2191–9. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schwartzentruber J, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–31. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 119.Tefferi A, et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood. 2007;110:1092–7. doi: 10.1182/blood-2007-04-083501. [DOI] [PubMed] [Google Scholar]
- 120.Grimwade D, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116:354–65. doi: 10.1182/blood-2009-11-254441. [DOI] [PubMed] [Google Scholar]
- 121.Greenberg PL, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454–65. doi: 10.1182/blood-2012-03-420489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bejar R, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364:2496–506. doi: 10.1056/NEJMoa1013343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bejar R, et al. Validation of a Prognostic Model and the Impact of Mutations in Patients With Lower-Risk Myelodysplastic Syndromes. J Clin Oncol. 2012 doi: 10.1200/JCO.2011.40.7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Patel JP, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–89. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schlenk RF, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909–18. doi: 10.1056/NEJMoa074306. [DOI] [PubMed] [Google Scholar]
- 126.Rosenbloom KR, et al. ENCODE Data in the UCSC Genome Browser: year 5 update. Nucleic acids research. 2012 doi: 10.1093/nar/gks1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Horn S, et al. TERT Promoter Mutations in Familial and Sporadic Melanoma. Science. 2013 doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- 128.Huang FW, et al. Highly Recurrent TERT Promoter Mutations in Human Melanoma. Science. 2013 doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yates LR, Campbell PJ. Evolution of the cancer genome. Nature reviews. Genetics. 2012;13:795–806. doi: 10.1038/nrg3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Carter SL, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–21. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Greenman C, Wooster R, Futreal PA, Stratton MR, Easton DF. Statistical analysis of pathogenicity of somatic mutations in cancer. Genetics. 2006;173:2187–98. doi: 10.1534/genetics.105.044677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Parmigiani GL, J., Simina, Sjoblom T, Kinzler KW, Velculescu VE, Vogelstein B. Working Paper. Vol. 126. Johns Hopkins University, Dept. of Biostatistics Working Papers.; 2007. Statistical methods for the analysis of cancer genome sequencing data. [Google Scholar]
- 133.Youn A, Simon R. Identifying cancer driver genes in tumor genome sequencing studies. Bioinformatics. 2011;27:175–81. doi: 10.1093/bioinformatics/btq630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ding L, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–75. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kan Z, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–73. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 136.Getz G, et al. Comment on “The consensus coding sequences of human breast and colorectal cancers”. Science. 2007;317:1500. doi: 10.1126/science.1138764. [DOI] [PubMed] [Google Scholar]
- 137.Dees ND, et al. MuSiC: identifying mutational significance in cancer genomes. Genome Res. 2012;22:1589–98. doi: 10.1101/gr.134635.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pleasance ED, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–6. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Schuster-Bockler B, Lehner B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature. 2012;488:504–7. doi: 10.1038/nature11273. [DOI] [PubMed] [Google Scholar]
- 140.Hellmann I, et al. Why do human diversity levels vary at a megabase scale? Genome research. 2005;15:1222–31. doi: 10.1101/gr.3461105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Stamatoyannopoulos JA, et al. Human mutation rate associated with DNA replication timing. Nature genetics. 2009;41:393–5. doi: 10.1038/ng.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nature methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic acids research. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sim NL, et al. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic acids research. 2012;40:W452–7. doi: 10.1093/nar/gks539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Carter H, et al. Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer research. 2009;69:6660–7. doi: 10.1158/0008-5472.CAN-09-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Verhaak RG, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Shibata T, et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:13568–73. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ciriello G, Cerami E, Sander C, Schultz N. Mutual exclusivity analysis identifies oncogenic network modules. Genome research. 2012;22:398–406. doi: 10.1101/gr.125567.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Vandin F, Upfal E, Raphael BJ. Algorithms for detecting significantly mutated pathways in cancer. Journal of computational biology : a journal of computational molecular cell biology. 2011;18:507–22. doi: 10.1089/cmb.2010.0265. [DOI] [PubMed] [Google Scholar]
- 150.Garraway LA, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–22. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- 151.Ruark E, et al. Mosaic PPM1D mutations are associated with predisposition to breast and ovarian cancer. Nature. 2013;493:406–10. doi: 10.1038/nature11725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Heyer J, Kwong LN, Lowe SW, Chin L. Non-germline genetically engineered mouse models for translational cancer research. Nature reviews. Cancer. 2010;10:470–80. doi: 10.1038/nrc2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kim M, et al. Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell. 2006;125:1269–81. doi: 10.1016/j.cell.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 154.Maser RS, et al. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature. 2007;447:966–71. doi: 10.1038/nature05886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zender L, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006;125:1253–67. doi: 10.1016/j.cell.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wartman LD, et al. Sequencing a mouse acute promyelocytic leukemia genome reveals genetic events relevant for disease progression. The Journal of clinical investigation. 2011;121:1445–55. doi: 10.1172/JCI45284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–4. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ding L, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–10. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tao Y, et al. Rapid growth of a hepatocellular carcinoma and the driving mutations revealed by cell-population genetic analysis of whole-genome data. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:12042–7. doi: 10.1073/pnas.1108715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Govindan R, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150:1121–34. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.