

Abstract
The prevalence of gastric cancer associated with Lynch syndrome (LS) is highly variable, and the underlying histologic pathway or molecular mechanisms remain unclear. From 1995 to 2012, 15 patients had been treated for both gastric and colonic adenocarcinomas and diagnosed as LS. In all cases, pathologic review, immunohistochemical analysis for mismatch-repair proteins, and microsatellite instability (MSI) tests were performed. To confirm LS, germline mutation tests and multiplex ligation-dependent probe amplification were performed. All gastric and colonic carcinomas were MSI-high and lost expressions of MLH1/PMS2 in 11 (73%) cases and MSH2/MSH6 in 4 (27%) cases. Remarkably, in a patient with LS and germline mutation of MLH1 gene, pyloric gland adenoma (PGA) transformed to adenocarcinoma during follow-up. In 2 additional cases, PGA was found adjacent to advanced gastric cancers. All PGAs in LS patients were MSI-high and lost expression of mismatch-repair proteins (MLH1/PMS2 in 2 cases and MSH2/MSH6 in 1 case), whereas none of the 14 sporadic PGAs was MSI-high or had lost expression of mismatch-repair proteins. On the basis of these observations, although very rare, we suggest the possibility that PGA may be a precursor lesion to gastric adenocarcinoma in LS and that the mismatch-repair deficient pathway of carcinogenesis is involved early in the gastric carcinogenesis pathway.
Key Words: gastric, pyloric gland adenoma, carcinoma, precursor, Lynch syndrome
Lynch syndrome (LS), also known as hereditary nonpolyposis colorectal cancer (CRC), is an autosomal dominant disorder associated with a germline mutation in one of the DNA mismatch-repair genes: MLH1, MSH2, MSH6, or PMS2.1–3 LS is characterized by the development of CRC, endometrial cancer, and various other cancers that are frequently diagnosed at an early age.4 In LS, the cumulative risk for developing gastric cancer by the age of 70 is approximately 5%, and there is no evidence for the clustering of gastric cancer in specific LS families.5,6 Although gastric cancer forms a part of the LS tumor spectrum, the risk for developing gastric cancer in LS families is unknown, resulting in a lack of clear guidelines for surveillance.5
The goal of surveillance for gastric cancer would be the detection of precursor lesions and gastric cancer at an early curable stage.4 A Finnish study reported “potential” precursor lesions in a substantial proportion of 73 carriers of mismatch-repair gene mutations: Helicobacter pylori infection in 26%, atrophy in 14%, and intestinal metaplasia in 14%.7 Few clinical or histologic details of gastric cancer in LS patients are available, and the prevalence of gastric cancer in LS varies from 0.8% to 30%.6,8,9 Moreover, these studies were conducted a long time ago when the incidence of gastric cancer in the background population was high, and histologic findings were described incompletely.5,6 Therefore, the histopathologic transformation pathway of gastric cancer in LS mutation carriers remains largely unclear.4,5
An additional challenge in LS is the lack of a defined gastric lesion such as those identified in patients with familial adenomatous polyposis (FAP), in which fundic gland polyp is the most common gastric abnormality.10 Fundic gland polyps in FAP can show foveolar dysplasia, and rarely invasive gastric adenocarcinoma has been reported in patients with FAP and fundic gland polyposis.10,11 Recently, we experienced a case of gastric pyloric gland adenoma (PGA) that showed malignant transformation within a 2-year span in an LS patient with germline mutation of the MLH1 gene. This case prompted us to evaluate the pathologic findings of gastric cancer in individuals with LS and determine whether PGAs are specific gastric lesions in LS patients.
MATERIALS AND METHODS
Case Selection
To identify LS patients with gastric cancer, we instituted 2 independent searches spanning a 17-year period (1995 to 2012). First, a computer search of the Samsung Comprehensive Cancer Centre identified 392 patients who had both gastric and colonic adenocarcinomas. In addition, data were retrieved for 250 patients who fulfilled revised Bethesda guidelines for LS12 and were enrolled in the familial cancer clinic. In total, 261 patients met the Amsterdam II criteria, and 15 patients (5.7%) developed gastric adenocarcinoma. In these 15 cases, archival formalin-fixed paraffin-embedded (FFPE) tissues were available for pathologic review and molecular analysis. To confirm LS, germline mutation tests for mismatch-repair genes were performed in 4 cases and MLPA tests in the remaining 11. Written informed consent was obtained from all patients, and the institutional ethics committee approved this study.
Immunohistochemistry for Mismatch-repair Proteins and Mucins
Immunohistochemical analysis for mismatch-repair proteins was performed in all gastric and colonic cancers. For mucin proteins, only gastric cancers were used with FFPE tissue sections. The primary antibodies, clone, manufacturer, and dilutions are listed in Table 1.
TABLE 1.
Antibodies Used for Immunohistochemistry
Immunohistochemical analysis was performed using a Ventana BenchMark XT autoimmunostainer (Ventana Medical Systems, Tucson, AZ), with incubation with primary antibodies at 37°C for 30 minutes, followed by standard Ventana signal amplification and counterstaining with hematoxylin as previously described.13 Slides were mounted and examined by light microscopy. For interpretation of data, only nuclear staining was considered.
Microsatellite Instability
Microsatellite instability (MSI) analysis was performed in all cases using multiplex polymerase chain reaction (PCR) with 5 quasimonomorphic mononucleotide repeat markers as previously described.14,15 Briefly, genomic DNA was isolated from FFPE tumor samples using a QIAamp DNA mini kit (Qiagen, Valencia, CA) with the help of a microscope. Each sense primer was end-labeled with one of the fluorescent markers—FAM, HEX, or NED. Pentaplex PCR was performed with an initial 15-minute denaturation at 94°C, followed by 35 cycles at 94°C for 30 seconds, 55°C for 30 seconds, and 72°C for 30 seconds with final extension at 72°C for 10 minutes. Amplified PCR products were run on an ABI Prism 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA). Allelic sizes were estimated using Genemapper 4.1 software (Applied Biosystems). Samples with no allelic size variations in any of the microsatellites were classified as microsatellite stable. Tumors with allelic size variations in <2 of the microsatellites were classified as MSI-low, whereas those with allelic size variations in ≥2 of the microsatellite markers were considered MSI-high.
Multiplex Ligation-dependent Probe Amplification Analysis
To detect large genomic alterations in the MLH1 and MSH2 genes, we used the SALSA MLPA kit P003-B1 (MRC-Holland, Amsterdam, The Netherlands) with genomic DNA from FFPE tissue samples according to the manufacturer’s instructions. Results were analyzed with ABI Prism 3130 Genetic Analyzer and Genemapper v.4.0 software (Applied Biosystems) to confirm deletions/duplications. Fourteen negative reference samples (gastric mucosa of healthy subjects) were included in each MLPA run to normalize MLPA ratios. For genes with >1 probe present in the kit, the mean of all the probe peaks of this gene in duplicate was calculated.
MethyLight for MLH1 Promoter Hypermethylation
MethyLight analysis was performed as previously described in all gastric and colonic carcinoma cases with MLH1-deficient carcinomas to detect promoter hypermethylation of the MLH1 in genomic DNAs extracted from FFPE tumor samples.16 To target the proximal region of the MLH1 promoter, primers located at −267 to −176 bp from the transcription start site of the MLH1 gene were used.17 The percentage methylation ratio ≥5.0 was considered positive for methylation.
Germline Mutation Testing
Genomic DNA was extracted from peripheral blood mononuclear cells with the Wizard Genomic DNA Purification kit according to the manufacturer’s instructions (Promega, Madison, WI). All coding exons and flanking intronic regions of the MLH1, MSH2, and MSH6 genes were amplified using specific primer sets (available upon request). PCR was performed using a thermal cycler (Applied Biosystems), and sequencing was performed on an ABI Prism 3730 Genetic Analyzer using a BigDye Terminator Cycle Sequencing Reaction Kit (Applied Biosystems) in a College of American Pathologists–certified laboratory. Germline MLH1 and MSH2 mutation analysis was performed in patients with loss of MLH1 or MSH2 staining, respectively. MSH6 genetic testing was performed in patients with lack of MSH6 expression or combined lack of MSH2 and MSH6 expression but did not have MSH2 mutations.
RESULTS
Clinical and Pathologic Findings of 15 Gastric Carcinomas in Patients With LS
The 15 patients included 13 male and 2 female individuals. Four patients presented with metachronous gastric and colonic cancers, and, in those cases, LS was not suspected clinically. The detailed clinical and pathologic findings are summarized in Table 2.
TABLE 2.
Clinicopathologic Findings of 15 Gastric Cancer Patients Associated With LS
The locations of the gastric cancer were cardia (n=3), body or fundus (n=5), and antrum (n=7). Histologic types were differentiated tubular adenocarcinoma in 13 cases and mucinous adenocarcinoma in 2 cases. In 1 patient (#13), 3 synchronous gastric cancers occurred in the body of the stomach, and all of them were early in pT stages. In 1 case (#14), recurrent gastric cancer developed in the cardia 22 years after subtotal gastrectomy, and many of his relatives suffered from cancers of the colon, in addition to endometrium, prostate, and thyroid cancer.
H. pylori infection was observed in 4 cases (26.7%). Gastric atrophy and intestinal metaplasia were observed in 13 cases (86.7%) and 14 cases (93.3%), respectively. Tumor stage was stage I in 12 cases, stage II in 2 cases, and stage III in 1 case by AJCC seventh edition.
PGAs Identified in Patients With LS
PGAs were observed in 3 patients (#1, #8, and #14) of the 15 LS patients (20%). Patient #1 was a healthy male without any significant medical history. A 0.3-cm-sized polypoid lesion was noted in the anterior wall of the gastric cardia during routine health checkup endoscopy. The lesion was removed by polypectomy and was mainly composed of closely packed complex pyloric-type glands (Fig. 1A). The glands were focally fused or irregularly branched (Fig. 1B). Most tumor glands are strongly positive for MUC6 protein (Fig. 1C), and the superficial epithelial layer was positive for MUC5AC (Fig. 1D). The pathologic diagnosis was PGA with high-grade dysplasia. After polypectomy, follow-up at 3 months and 1 year revealed no residual tumor. After 2 years, follow-up endoscopy revealed a poorly defined mass with central depression at the previous polypectomy site (Fig. 2A). Endoscopic submucosal dissection was performed to remove the mass. PGA showed closely packed glands with round nuclei and amphophilic cytoplasm (Fig. 2B). There was direct transformation of PGA to invasive adenocarcinoma (Fig. 2C). The adenocarcinoma invaded into the deep portion of the lamina propria, and the neoplastic glands were surrounded by plump loose myxoid stroma (Fig. 2D). Both gastric adenocarcinoma and adjacent PGA had lost MLH1 (Fig. 2E) and PMS2 expression.
FIGURE 1.
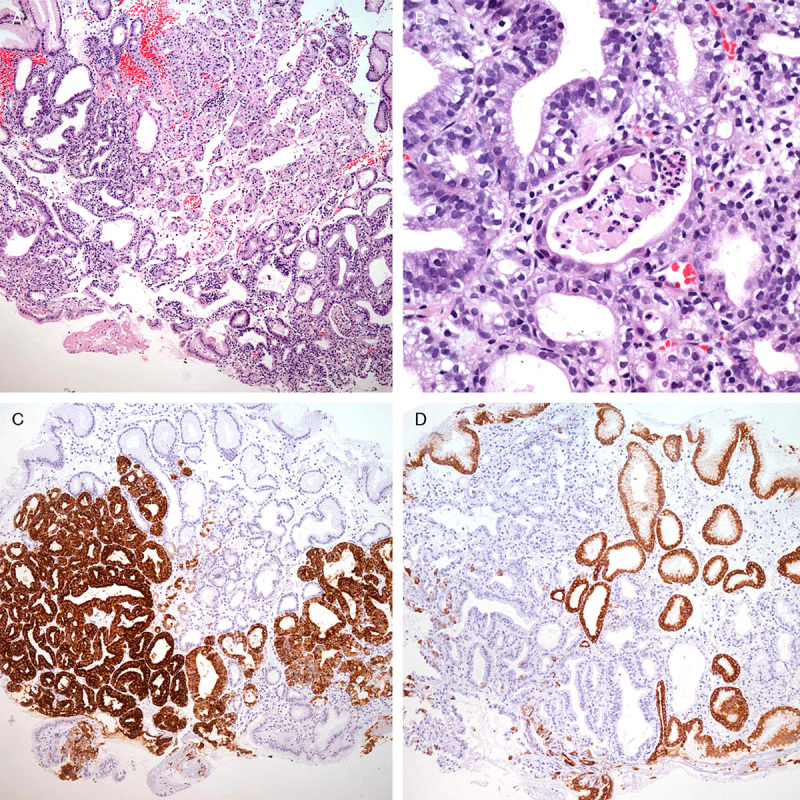
Initial endoscopy findings of patient #1. A, The lesion was composed of packed complex pyloric-type glands lined by cuboidal or columnar cells. B, The tumor cells showed enlarged rounded nuclei and loss of polarity with pale to eosinophilic cytoplasm. C, Most glands were strongly positive for MUC6 protein. D, The superficial layer was positive for MUC5AC.
FIGURE 2.
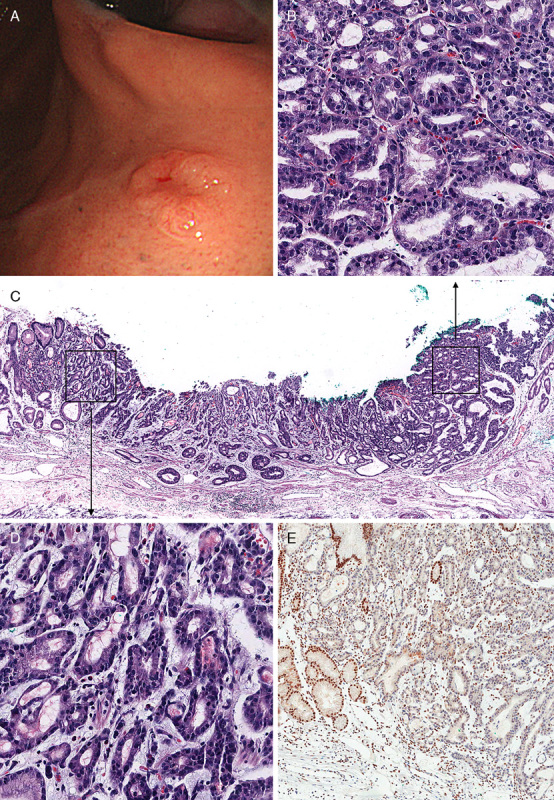
A, Endoscopic findings at the 2-year follow-up for patient #1. A, Dome-like polypoid lesion measuring 1.0×0.6 cm with central ulceration was noted. B, Pathologic examination of endoscopic submucosal dissection specimen showed PGA with round nuclei and amphophilic cytoplasm, which is similar to that of the original biopsy. C, Direct transformation of PGA to invasive adenocarcinoma. D, Adenocarcinoma with invasion of glands into lamina propria. E, Loss of expression of MLH1 in adenocarcinoma and PGA.
In patients #8 and #14, PGA was also identified adjacent to the gastric adenocarcinoma. In patient #8, a direct transition from PGA to carcinoma was noted (Fig. 3). In patient #14, the carcinoma developed in the cardia of the remnant stomach, and direct transition from PGA to invasive carcinoma was also noted (Figs. 4A, B). Although PGA and the adjacent deep portion of the associated gastric adenocarcinoma were positive for MUC6 (Fig. 4C), the superficial layers of PGA and associated gastric adenocarcinoma were positive for MUC5AC (Fig. 4D). In both lesions, MUC2 and CD10 were negative.
FIGURE 3.
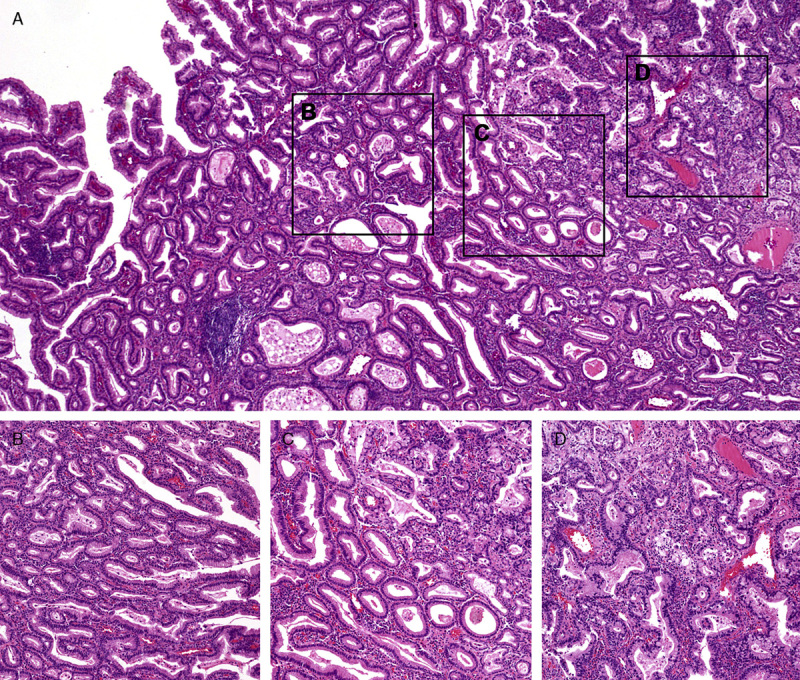
Pathologic findings of gastric tumor in patient #8. A, Low-power examination shows a direct transition from PGA to carcinoma. B, Higher magnifications of PGA, (C) transition from PGA to carcinoma and (D) adenocarcinoma.
FIGURE 4.
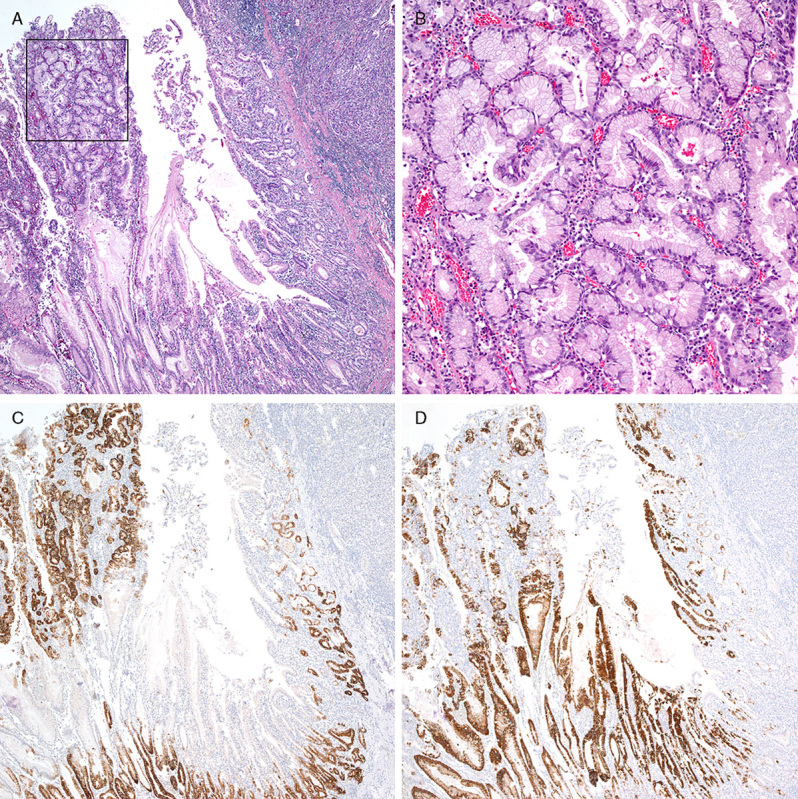
Histologic features of patient #14. A, PGA adjacent to adenocarcinoma. B, Higher magnification of the square in (A) showing typical histologic features of PGA. C, Immunohistochemistry for MUC6. D, Immunohistochemistry for MUC5AC.
Immunohistochemistry for Mismatch-repair Protein and Mucin Phenotypes
In both gastric and colonic carcinomas, expression of MLH1/PMS2 protein was lost in 11 (73.3%) cases and that of MSH2/MSH6 protein in the remaining 4 cases. In gastric cancer patients associated with PGA, gastric adenocarcinoma and adjacent PGA were separately evaluated, and both lesions had lost MLH1/PMS2 expression in patients #1 and #14 and MSH2/MSH6 expression in patient #8. In 14 sporadic PGAs, there were no losses of mismatch-repair proteins in any of the cases.
Of 15 gastric cancers, MUC6, MUC5AC, MUC2, and CD10 were positive in 13 (86.7%), 11 (73.3%), 8 (53.3%), and 1 (6.7%) cases, respectively. Seven (46.7%) cancers were classified as gastric mucin phenotype, 6 (40%) as mixed mucin phenotype, and 2 (13.3%) as intestinal mucin phenotype.
Molecular Findings
All gastric and colonic carcinomas were MSI-high, and there was no promoter hypermethylation of the MLH1 gene in any of the cases. Three PGAs observed in patients with LS were MSI-high too. In all patients who agreed to germline mutation tests, direct sequencing of a peripheral blood sample showed MLH1 c.1447_1469dup23 mutation in patient #1, c.1758_9insC mutation in patient #12, c.808_811delACTT mutation in patient #13, and c.303_304dupTG mutations in patients #14 and #15. In the remaining 11 cases tested by MLPA, gains of MSH2 were observed in 3 cases, and gains of MLH1 were found in 7 cases, and the genetic alterations matched perfectly to protein losses of the mismatch-repair gene.
DISCUSSIONS
In LS patients, the prevalence of gastric cancer is highly variable, and the molecular pathway underlying gastric cancer is not defined. The present study aimed to determine the detailed histology and molecular findings of gastric cancer in LS patients. We identified gastric cancer in 5.7% of suspected LS patients and identified PGA as one of the precursor lesion of gastric cancer in LS by demonstrating transformation of PGA to carcinoma.
LS is caused by germline mutations in a mismatch-repair gene. The function of the mismatch-repair genes is to maintain genomic stability by correcting any mismatches that might be generated during DNA replication. Mismatch-repair malfunction leads to MSI-high, and 4 mismatch-repair genes, MLH1, MSH2, MSH6, or PMS2, account for almost all of the identified mutations in LS.18 Patients with LS are at high risk for developing CRC at an early age (mean age 45 y) in addition to extracolonic cancers. The most common locations of extracolonic cancer in LS are endometrium, ovary, stomach, small bowel, pancreas, hepatobiliary, brain, and upper genitourinary tract.19 The cumulative risk for developing gastric cancer has been estimated as 5%4,5 and 19%.8 The prevalence of gastric cancer in LS also varies from 0.8% to 30%,5,6,8,9 suggesting a close relationship between the phenotype of LS and the cancer spectrum of the background population. However, recent studies have shown that there is no evidence for clustering of gastric cancer in specific LS families.3–5 In the present study conducted at a comprehensive cancer center located in a gastric cancer-prevalent area, we identified gastric cancer in 5.7% of patients with suspected LS, which is similar to previous prevalence estimates in western populations.20 Moreover, most gastric cancers in LS patients were of gastric mucin phenotype. Given that PGA is a gastric-type adenoma,21 our findings also support PGA as one of the precursor lesions of gastric cancer in LS.
Gastric polyps are relatively uncommon in the general population, with a reported incidence of 2.7% in nonsyndromic patients.22 PGA is an uncommon histologic subtype of gastric polyps. Until recently, it was not well established as a distinct entity, especially in the literature from the United States.21,23 PGA occurs more commonly in older women and predominantly in the cardia and fundic mucosa in H. pylori or autoimmune gastritis.21,23,24 Although most PGAs show a bland histologic appearance lacking distinct dysplasia, PGA is a truly neoplastic lesion with frequent p53 overexpression and chromosomal abnormalities25,26 and a higher rate of transition to carcinoma compared with conventional adenomas of similar size (11% to 42.7% vs. 1% to 5%).21,24,27,28 In this study, we identified transformation of a PGA to carcinoma during follow-up of a patient with germline MLH1 mutation identified 2 years before diagnosis of LS due to colonic cancer. Moreover, after thorough pathologic examinations, we found 2 additional cases of PGA located adjacent to gastric cancer in LS patients. All 3 PGAs were located in the cardia or high body of the stomach, where PGA is frequently found. In LS patients, most gastric cancers were intestinal in histology, and almost half of them were located in the antrum.6 In our study, all of the gastric cancer cases with PGA occurred in relatively young male patients aged 32, 53, and 57 years and had already lost expression of mismatch-repair proteins and were MSI-high, supporting the hypothesis that mismatch-repair deficiency and the resulting MSI-high is an early event in gastric carcinogenesis by PGA in almost 20% of patients with LS. Furthermore, as none of the tumors with MLH1 deficiency showed MLH1 methylation, methylation is not a “second hit” mechanism in biallelic inactivation of MLH1. These observations are similar to the molecular abnormalities described in the adenoma-carcinoma sequence of the colon in LS.29–31 However, sporadic PGA occurs predominantly in old age (mean 73 y) and more frequently in women (75%) than in men.25 Moreover, none of the 14 sporadic PGAs in our study showed loss of mismatch-repair proteins or MSI-high. In a recent study on 35 sporadic PGAs, frequent GNAS (63%) and KRAS (41%) mutations were observed, whereas loss of mismatch-repair protein was found in only 1 PGA (3%). These findings suggest that the chromosomal instability pathway involves sporadic PGA in transformation into adenocarcinoma rather than mismatch-repair deficiency.32
In conclusion, we found 15 gastric cancers developed in 261 patients who met the Amsterdam II clinical criteria for LS. Three of these gastric cancers arose from PGA that had lost mismatch-repair protein expression and had MSI-high. Our findings strongly support PGA as one of the precursor lesions of gastric cancer in LS, and mismatch-repair deficiency is involved early in the pathogenesis of stomach cancer. Further studies are needed to support our observations.
Footnotes
Conflicts of Interest and Source of Funding: Funded by a grant from the Korea Healthcare Technology R&D project, Ministry of Health & Welfare, Republic of Korea (A101130). The authors have disclosed that they have no significant relationships with, or financial interest in, any commercial companies pertaining to this article.
REFERENCES
- 1.Jass JR, Stewart SM.Evolution of hereditary non-polyposis colorectal cancer.Gut. 1992;33:783–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lynch HT, Smyrk T.An update on Lynch syndrome.Curr Opin Oncol. 1998;10:349–356 [DOI] [PubMed] [Google Scholar]
- 3.Watson P, Lynch HT.Extracolonic cancer in hereditary nonpolyposis colorectal cancer.Cancer. 1993;71:677–685 [DOI] [PubMed] [Google Scholar]
- 4.Vasen HF, Blanco I, Aktan-Collan K, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts.Gut. 2013;62:812–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Capelle LG, Van Grieken NC, Lingsma HF, et al. Risk and epidemiological time trends of gastric cancer in Lynch syndrome carriers in the Netherlands.Gastroenterology. 2010;138:487–492 [DOI] [PubMed] [Google Scholar]
- 6.Aarnio M, Salovaara R, Aaltonen LA, et al. Features of gastric cancer in hereditary non-polyposis colorectal cancer syndrome.Int J Cancer. 1997;74:551–555 [DOI] [PubMed] [Google Scholar]
- 7.Renkonen-Sinisalo L, Sipponen P, Aarnio M, et al. No support for endoscopic surveillance for gastric cancer in hereditary non-polyposis colorectal cancer.Scand J Gastroenterol. 2002;37:574–577 [DOI] [PubMed] [Google Scholar]
- 8.Aarnio M, Mecklin JP, Aaltonen LA, et al. Life-time risk of different cancers in hereditary non-polyposis colorectal cancer (HNPCC) syndrome.Int J Cancer. 1995;64:430–433 [DOI] [PubMed] [Google Scholar]
- 9.Cristofaro G, Lynch HT, Caruso ML, et al. New phenotypic aspects in a family with Lynch syndrome II.Cancer. 1987;60:51–58 [DOI] [PubMed] [Google Scholar]
- 10.Abraham SC, Nobukawa B, Giardiello FM, et al. Fundic gland polyps in familial adenomatous polyposis: neoplasms with frequent somatic adenomatous polyposis coli gene alterations.Am J Pathol. 2000;157:747–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Odze RD, Marcial MA, Antonioli D.Gastric fundic gland polyps: a morphological study including mucin histochemistry, stereometry, and MIB-1 immunohistochemistry.Hum Pathol. 1996;27:896–903 [DOI] [PubMed] [Google Scholar]
- 12.Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability.J Natl Cancer Inst. 2004;96:261–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boland CR, Koi M, Chang DK, et al. The biochemical basis of microsatellite instability and abnormal immunohistochemistry and clinical behavior in Lynch syndrome: from bench to bedside.Fam Cancer. 2008;7:41–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nardon E, Glavac D, Benhattar J, et al. A multicenter study to validate the reproducibility of MSI testing with a panel of 5 quasimonomorphic mononucleotide repeats.Diagn Mol Pathol. 2010;19:236–242 [DOI] [PubMed] [Google Scholar]
- 15.Park HY, Kang SY, Kang GH, et al. EBV infection and mismatch repair deficiency mediated by loss of hMLH1 expression contribute independently to the development of multiple synchronous gastric carcinomas.J Surg Oncol. 2012;106:777–782 [DOI] [PubMed] [Google Scholar]
- 16.Eads CA, Danenberg KD, Kawakami K, et al. MethyLight: a high-throughput assay to measure DNA methylation.Nucleic Acids Res. 2000;28:E32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park SJ, Rashid A, Lee JH, et al. Frequent CpG island methylation in serrated adenomas of the colorectum.Am J Pathol. 2003;162:815–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lynch HT, de la Chapelle A.Hereditary colorectal cancer.N Engl J Med. 2003;348:919–932 [DOI] [PubMed] [Google Scholar]
- 19.Nair N, Curtin JP, Mittal K, et al. Cervical adenocarcinoma in a patient with Lynch syndrome, Muir-Torre variant.J Clin Oncol. 2012;30:e5–e6 [DOI] [PubMed] [Google Scholar]
- 20.Vasen HF, Offerhaus GJ, den Hartog Jager FC, et al. The tumour spectrum in hereditary non-polyposis colorectal cancer: a study of 24 kindreds in the Netherlands.Int J Cancer. 1990;46:31–34 [DOI] [PubMed] [Google Scholar]
- 21.Chen ZM, Scudiere JR, Abraham SC, et al. Pyloric gland adenoma: an entity distinct from gastric foveolar type adenoma.Am J Surg Pathol. 2009;33:186–193 [DOI] [PubMed] [Google Scholar]
- 22.Oberhuber G, Stolte M.Gastric polyps: an update of their pathology and biological significance.Virchows Arch. 2000;437:581–590 [DOI] [PubMed] [Google Scholar]
- 23.Vieth M, Kushima R, Mukaisho K, et al. Immunohistochemical analysis of pyloric gland adenomas using a series of Mucin 2, Mucin 5AC, Mucin 6, CD10, Ki67 and p53.Virchows Arch. 2010;457:529–536 [DOI] [PubMed] [Google Scholar]
- 24.Vieth M, Kushima R, Borchard F, et al. Pyloric gland adenoma: a clinico-pathological analysis of 90 cases.Virchows Arch. 2003;442:317–321 [DOI] [PubMed] [Google Scholar]
- 25.Kushima R, Vieth M, Borchard F, et al. Gastric-type well-differentiated adenocarcinoma and pyloric gland adenoma of the stomach.Gastric Cancer. 2006;9:177–184 [DOI] [PubMed] [Google Scholar]
- 26.Kushima R, Muller W, Stolte M, et al. Differential p53 protein expression in stomach adenomas of gastric and intestinal phenotypes: possible sequences of p53 alteration in stomach carcinogenesis.Virchows Arch. 1996;428:223–227 [DOI] [PubMed] [Google Scholar]
- 27.Hay LJ.Surgical management of gastric polyps and adenomas.Surgery. 1956;39:114–119 [PubMed] [Google Scholar]
- 28.Kushima R, Ruthlein HJ, Stolte M, et al. “Pyloric gland-type adenoma” arising in heterotopic gastric mucosa of the duodenum, with dysplastic progression of the gastric type.Virchows Arch. 1999;435:452–457 [DOI] [PubMed] [Google Scholar]
- 29.Aaltonen LA, Peltomaki P, Mecklin JP, et al. Replication errors in benign and malignant tumors from hereditary nonpolyposis colorectal cancer patients.Cancer Res. 1994;54:1645–1648 [PubMed] [Google Scholar]
- 30.Iino H, Simms L, Young J, et al. DNA microsatellite instability and mismatch repair protein loss in adenomas presenting in hereditary non-polyposis colorectal cancer.Gut. 2000;47:37–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim KM, Salovaara R, Mecklin JP, et al. PolyA deletions in hereditary nonpolyposis colorectal cancer: mutations before a gatekeeper.Am J Pathol. 2002;160:1503–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsubara A, Sekine S, Kushima R, et al. Frequent GNAS and KRAS mutations in pyloric gland adenoma of the stomach and duodenum.J Pathol. 2013;229:579–587 [DOI] [PubMed] [Google Scholar]