

Abstract
Thermoplastic biodegradable polymers displaying elastomeric behavior and mechanical consistency are greatly appreciated for the regeneration of soft tissues and for various medical devices. However, while the selection of a suitable base material is determined by mechanical and biodegradation considerations, it is the surface properties of the biomaterial that are responsible for the biological response. In order to improve the interaction with cells and modulate their behavior, biologically active molecules can be incorporated onto the surface of the material. With this aim, the surface of a lactide and caprolactone based biodegradable elastomeric terpolymer was modified in two stages. First, the biodegradable polymer surface was aminated by atmospheric pressure plasma treatment and second a crosslinker was grafted in order to covalently bind the biomolecule. In this study, albumin was used as a model protein. According to X-ray photoelectron spectroscopy (XPS) and atomic force microscopy (AFM), albumin was efficiently immobilized on the surface of the terpolymer, the degree of albumin surface coverage (ΓBSA) reached ~35%. Moreover, gel permeation chromatography (GPC) studies showed that the hydrolytic degradation kinetic of the synthesized polymer was slightly delayed when albumin was grafted. However, the degradation process in the bulk of the material was unaffected, as demonstrated by Fourier transform infrared (FTIR) analyses. Furthermore, XPS analyses showed that the protein was still present on the surface after 28 days of degradation, meaning that the surface modification was stable, and that there had been enough time for the biological environment to interact with the modified material.
Keywords: albumin, grafting, hydrolytic degradation, poly(lactide/ε-caprolactone), surface modification, terpolymer
Introduction
The synthesis of lactide and caprolactone based copolymers (PLCL) could play a role in the growing demand for highly elastic and biodegradable materials in the biomedical field. The mechanical properties displayed by these copolymers are of key importance regarding cardiac tissue engineering applications, where biomaterials should have enough elasticity to respond synchronically with the myocardium contraction and to transfer effectively the mechanical stimuli from the myocardial microenvironment to the transported cells.1 Moreover, the elastomeric behavior and shape recovery capacity of these materials create a dynamic cell culture with mechanical stimulation, this promotes cell proliferation, extracellular matrix production and gene expression for the regeneration of muscles, tendons or ligaments.2 The degradability of these copolyesters is also highly appreciated for its use in tissue engineering applications. For example, in urothelium tissue engineering, the use of these resorbable polymers could be a promising alternative to the techniques currently employed, such as the use of the patient’s own genital tissue or buccal mucosal grafts.3
Both the mechanical properties and degradation behavior of these materials can be tuned by controlling the composition and chain microstructure.4-6 For example, the presence of ε-caprolactone improved the elastomeric behavior of the obtained material, avoiding problems related to brittleness associated with poly (L-lactide) (PLLA), poly(D,L-lactide) (PDLLA) or poly(lactide-co-glycolyde) (PLGA) polymers, making them unsuitable for biomedical applications requiring elastomeric behavior. On the other hand, some PLCL copolymers found in the literature present initially elastomeric behavior but, during hydrolytic degradation, they become glassy plastics.6,7 Amorphous character is also very important for the use of this material in tissue engineering and biomedical applications. Commonly employed synthetic biodegradable polymers, such as PLLA, poly(ε-caprolactone) (PCL) or other PLCL copolymers are highly crystalline or develop crystalline domains during hydrolytic degradation, thus displaying slow degradation rates.6-8 The aforementioned crystalline residues are highly resistant to hydrolytic degradation and can remain in the body for years, causing foreign body reactions.9,10 In contrast to other commonly employed lactide and caprolactone based copolymers (PLCLs),7 the PLCLs developed in our laboratory, due to a specific lactide/caprolactone ratio, exhibited minor changes in their microstructure during hydrolytic degradation, retaining their elastomeric behavior and amorphous character during application.8 The formation of non-desirable crystalline residues is avoided thanks to the random distribution of polymer chains and the corresponding shortening of the average-sequence length of crystallizable units.
As an illustration, in our previous work we synthesized several PLCLs displaying different average-sequence lengths of L-lactide (lLA), that were hydrolytically degraded for a period up to 98 d.8 All the studied PLCLs presented initially a completely amorphous character, showing a single glass transition temperature and no melting peaks. However, while those PLCLs that showed lLA shorter than ~3.2 were able to maintain their amorphous character during the hydrolytic degradation study, PLCLs that displayed longer values of lLA slightly crystallize in the late stages of degradation. In this sense, the PLCL that had a lLA of 3.62 showed a melting peak at day 35 of degradation that gradually increased as the time submerged in phosphate-buffered saline increased. On the other hand, the PLCL that had a lLA of 3.28 was able to preserve the amorphous character until day 49. In order to avoid the formation of hydrolytically resistant crystalline residues and the brittleness problems associated to the development of crystalline domains, we synthesized in this work a PLCL with reduced lLA (<3.28). This polymer, having a very similar composition and chain microstructure of one of the polymers studied previously,8 does not crystallize during degradation and keeps its elastomeric behavior and structural properties during the first 3–4 wk of degradation.
Biodegradable materials for biomedical applications should not only have appropriate mechanical and biodegradation properties, but also good interactions with the surrounding tissues of the implantation site. Indeed, the biological response is mainly influenced by the surface properties of the biomaterial. Therefore, incorporation of biologically active molecules, such as proteins or peptides, on the surface of biomaterials is recognized as a promising strategy to improve the bioactivity of the materials employed in the medical field.11,12 In order to obtain a stable protein layer, the biomolecule should be covalently bonded on the surface of the material. However, PLCL copolymers do not have any reactive groups in their chemical structure for further covalent attachment of proteins. Thus, they should be functionalized, which is a major challenge for this kind of material. Indeed, special care has to be taken when trying to modify the surface of biodegradable polymers showing glass transition temperatures similar to room or body temperatures such as the PLCL used in this work. Moreover, the functionalization should not affect the copolymer structural composition in order to keep all its initial properties.
Among the numerous techniques described in literature for the introduction of reactive groups,13,14 surface modification by plasma treatment is the most adequate15,16 since it allows the efficient incorporation of functional groups onto the biomaterial surface without affecting their bulk properties (chain reorganization, crystallinity, faster degradation).17,18 Regarding plasma treatments applied on biodegradable polymers, to our knowledge, no study on PLCL has been reported whereas such treatments have already been done on PLLA, poly(glycolide) (PGA), PLGA or PCL.19-22
In this work a terpolymer composed of ε-caprolactone (CL), D-lactide (D-LA) and L-lactide (L-LA) was used as base material and fully characterized in terms of molecular weight, molecular organization, chemical structure and mechanical properties before surface modification. The terpolymer functionalization was done under N2 and H2 atmospheric pressure plasma discharge. The amino groups created were used as the anchoring point for the covalent grafting of albumin by using a heterobifunctional crosslinker. X-ray photoelectron spectroscopy (XPS), contact angle and atomic force microscopy (AFM) were used to characterize the surface modifications of the material. Finally, a hydrolytic degradation study of untreated terpolymer (PLCL), plasma modified PLCL (P-PLCL) and albumin grafted PLCL (BSA-PLCL) in phosphate-buffered saline (PBS) at 37 °C was performed for a period up to 63 d. The changes in crystallinity, molecular weight and chemical composition of the samples were followed by using differential scanning calorimetry (DSC), gel permeation chromatography (GPC), Fourier Transform Infrared spectroscopy (FTIR) and XPS.
Results and Discussion
Initial characterization of the synthesized polymer
As previously described, the chain microstructural and mechanical properties as well as the amorphous character of PLCL materials should be suitable for use in many biomedical applications. Therefore, the terpolymer employed in this work was fully characterized (Table 1). The obtained material showed a single glass transition temperature at ~34 °C and was completely amorphous, limiting the foreign body reactions due to the highly resistant crystalline residues.9,10 The initial molecular weight was 101.6 × 103 g/mol with a dispersity of 2.3. From 1H NMR, it was concluded that the terpolymer was composed of 66.9% of L-lactide, 17.4% of D-lactide and 15.7% of ε-caprolactone. The high randomness character (R close to 1) and the incorporation of D-lactide, which disrupts the chain microstructural arrangements of crystallizable L-lactide, allow its average sequence length to be shortened (lL-LA = 3.18).
Table 1. Main characteristics of the lactide and caprolactone based terpolymer.
| Structural properties | Mw (x103) | 101.6 g/mol | |
| Dispersity (D) | 2.3 | ||
| %L-lactide | 66.9% | ||
| Composition1 | %D-lactide | 17.4% | |
| %ε-caprolactone | 15.7% | ||
| Microstructural magnitudes2 | lLA | 6.68 | |
| lL-LA3 | 3.18 | ||
| lD-LA3 | 1.27 | ||
| lCL | 1.25 | ||
| R | 0.95 | ||
| Physical properties | Tg | ~34°C | |
| Mechanical properties | Secant modulus (at 2%) | 21°C | 763.2 ± 68.3 MPa |
| 37°C | 10.5 ± 1.4 MPa | ||
| Yield strength | 21°C | 21.1 ± 1.8 MPa | |
| 37°C4 | 0.7 ± 0.1 MPa | ||
| Tensile strength | 21°C5 | 17.1 ± 0.9 MPa | |
| 37°C6 | 1.6 ± 0.1 MPa | ||
| Elongation at break | 21°C | 354 ± 35% | |
| 37°C7 | >300% | ||
| Strain recovery | 21°C | 79 ± 4% |
1 Calculated from 1H NMR spectra. Because of the impossibility of offering the exact L-LA and D-LA molar content (indistinguishable in the NMR spectrum), approximate values are given under the assumption that the reactivity of both L-LA and D-LA are the same. 2lLA and lCL are the LA and CL number average sequence lengths obtained from 1H NMR. These values are compared with the Bernoullian random number-average sequence lengths (lLA = 1/CL and lCL = 1/LA), obtaining the randomness character value (R). 3lL-lA and lD-LA are the L-LA and D-LA approximate values of number average sequence lengths under the assumption that the reactivity of both L-LA and D-LA are the same. 4Offset Yield Strength was calculated at a 10% of strain using the secant modulus at 2% as elastic modulus (E) for the material at 37 °C since no yield point was observed in the stress-strain plot. 5The tensile strength was determined as ultimate stress value (σu). 6The tensile strength was determined as the stress value at 300% of deformation. 7At 37 °C, the maximum permitted distance between clamps corresponds to a sample deformation of 300%.
This terpolymer exhibits excellent mechanical properties, showing a neat yield point at 21.1 MPa, a secant modulus of 763.2 MPa and elongation at break of 354% at room temperature (Table 1). Moreover, the polymer presented 79% of strain recovery after break. For this reason, the behavior of the material at room temperature can be considered as that of a thermoplastic elastomer (TPE), showing an intermediate behavior between a glassy plastic with yield point and an elastomer of high strain capability and elastic recovery.8 Please note that TPEs do not need covalent crosslinking but soft and hard domains in the chain microstructure that will interact through entanglements (physical crosslinks) providing elastomeric behavior. At body temperature (37 °C), the material also showed an elastomeric behavior having a secant modulus of 10.5 MPa, no neat yield point and elongation at break > 300%. This improved elastomeric behavior of the material is mainly due to the presence of ε-caprolactone, which prevented the brittleness related problems associated with PLLA, PDLLA or PLGA polymers. Indeed, these materials usually show a high Young’s modulus (~3 GPa) but low elongation at break (~4%).23,24 On the other hand, some PLCL copolymers found in the literature present initially elastomeric behavior but, during hydrolytic degradation, they are submitted to microstructural rearrangements and their mechanical properties shift from elastomeric to glassy plastics.6,7 For example, Fernández et al.6 found that a PLCL with a composition of 74% of L-lactide and 26% of ε-caprolactone exhibited initially a low Young’s modulus (19.4 MPa) and high elongation at break (~324%). However, after two weeks immersed in PBS at 37 °C, the material lost its elastomeric character presenting a Young’s modulus of 135 MPa and a elongation at break of ~121% due to the development of crystalline domains. Karjalainen et al.7 also found this transition from elastomeric to glassy plastic in a PLCL copolymer composed of 40% of L-lactide and 60% of ε-caprolactone. In this sense, this material became more brittle in the course of hydrolysis and it exhibited plastic-like rather than rubber-like deformation.
The mechanical properties of this PLCL at 37 °C make the obtained material into a promising candidate for application in the regeneration of soft tissues and the fabrication of medical devices requiring elastomeric behavior and mechanical consistency at body temperature.
Albumin grafting
Surface chemical characterization
In order to improve the interaction between the PLCL and the biological environment, surface modifications were investigated. In this work we used atmospheric pressure plasma and sulfo-SMPB (sulfosuccinimidyl 4-[p-maleimidophenyl]butyrate) in order to graft the albumin, as biomolecule model, onto PLCL film (Fig. 1). Table 2 and Figure 2 show the XPS survey data and the C1s high resolution (HR) spectra of the samples after each surface modification step. The surface of the untreated terpolymer contained 61.5 ± 0.7% of carbon and 38.5 ± 0.7% of oxygen, which correspond to a C/O ratio of 1.6, that is very close to the theoretically calculated value of 1.7. Furthermore, the C1s HR spectrum of the pristine PLCL (Fig. 2A) can be satisfactorily fitted with three peaks at 285.0, 286.9 and 289.0 eV, which correspond to C–C/C–H, C–O and O–C = O groups respectively.25 After plasma treatment, the C/O ratio increased to 2.4, meaning that oxygen containing chemical groups were reduced during the plasma treatment, due to PLCL surface degradation. This observation can be explained by ester group scissions followed by decarboxylation step as already described in literature on polyesters.26-28 This hypothesis was also correlated by the significant drop in the relative area of O–C = O peak in the C1s HR spectrum (Fig. 2B) while the contribution at 285.0 eV (C-C and C-H) increased. The contribution at 286.9 eV (Fig. 2B), associated with C-O and C-N bonds, and the nitrogen peak in the surveys (3.5 ± 1.0%) increased after the plasma treatment, indicating the presence of nitrogen containing chemical groups on the surface of the material. In order to differentiate amino groups from the other nitrogen containing groups (imine, nitrile, etc.) formed during the plasma treatment and to quantify them, a derivatization reaction with 4-(trifluoromethyl)benzaldehyde (TFBA) was performed.29 Indeed, the aldehyde group of TFBA reacts specifically and quantitatively with NH2 groups to form azomethines. The detection of fluorine in the XPS survey spectrum confirmed the presence of amino groups (data not shown), and also allowed the percentage of these groups to be calculated. According to equation 8 (see Materials and Methods part), the surface amino content reached ~0.5%, producing enough reactive sites for further biomolecule immobilization.

Figure 1. Synthesis of the PLCL (above) and schematic representation of the covalent grafting of BSA on its surface (below).
Table 2. Surface compositions determined by XPS survey analyses and contact angle (CA) values after each surface modification step.
| C(%) | O(%) | N(%) | C/O ratio | %NH2 | CA (°) | |
|---|---|---|---|---|---|---|
| Pristine | 61.5 ± 0.7 | 38.5 ± 0.7 | - | 1.6 | - | 73 ± 2 |
| Plasma treated | 67.7 ± 0.9 | 28.8 ± 1.9 | 3.5 ± 1.0 | 2.4 | 0.4 ± 0.1 | 29 ± 2 |
| SMPB grafting | 62.0 ± 0.5 | 37.8 ± 0.4 | 0.2 ± 0.1 | 1.6 | - | - |
| Albumin grafting | 65.8 ± 1.2 | 29.8 ± 1.3 | 4.4 ± 0.6 | 2.2 | - | 52 ± 2 |

Figure 2. High-resolution C 1s spectra of (A) pristine PLCL, (B) plasma treated PLCL, (C) sulfo-SMPB grafted PLCL and (D) albumin grafted PLCL.
After the grafting of sulfo-SMPB, the nitrogen signal drastically decreased as this molecule provides only one atom of nitrogen on the polymer surface after surface conjugation. However, it was difficult to demonstrate the grafting of the crosslinker with the survey data and the C1s HR spectrum (Fig. 2C) since this molecule contains C, O and N, all of which were previously found on the surface of the plasma treated sample. Therefore, a similar crosslinker with succinimidyl end group as sulfo-SMPB and containing a specific atom, iodine (N-succinimidyl iodoacetate, SIA), was grafted onto the surface of the plasma treated sample. The presence of iodine in the XPS surveys confirms without any doubt the crosslinker grafting (data not shown), meaning surface amino groups were able to react with succinimydyl derivatives.
Once the SMPB was grafted onto the aminated surface, its free maleimidyl functionality can be further used as an anchoring point for biomolecule immobilization as it reacts with cystein from protein (Fig. 1). After this reaction, 4.4 ± 0.6% of nitrogen was detected in XPS whereas only 0.2 ± 0.1% was detected after SMPB grafting. As proteins are mainly composed of C, O and N, this increase in nitrogen was associated with albumin grafting, which was ascertained by C1s HR spectrum (Fig. 2D). Indeed, in order to fit the C1s spectrum correctly, an additional peak located at 287.6 eV, assigned to O = C–NH groups was necessary. These functionalities are typically associated with peptidic bonds coming from the albumin immobilization on the terpolymer surface. It should also be mentioned that albumin adsorbed on the PLCL sample in the same conditions as in the grafting process (with SMPB) was efficiently removed by rinsing the surface thoroughly: no nitrogen detected in XPS. Therefore, the nitrogen content measured from the XPS survey spectra after grafting can be used to roughly estimate the degree of albumin surface coverage (ΓBSA) employing the following equation:30
| ΓBSA = (AmN/ApN) × 100 | (1) |
where AmN is the nitrogen atomic percentage on the surface of the material measured by XPS and ApN is the nitrogen atomic percentage of albumin under complete coverage of the surface with the protein. ApN is 12.95% based on the total atomic composition of C, O, N, and S elements in pure albumin.30 According to the obtained results, the protein surface coverage is estimated at ~36%, which means that more than one third of the surface is covered with albumin.
Surface topography and hydrophilicity
The influence of each surface modification step was also followed by AFM (Fig. 3) and CA on pristine PLCL, plasma-treated PLCL and albumin grafted PLCL. Pristine PLCL displayed a smooth surface with no defects and presented a root mean squared roughness (RRMS) of 0.6 nm for 20 × 20 µm2. After plasma treatment, the surface was slightly damaged and the RRMS increased to 4.5 nm. The filamentary discharge generated with the plasma conditions used in this work favors amine formation on the surface but causes higher damage in comparison to the glow discharge regime.31,32 Plasma treatment should be thus a good compromise between amine grafting and fragmentations (polymer chain scission). For this PLCL, a treatment time of one minute seems to be the best option. Indeed a longer treatment time gave a high degree of fragmentation (exhibited by AFM images) and degradation (evidenced by the C/O ratio increase in XPS) (data not shown).

Figure 3. AFM images (20 × 20 µm2) of pristine PLCL (top-left), plasma treated PLCL (top-right), albumin-grafted PLCL (bottom-left) and albumin-grafted PLCL treated with Image J software (bottom-right). In the image treated by Image J, white spots correspond to albumin deposits and where used to estimate albumin surface coverage.
AFM images on the albumin grafted sample showed that some aggregates can be ascribed to the presence of albumin deposits (Fig. 3), bringing an increase in the surface roughness: RRMS ~7.3 nm. From these deposits, and by using ImageJ software, the surface coverage can be estimated. The albumin surface coverage deduced was ~30%, which is in accordance with the value obtained from XPS analysis, ~36%.
From water contact angle measurements (Table 2) it was observed that the hydrophilicity of the surface considerably increased after plasma treatment. In fact, the water contact angle decreased from 73 ± 2° for the untreated polymer to 29 ± 2° for the plasma-treated polymer due to the incorporation of polar functionalities. Finally, after the albumin grafting the value of the water contact angle increased to 52 ± 2°, therefore indicating the surface chemistry modification resulting from the albumin conjugation.
Hydrolytic degradation study
The control over the chain microstructure and the composition of the polymers allows the design of materials with a predictable hydrolytic degradation rate. However, the surface modification and the grafting of biologically active molecules may alter the expected degradation behavior.33,34 In order to evaluate the effect of surface modification, the hydrolytic degradation of the pristine terpolymer (PLCL), plasma modified PLCL (P-PLCL) and albumin grafted PLCL (BSA-PLCL) were studied in PBS at 37 °C for a period up to 63 d.
As determined by DSC (Fig. 4), all the samples maintained their amorphous structure during the in vitro degradation study, displaying a single glass transition and no melting peaks in their thermograms. It has been previously reported that those lactide and caprolactone based terpolymers showing L-lactide average sequence length (lLA) shorter than 3.62 underwent minor changes in their thermal transitions and maintained their amorphous nature during degradation, avoiding the production of non-desirable crystalline residues.8 Neither the plasma modification (P-PLCL) nor the albumin grafting (BSA-PLCL) influenced the observed thermograms during the course of this study.
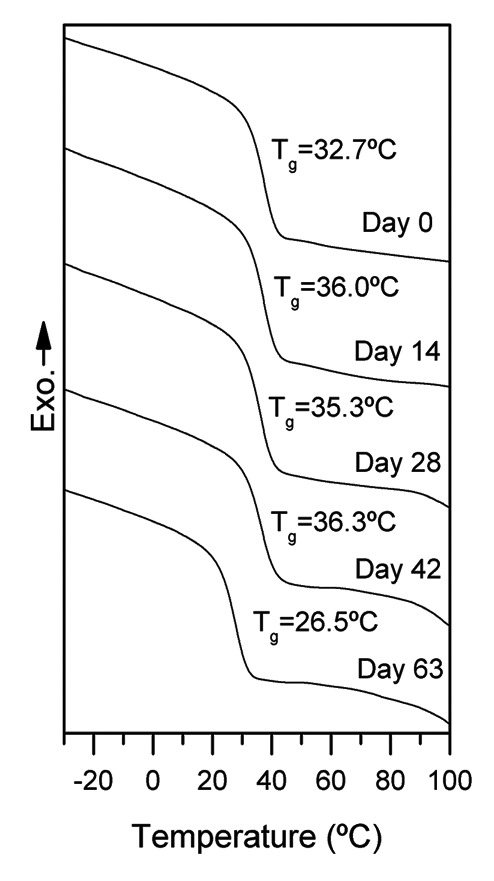
Figure 4. DSC curves of the 1st scans of PLCL at different degradation times.
Figure 5 shows the evolution of weight-averaged molecular weight (Mw) for the studied systems and gravimetric data for PLCL and P-PLCL.
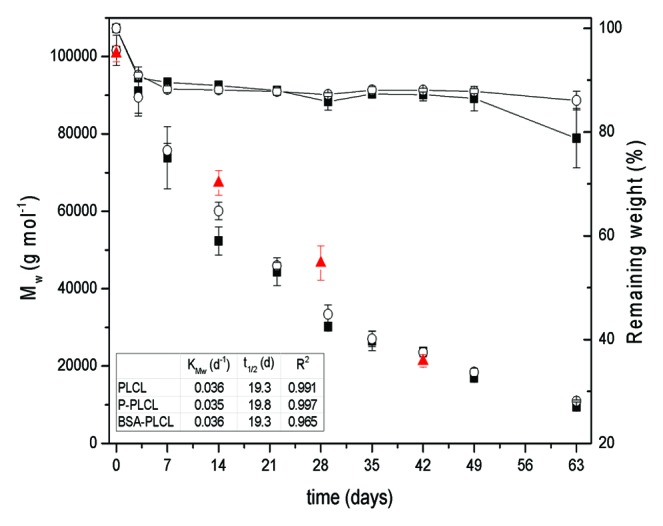
Figure 5. Evolution of Mw at different times of degradation for PLCL (■), P-PLCL (○) and BSA-PLCL (▲) (left axis) and gravimetric data for PLCL (■) and P-PLCL (○) (right axis).
As it can be seen, Mw for both PLCL and P-PLCL followed (R2 > 0.99) the exponential relationship previously described for biodegradable polyesters degrading under bulk degradation satisfactorily:35
| lnMw = lnMw0 – KMw t | (2) |
| t1/2 = ln 2/KMw | (3) |
where Mw is the weight-averaged molecular weight, Mw0 is the initial weight-averaged molecular weight, KMw represents the apparent degradation rate and t1/2 is the half degradation time.
KMw for PLCL and P-PLCL were respectively 0.036 and 0.035 d−1 and their corresponding t1/2 were 19.3 and 19.8 d. In view of these results, it can be concluded that the incorporation of amino functionalities on the surface of the pristine polymer did not modify its degradation behavior to a large extent. Amino functionalities are relatively small, so they can easily migrate from the surface to the bulk material due to the surface reorganization occurring at 37 °C and in aqueous medium. In fact, no nitrogen signals were observed in XPS surveys after immersing the plasma treated sample during 1 h in PBS at room temperature (not shown), indicating the migration of amino functionalities from the surface of the sample to the bulk and/or the solubilisation of small polymer chains containing amino groups in the aqueous medium. Therefore, the effect of plasma treatment on the degradation behavior of the studied terpolymer was almost negligible.
GPC results exhibited different degradation behavior between pristine or plasma-treated PLCL and BSA-PLCL samples. In fact, during the first 4 wk of degradation, the Mw for BSA-PLCL was maintained well above the Mw of both PLCL and P-PLCL. For example, after 28 d immersed in PBS, the Mw of PLCL and P-PLCL were respectively (30.2 ± 0.9) × 103 and (33.4 ± 2.4) × 103 g/mol whereas the Mw of BSA-PLCL was (46.6 ± 4.5) × 103 g/mol. At day 42, the observed Mw for BSA-PLCL was very similar to those observed for PLCL and P-PLCL. On this day, the Mw of PLCL, P-PLCL and BSA-PLCL were respectively (23.7 ± 0.8) × 103, (23.6 ± 1.2) × 103 and (21.3 ± 1.6) × 103 g/mol.
According to the GPC results, the bulk degradation was different in the first 4 wk for the PLCL or P-PLCL and the BSA-PLCL. However, FTIR analyses clearly showed that the degradation process of BSA-PLCL samples was almost identical to PLCL and P-PLCL samples. Figure 6 shows FTIR spectra of PLCL samples after 0, 14, 28, 42 and 63 d immersed in PBS at 37 °C. The main bands appearing in the spectrum of PLCL at day 0 are assigned to the carbonyl (C = O) stretching at 1750 cm−1, the –CH3 bending at 1452 and 1370 cm−1 and C–O stretching at 1185 and 1046 cm−1.36 Few changes were observed in the spectra until 21 d of degradation. However, at day 28, a new band at 1600 cm−1 appeared and gradually increased as the degradation occurred. This band may have been associated with the carboxylate (–COO-) groups formed due to the hydrolytic scission of the ester groups present in the PLCL structure.37,38 The ratio between the (–COO-) and the (C = O) band was employed as an indicator of water absorption and subsequent hydrolysis of ester bonds. As seen in Figure 7, this ratio was almost identical for the PLCL and BSA-PLCL samples, indicating that the hydrolysis of ester bonds and the resulting chain-scission was occurring in both samples in a similar way.
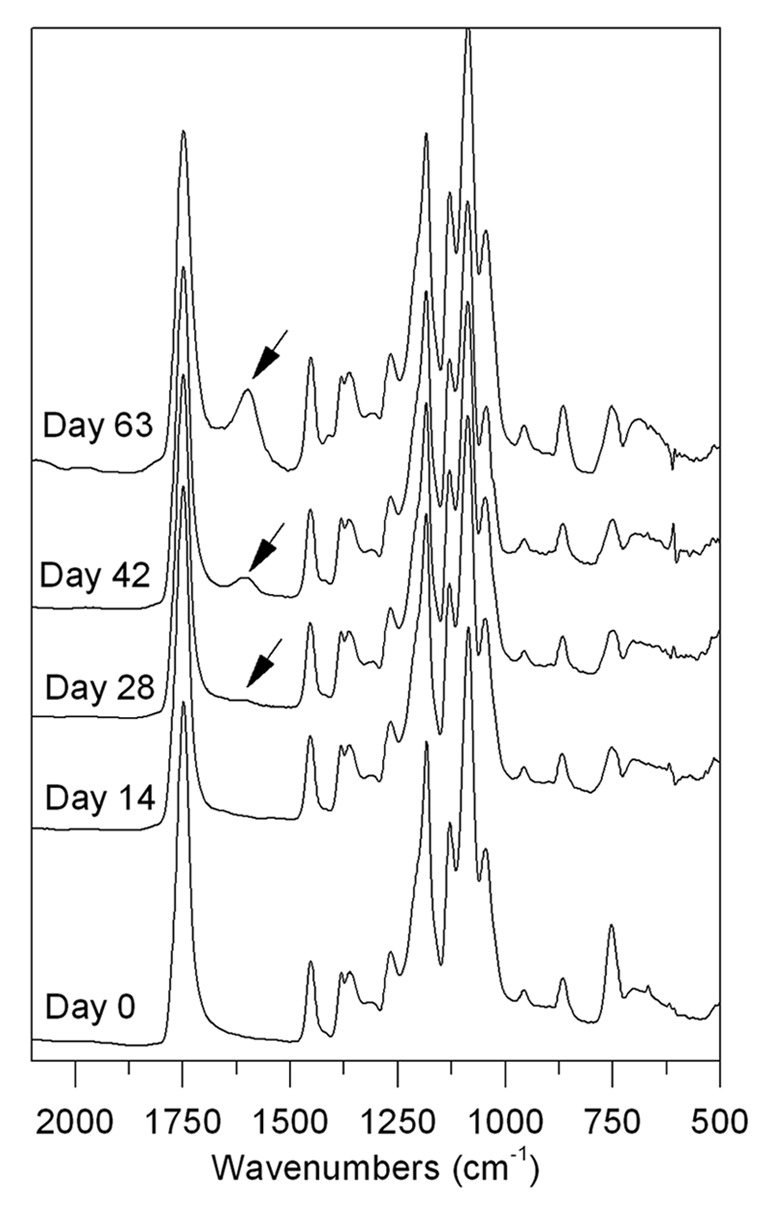
Figure 6. FTIR spectra for PLCL at different degradation times.
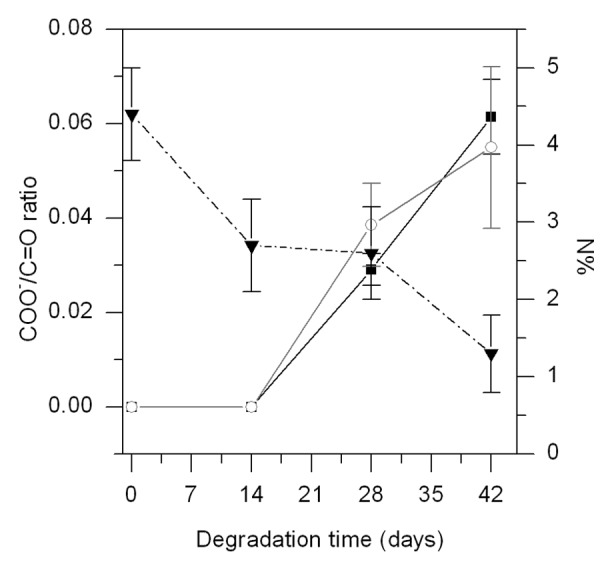
Figure 7. COO-/C = O ratio for PLCL (―○—) and BSA-PLCL (―■—) samples (left axis) and %N detected by XPS on the surface of BSA-PLCL sample during degradation (- - ▼ - -) (right axis).
In view of these results, it can be concluded that the incorporation of BSA on the surface of the polymer did not affect the degradation of the bulk polymer to a great extent. The differences observed by GPC measurements could be associated with the presence of albumin on the surface of the BSA-PLCL samples during the first 28 d of degradation. In fact, as determined by XPS (Fig. 7), the nitrogen content associated with the presence of albumin on the surface of the sample dropped from the initial value of 4.4 ± 0.6% to 2.6 ± 0.6% on day 28 of degradation and further decreased to 1.3 ± 0.5% at day 42. These results demonstrate that, despite the polymer degradation, the albumin was still present after 28 d, confirming the stability of the surface modification. Therefore, this could lead more time for the biological environment to interact with the modified materials, favoring the biological activity and response of surrounding cells.
The mechanical properties and degradation times of the material employed indicate that it may be a good candidate for use in vascular related tissue engineering applications. According to other studies, elastomeric behavior and relatively short degradation times are required for these applications.39-41 Indeed, Pektok et al.39 found almost complete neointima formation in a PCL scaffold implanted in a rat’s arteries after 6 wk of implantation. Finn et al.40 found 80% of endothelialisation in a bare metal stent implanted in the artery of 38-y-old woman after 2 mo of implantation. Finally, Kotani et al.41 reported complete neointimal coverage in bare metal stents implanted in humans after 3 to 6 mo of implantation. The material employed in this work, presenting an in vitro degradation time of 2 to 3 mo, could induce tissue regeneration and gradually degrade as the new tissue (neoendothelium) is developing.
Materials and Methods
Synthesis of the PLCL terpolymer
The statistical poly(lactide-co-ε-caprolactone) (PLCL) terpolymer containing L-lactide, D,L-lactide (>99.5%, Purac Biochem BV) and ε-caprolactone (>98%, Merck KGaA) was synthesized following the procedure described in a previous work.42 In brief, the polymer was synthesized by ring opening polymerization (ROP) catalyzed by bismuth (III) subsalicylate (BiSS, 99.9% trace metals basis, Sigma-Aldrich, 1500: 1 comonomers: catalyst molar ratio) for 72 h at 130 °C, adding 50 wt% of L-lactide, 35% of D,L-lactide and 15% of ε-caprolactone. The obtained product was dissolved in chloroform (Labbox) and precipitated by pouring the polymer solution into an excess of methanol (Labbox) in order to remove the catalyst impurities and the monomers that did not react. The chemical structure of a lactide and caprolactone based polymer is displayed in Figure 1.
Surface modification and albumin grafting
The surface modification procedure followed in this work is illustrated in Figure 1. First, the polymer was dissolved in chloroform (Laboratoire MAT, 15 wt%) and poured onto glass slides (1.25 × 1.25 cm2). To evaporate the solvent, the samples were placed under ambient conditions for 24 h and thoroughly dried in a vacuum oven for another 72 h.
In order to introduce amino functionalities onto the surface of the samples, an atmospheric plasma treatment was performed.31 Samples were placed in a conventional parallel-plate dielectric barrier discharge (DBD) reactor on the grounded electrode. Gas flow (95% N2 + 5% H2) was introduced directly between the electrodes through a diffuser and was maintained constant at 10 L/min. The frequency, the applied voltage, the gas gap and the treatment time were kept constant (3 kHz, 15 kV, 1 mm and 60 s). Before and after each plasma treatment, the plasma chamber was purged for 5 min to ensure homogeneity and gas purity for the discharge and to avoid post-plasma oxidation reactions with free radicals or unstable functional groups present on the surface.
Plasma treated samples were then submerged in phosphate buffered saline (PBS, pH 7.4, Sigma) containing 2–3 mg/mL of sulfo-SMPB (G-Biosciences) for 2 h at 4 °C under stirring and sheltered from light to prevent light-induced degradation of the crosslinker. It should be mentioned that, when trying to perform the grafting reaction between the amino groups and sulfo-SMPB at room temperature, no maleimidyl group conjugation was achieved. Considering the glass transition temperature of our material close to room temperature, working at 21 °C and in the presence of water may have favored the chain mobility and the resulting surface reorganization, causing the migration of amino functionalities from the surface to the bulk material or the solubilization of some short polymer chains bearing amino groups. After the grafting of sulfo-SMPB, films were washed with PBS and subsequently immersed in a bovine serum albumin solution (BSA, ≥ 96%, Sigma) overnight at 4 °C under stirring, allowing the reaction between the maleimidyl functionalities present in sulfo-SMPB and the sulfhydryl groups contained in the free cystein residues of the protein.
After the immobilization of BSA, the samples were vigorously rinsed with nanopure water and 0.1% Triton X-100 (Sigma-Aldrich) aqueous solution in a vortex. This washing procedure was demonstrated to effectively remove the physically adsorbed BSA. In fact, pristine PLCL samples were also immersed in BSA solution to physically adsorb the protein on the surface of the material. After the rinsing method mentioned above, no BSA associated signals were observed in the XPS survey and high resolution spectra, indicating the complete removal of physically adsorbed molecules from the surface.
Hydrolytic degradation study
For the in vitro degradation study, square samples [initial weight (W0) = 28 ± 2 mg and initial thickness = 156 ± 24 µm (n = 3)] of pristine terpolymer (PLCL), plasma modified terpolymer (P-PLCL) and albumin grafted terpolymer (BSA-PLCL) were placed in Falcon tubes containing PBS (pH 7.2, Fluka) maintaining a surface area to volume ratio equal to 0.1 cm−1. The samples were stored in an oven at 37 °C. At different periods of time, three samples of each group were removed from the PBS, rinsed with nanopure water and vacuum-dried for 48 h prior to their characterization.
Characterization
Characterization of the synthesized polymer
Proton nuclear magnetic resonance (1H NMR) spectra of the synthesized polymer were recorded in a Bruker Avance DPX 300 at 300 MHz, using 5 mm O.D. sample tubes following the experimental conditions described in the previous work.5 The signals of the lactide methine, centered at 5.15 ppm, and the signals of the α and ε methylenes of the ε-caprolactone, around 2.35 and 4.1 ppm, from the 1H NMR spectrum can be assigned to the different dyads.4 The lactide and ε-caprolactone molar content and the microstructural magnitudes of the terpolymer were obtained from the average dyad relative molar fractions. L-LA and D-LA are indistinguishable in the NMR spectrum so their contents and average sequence lengths are approximate values calculated under the assumption that the reactivities of both L-LA and D-LA are the same (taking into account the L-LA:D-LA feed ratios). Equations 4–7 were employed to obtain the number-average sequence lengths (li), the Bernoullian random number-average sequence lengths (li) and the randomness character (R):43
(4)
(5)
(6)
(7)
where (LA) and (CL) are the lactide and ε-caprolactone molar fractions obtained from the integration of the lactide methine signals and the ε-caprolactone methylene signals, (L-LA) and (D-LA) are the L-lactide and D-lactide approximate molar fractions and (LA-CL) is the LA-CL average dyad relative molar fraction.
For the mechanical characterization of the synthesized polymer, films of 150–200 µm were prepared by compression molding in a Collin’s P200E hydraulic press at 180 °C and 240 bar followed by water quenching in order to guarantee an amorphous state. From these films repetitive samples for mechanical characterization (10 × 1 cm2) were obtained. The mechanical properties of these samples were determined by tensile tests with an Instron 5565 (Instron) testing machine at a crosshead displacement rate of 10 mm/min. These tests were performed both at room (21 ± 2 °C) and at body (37 °C) temperature following ISO 527-3/1995. The mechanical properties reported correspond to average values of at least 5 determinations.
Characterization of PLCL after surface modifications
XPS analyses were performed on samples after each surface modification step. A PHI 5600-ci spectrometer (Physical Electronics) was used to record the spectra. A standard aluminum X-ray source (1486.6 eV) at 200 W was used along with a charge neutralizer to record survey spectra while a standard magnesium X-Ray source (1253.6 eV) at 150 W without charge neutralization was used to record high resolution spectra. Detection was performed with a take-off angle of 45° on a 0.5 mm2 area. The binding energies were referenced to the C1s peak at 285.0 eV to compensate for charging effects. Three measurements were made on each sample to ascertain the homogeneity of the surface chemistry.
To quantify the amount of amino functionalities on the surface of our material after plasma treatment, a vapor-phase derivatization reaction with 4-(trifluoromethyl)benzaldehyde (TFBA, Aldrich) was performed at room temperature for 2 h. TFBA specifically reacts with the amino functionalities present on the surface of the material and contains one atom (fluorine) in its structure that is neither a part of the polymer structure nor the gas used to perform the plasma. In this way, the XPS signal from this particular atom can be used to quantify the surface concentration of the derivatized surface moieties according to the following equation:29,44
(8)
Atomic force microscopy (AFM) investigations were performed using the tapping mode on a DimensionsTM 3100 atomic force microscope (Digital Instruments, Bruker) with an etched silicon tip (model NCHV, tip radius = 10 nm, Bruker). Surface topography was evaluated for areas of 20 × 20 µm2 and the surface roughness was evaluated using root mean square average of height deviations (RRMS) using NanoScope Analysis software.
Finally, static contact angle measurements were recorded on the samples using a VCA 2500 XE system (AST, Billerica, MA, USA). At least three nanopure water drops (3 µL) were deposited on different parts of each sample.
Characterization of the samples during hydrolytic degradation
The thickness of the initial PLCL films employed for the hydrolytic degradation study (see section 3.3.) was determined by using a DektakXT Advanced System (Bruker). A force of 0.3 mg was applied on the 12.5 µm stylus to limit the damage of the samples.
The thermal properties of the samples during hydrolytic degradation were determined by DSC823e (Mettler Toledo). Samples of ~10 mg were heated from -50 °C to 185 °C at 20 °C/min. After this first scan the sample was cooled at 20 °C/min and reheated from -50 °C to 185 °C at 20 °C/min. In this second scan the glass transition temperature (Tg) was determined from the inflection point of the heat flow curve.
The molecular weight evolution of the samples during hydrolytic degradation was measured by GPC using two Shodex KF804 columns (Waters) and the RI detector 401 (Waters) at 35 °C. Tetrahydrofuran (HPLC grade, Fisher) was used as an eluent at a flow rate of 1 mL/min and polystyrene standards (Shodex Standards, SM-105, Waters) were used to obtain a primary calibration curve.
Changes in the chemical structures of these samples during degradation were analyzed by ATR-FTIR (Cary 660 FTIR, Agilent Technologies) equipped with a DLaTGS detector, a Ge coated KBr beam splitter and a Si crystal. Spectra were taken with a resolution of 4 cm−1 and were averaged over 64 scans.
Conclusions
In this work, a lactide and caprolactone based biodegradable elastomeric terpolymer with specific ratio has been first aminated by atmospheric pressure plasma treatment. Then, the albumin was efficiently immobilised on the PLCL surface, as demonstrated by XPS and AFM, with a surface coverage (ΓBSA) around 30–35%.
Although slight differences were observed in the degradation behavior between BSA-PLCL and pristine PLCL by GPC, the carboxylate/ester ratio, estimated by FTIR, did not show any evidence difference in the degradation process of the bulk PLCL between these samples.
This polymer presents controlled degradability and adequate mechanical properties for the regeneration of soft tissues and the fabrication of medical devices requiring elastomeric character. Furthermore, as demonstrated by XPS, albumin was still present after 28 d of degradation, giving more time for the biological environment to interact with the modified materials. Depending on the biomedical application, specific proteins could be covalently grafted on PLCL surfaces using this procedure in order to obtain stable and bioactive interfaces.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors are grateful for funds from the Basque Government, Department of Education, Universities and Research (GIC10/152-IT-334-10) and Dept. of Industry (IE10/276). A.L. thanks the University of the Basque Country (UPV-EHU) for a predoctoral and mobility grant. A.L.’s work in Quebec City was supported by the Centre Québécois sur les Matériaux Fonctionnels (CQMF). Rodica Plesu is gratefully acknowledged for GPC and DSC support.
Footnotes
Previously published online: www.landesbioscience.com/journals/biomatter/article/27979
References
- 1.Karam J-P, Muscari C, Montero-Menei CN. Combining adult stem cells and polymeric devices for tissue engineering in infarcted myocardium. Biomaterials. 2012;33:5683–95. doi: 10.1016/j.biomaterials.2012.04.028. [DOI] [PubMed] [Google Scholar]
- 2.Lee J, Guarino V, Gloria A, Ambrosio L, Tae G, Kim YH, Jung Y, Kim S-H, Kim SH. Regeneration of Achilles’ tendon: the role of dynamic stimulation for enhanced cell proliferation and mechanical properties. J Biomater Sci Polym Ed. 2010;21:1173–90. doi: 10.1163/092050609X12471222313524. [DOI] [PubMed] [Google Scholar]
- 3.Sartoneva R, Haimi S, Miettinen S, Mannerström B, Haaparanta AM, Sándor GK, Kellomäki M, Suuronen R, Lahdes-Vasama T. Comparison of a poly-L-lactide-co-ε-caprolactone and human amniotic membrane for urothelium tissue engineering applications. J R Soc Interface. 2011;8:671–7. doi: 10.1098/rsif.2010.0520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fernández J, Etxeberría A, Sarasua JR. Synthesis, structure and properties of poly(L-lactide-co-ε-caprolactone) statistical copolymers. J Mech Behav Biomed Mater. 2012;9:100–12. doi: 10.1016/j.jmbbm.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 5.Fernández J, Etxeberría A, Ugartemendia JM, Petisco S, Sarasua JR. Effects of chain microstructures on mechanical behavior and aging of a poly(L-lactide-co-ε-caprolactone) biomedical thermoplastic-elastomer. J Mech Behav Biomed Mater. 2012;12:29–38. doi: 10.1016/j.jmbbm.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 6.Fernández J, Larrañaga A, Etxeberría A, Sarasua JR. Effects of Chain Microstructures and Derived Crystallization Capability on Hydrolytic Degradation of Poly(L-lactide/ε-caprolactone) Copolymers. Polym Degrad Stabil. 2013;98:481–9. doi: 10.1016/j.polymdegradstab.2012.12.014. [DOI] [Google Scholar]
- 7.Karjalainen T, Hiljanen-Vainio M, Mailin M, Seppälä J. Biodegradable Lactone Copolymers. III: Mechanical Properties of ε-caprolactone and Lactide Copolymers After Hydrolysis In Vitro. J Appl Polym Sci. 1996;59:1299–304. doi: 10.1002/(SICI)1097-4628(19960222)59:8<1299::AID-APP13>3.0.CO;2-1. [DOI] [Google Scholar]
- 8.Fernández J, Larrañaga A, Etxeberría A, Wang W, Sarasua JR. A new generation of poly(lactide/ε-caprolactone) polymeric biomaterials for application in the medical field. J Biomed Mater Res A. 2013 doi: 10.1002/jbma.35036. In press. [DOI] [PubMed] [Google Scholar]
- 9.Tsuji H, Ikarashi K. In vitro hydrolysis of poly(L-lactide) crystalline residues as extended-chain crystallites. Part I: long-term hydrolysis in phospate-buffered solution at 37°C. Biomaterials. 2004;25:5499–55. doi: 10.1016/j.biomaterials.2003.12.053. [DOI] [PubMed] [Google Scholar]
- 10.Bergsma JE, de Bruijn WC, Rozema FR, Bos RRM, Boering G. Late degradation tissue response to poly(L-lactide) bone plates and screws. Biomaterials. 1995;16:25–31. doi: 10.1016/0142-9612(95)91092-D. [DOI] [PubMed] [Google Scholar]
- 11.Vallières K, Petitclerc E, Laroche G. Covalent grafting of fibronectin onto plasma-treated PTFE: influence of the conjugation strategy on fibronectin biological activity. Macromol Biosci. 2007;7:738–45. doi: 10.1002/mabi.200600267. [DOI] [PubMed] [Google Scholar]
- 12.Crombez M, Chevallier P, -Gaudreault RC, Petitclerc E, Mantovani D, Laroche G. Improving arterial prosthesis neo-endothelialization: application of a proactive VEGF construct onto PTFE surfaces. Biomaterials. 2005;26:7402–9. doi: 10.1016/j.biomaterials.2005.05.051. [DOI] [PubMed] [Google Scholar]
- 13.Zhu Y, Chian KS, Chan-Park MB, Mhaisalkar PS, Ratner BD. Protein bonding on biodegradable poly(L-lactide-co-caprolactone) membrane for esophageal tissue engineering. Biomaterials. 2006;27:68–78. doi: 10.1016/j.biomaterials.2005.05.069. [DOI] [PubMed] [Google Scholar]
- 14.Edlund U, Dånmark S, Albertsson AC. A strategy for the covalent functionalization of resorbable polymers with heparin and osteoinductive growth factor. Biomacromolecules. 2008;9:901–5. doi: 10.1021/bm701267u. [DOI] [PubMed] [Google Scholar]
- 15.Desmet T, Morent R, De Geyter N, Leys C, Schacht E, Dubruel P. Nonthermal plasma technology as a versatile strategy for polymeric biomaterials surface modification: a review. Biomacromolecules. 2009;10:2351–78. doi: 10.1021/bm900186s. [DOI] [PubMed] [Google Scholar]
- 16.Morent R, De Geyter N, Desmet T, Dubruel P, Leys C. Plasma Surface Modification of Biodegradable Polymers: A Review. Plasma Process Polym. 2011;8:171–90. doi: 10.1002/ppap.201000153. [DOI] [Google Scholar]
- 17.Pu FR, Williams RL, Markkula TK, Hunt JA. Effects of plasma treated PET and PTFE on expression of adhesion molecules by human endothelial cells in vitro. Biomaterials. 2002;23:2411–28. doi: 10.1016/S0142-9612(01)00377-5. [DOI] [PubMed] [Google Scholar]
- 18.Barradas AMC, Lachmann K, Hlawacek G, Frielink C, Truckenmoller R, Boerman OC, van Gastel R, Garritsen H, Thomas M, Moroni L, et al. Surface modifications by gas plasma control osteogenic differentiation of MC3T3-E1 cells. Acta Biomater. 2012;8:2969–77. doi: 10.1016/j.actbio.2012.04.021. [DOI] [PubMed] [Google Scholar]
- 19.Park K, Ju YM, Son JS, Ahn KD, Han DK. Surface modification of biodegradable electrospun nanofiber scaffolds and their interaction with fibroblasts. J Biomater Sci Polym Ed. 2007;18:369–82. doi: 10.1163/156856207780424997. [DOI] [PubMed] [Google Scholar]
- 20.Khorasani MT, Mirzadeh H, Irani S. Plasma Surface Modification of Poly(L-lactic Acid) and Poly(Lactic-co-Glycolic acid) Films for Improvement of Nerve Cells Adhesion. Radiat Phys Chem. 2008;77:280–7. doi: 10.1016/j.radphyschem.2007.05.013. [DOI] [Google Scholar]
- 21.Oyane A, Uchida M, Yokoyama Y, Choong C, Triffitt J, Ito A. Simple surface modification of poly(epsilon-caprolactone) to induce its apatite-forming ability. J Biomed Mater Res A. 2005;75:138–45. doi: 10.1002/jbm.a.30397. [DOI] [PubMed] [Google Scholar]
- 22.Martins A, Pinho ED, Faria S, Pashkuleva I, Marques AP, Reis RL, Neves NM. Surface modification of electrospun polycaprolactone nanofiber meshes by plasma treatment to enhance biological performance. Small. 2009;5:1195–206. doi: 10.1002/smll.200801648. [DOI] [PubMed] [Google Scholar]
- 23.Garlotta G. A Literature Review of Poly(Lactic Acid) J Polym Environ. 2001;9:63–84. doi: 10.1023/A:1020200822435. [DOI] [Google Scholar]
- 24.Södergard A, Stolt M. Properties of Lactic Acid Based Polymers and Their Correlation with Composition. Prog Polym Sci. 2002;27:1123–63. doi: 10.1016/S0079-6700(02)00012-6. [DOI] [Google Scholar]
- 25.Beamson G, Briggs D. High Resolution XPS of Organic Polymers: The Scienta ESCA300 Database (Chichester, New-York, Brisbane, Toronto, Singapore: John Whiley & Sons) 1992. [Google Scholar]
- 26.Inagaki N, Narushima K, Lim SK. Effects of Aromatic Groups in Polymer Chains on Plasma Surface Modification. J Appl Polym Sci. 2003;89:86–103. doi: 10.1002/app.12160. [DOI] [Google Scholar]
- 27.Narushima K, Yamashita N, Fukuoka M, Inagaki N, Isono Y, Islam MR. Surface Modification of Polyester Films by Ammonia Plasma. Jpn J Appl Phys. 2007;46:4238–45. doi: 10.1143/JJAP.46.4238. [DOI] [Google Scholar]
- 28.Oteyaka MO, Chevallier P, Turgeon S, Robitaille L, Laroche G. Low Pressure Radio Frequency Ammonia Plasma Surface Modification on Poly(Ethylene Terephtalate) Films and Fibers: Effect of the Polymer Forming Process. Plasma Chem Plasma Process. 2012;32:17–33. doi: 10.1007/s11090-011-9330-3. [DOI] [Google Scholar]
- 29.Favia P, Stendardo MV, d’Agostino R. Selective Grafting of Amine Groups on Polyethylene by Means of NH3-H2 RF Glow Discharges. Plasmas Polym. 1996;1:91–112. doi: 10.1007/BF02532821. [DOI] [Google Scholar]
- 30.Zhang C, Jin J, Zhao J, Jiang W, Yin J. Functionalized polypropylene non-woven fabric membrane with bovine serum albumin and its hemocompatibility enhancement. Colloids Surf B Biointerfaces. 2013;102:45–52. doi: 10.1016/j.colsurfb.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 31.Sarra-Bournet C, Turgeon S, Mantovani D, Laroche G. A Study of Atmospheric Pressure Plasma Discharges for Surface Functionalization of PTFE Used in Biomedical Applications. J Phys D Appl Phys. 2006;39:3461–9. doi: 10.1088/0022-3727/39/16/S03. [DOI] [Google Scholar]
- 32.Sarra-Bournet C, Ayotte G, Turgeon S, Massines F, Laroche G. Effects of chemical composition and the addition of H2 in a N2 atmospheric pressure dielectric barrier discharge on polymer surface functionalization. Langmuir. 2009;25:9432–40. doi: 10.1021/la900652y. [DOI] [PubMed] [Google Scholar]
- 33.Mattioli S, Kenny JM, Armentano I. Plasma Surface Modification of Porous PLLA Films: Analysis of Surface Properties and In Vivo Hydrolytic Degradation. J Appl Polym Sci. 2012;125:239–47. doi: 10.1002/app.36827. [DOI] [Google Scholar]
- 34.Kim MC, Masuoka T. Degradation Properties of PLA and PHBV Films Treated with CO2-Plasma. React Funct Polym. 2009;69:287–92. doi: 10.1016/j.reactfunctpolym.2009.01.013. [DOI] [Google Scholar]
- 35.Wu L, Ding J. Effects of porosity and pore size on in vitro degradation of three-dimensional porous poly(D,L-lactide-co-glycolide) scaffolds for tissue engineering. J Biomed Mater Res A. 2005;75:767–77. doi: 10.1002/jbm.a.30487. [DOI] [PubMed] [Google Scholar]
- 36.Garkhal K, Verma S, Jonnalagadda S, Kumar N. Fast Degradable Poly(L-lactide-co-ε-caprolactone) Microspheres for Tissue Engineering: Synthesis, Characterization, and Degradation Behavior. J Polym Sci A Polym Chem. 2007;45:2755–64. doi: 10.1002/pola.22031. [DOI] [Google Scholar]
- 37.Larrañaga A, Sarasua JR. Effect of Bioactive Glass Particles on the Thermal Degradation Behavior of Medical Polyesters. Polym Degrad Stabil. 2013;98:751–8. doi: 10.1016/j.polymdegradstab.2012.12.015. [DOI] [Google Scholar]
- 38.Pamula E, Blazewicz M, Paluszkiewicz C, Dobrzynski P. FTIR Study of Degradation Products of Aliphatic Polyester-Carbon Fibres Composites. J Mol Struct. 2001;596:69–75. doi: 10.1016/S0022-2860(01)00688-3. [DOI] [Google Scholar]
- 39.Pektok E, Nottelet B, Tille J-C, Gurny R, Kalangos A, Moeller M, Walpoth BH. Degradation and healing characteristics of small-diameter poly(ε-caprolactone) vascular grafts in the rat systemic arterial circulation. Circulation. 2008;118:2563–70. doi: 10.1161/CIRCULATIONAHA.108.795732. [DOI] [PubMed] [Google Scholar]
- 40.Finn AV, Nakazawa G, Joner M, Kolodgie FD, Mont EK, Gold HK, Virmani R. Vascular responses to drug eluting stents: importance of delayed healing. Arterioscler Thromb Vasc Biol. 2007;27:1500–10. doi: 10.1161/ATVBAHA.107.144220. [DOI] [PubMed] [Google Scholar]
- 41.Kotani J, Awata M, Nanto S, Uematsu M, Oshima F, Minamiguchi H, Mintz GS, Nagata S. Incomplete neointimal coverage of sirolimus-eluting stents: angioscopic findings. J Am Coll Cardiol. 2006;47:2108–11. doi: 10.1016/j.jacc.2005.11.092. [DOI] [PubMed] [Google Scholar]
- 42.Fernández J, Meaurio E, Chaos A, Etxeberria A, Alonso-Varona A, Sarasua JR. Synthesis and Characterization of poly(L-lactide/ε-caprolactone) Statistical Copolymers with Well Resolved Chain Microstructures. Polymer (Guildf) 2013;54:2621–31. doi: 10.1016/j.polymer.2013.03.009. [DOI] [Google Scholar]
- 43.Herbert IR. Statistical Analysis of Copolymer Sequence Distribution. In NMR Spectroscopy of Polymers. Ibbet, R.N. Ed.: Blackie Academic & Professional, London, 1993: 50-79. (Chapter 2) [Google Scholar]
- 44.Chevallier P, Castonguay N, Turgeon S, Dubrulle N, Mantovani D, McBreen PH, Wittmann JC, Laroche G. Ammonia RF-Plasma on PTFE Surfaces: Chemical Characterization of the Species Created on the Surface by Vapor-Phase Chemical Derivatization. J Phys Chem B. 2001;105:12490–7. doi: 10.1021/jp011607k. [DOI] [Google Scholar]