

Abstract
The Anaphase Promoting Complex (APC) has been characterized to play pivotal roles in regulating the timely cell cycle progression by forming two functionally distinct E3 ubiquitin ligase sub-complexes, APCCdc20 and APCCdh1. Interestingly, recent studies have shown that Cdh1 is functioning as a tumor suppressor whereas Cdc20 may function as an oncoprotein to promote the development and progression of human cancers. In this review, we will discuss the physiological role of Cdc20 and its downstream substrates in vitro and in the transgenic mouse model reminiscent of the pathogenesis of human cancers. Furthermore, we summarize recent findings to indicate that Cdc20 may represent a promising therapeutic target, thus development of Cdc20 inhibitors could be useful for achieving better treatment outcome of cancer patients.
Keywords: cancer, Cdc20, cell growth, oncogene, ubiquitination, therapy
1. Introduction
Ubiquitination by the ubiquitin proteasome system (UPS) is a post-translational modification that regulates the abundance of key cellular proteins, which controls various cellular processes including cell proliferation, differentiation, cell apoptosis, cell cycle progression, DNA damage repair, cell migration and senescence [1]. Thus far, two related, multi-subunit E3 ubiquitin ligase enzymes, the Anaphase Promoting Complex (APC) and the Cullin Ring (CRLs) ubiquitin ligases have been considered as the major driving forces governing these cellular processes [2]. For example, the SCF (Skp1-Cullin1-F-box) complex regulates entry into S phase by degradation of G1 CKIs (casein kinase I) and G1 cyclins [2]. APC has been found to play critical roles in regulating timely cell cycle progression in both the M and G1 phases [3]. Moreover, the APC is essential for chromosome segregation, as well as exit from mitosis by degradation of its substrates including securin and mitotic cyclins [3].
It is known that APC is activated by the substrate-recruiting module Cdc20 (cell division cycle 20 homologue) and Cdh1 (also called Fzr1) [4]. These two activator proteins govern the APC/C to interact with specific ubiquitin substrates for their subsequent degradation by the 26S proteasome at different times during the cell cycle progression, thus governing cell cycle forward in a unidirectional manner. Cdc20 can recruit substrates with the Destruction Box (D-box) motif while Cdh1 recruits substrates with the D-box and/or KEN-box to the APC core for ubiquitination [5]. It is important to note that Cdc20 and Cdh1 have different functions during different phases of the cell cycle, in part by controlling the destruction of various substrates of APC/C [5]. It has been well documented that Cdc20 consists of 499 amino acids with WD40 repeats at its C-terminus for protein binding, serving as the substrate recognizing subunit of APC/C [6]. In addition to cell cycle control, accumulating evidence has demonstrated that Cdc20 also plays an important role in the development and progression of human cancers [7]. In the following paragraphs, we will mainly focus on the recent literature regarding the novel function and regulation of Cdc20 in the pathogenesis of human malignancies.
2. Downstream substrates of Cdc20
Cdc20 was discovered by Lee Hartwell around 40 years ago [8]. The elegant experiments from the Hartwell group showed that Cdc20 mutants arrested cell division in mitosis and failed to initiate anaphase and chromosome segregation [8]. Later on, further studies suggested that Cdc20 contains seven WD40 repeats that play a critical role in mediating protein-protein interactions [9]. So far, it has become clear that the activity of Cdc20 is to activate APC/C during mitosis, leading to coordinated mitotic progression through the orderly degradation of mitotic APC/C substrates.
Multiple studies from various groups have demonstrated that Cdc20 targets several key substrates including Securin [10], Cyclin B1 [11; 12], Cyclin A [13; 14], Nek2A [15], p21 [16], and Mcl-1 [17] for degradation to govern cell cycle progression. For example, it has been documented that APC/CCdc20 mediated the degradation of Cyclin A as cells enter mitosis, whereas the spindle assembly checkpoint (SAC) delays the destruction of Cyclin B1 until the metaphase to anaphase transition [13]. Hames et al. reported that the centrosomal kinase Nek2A is also destroyed upon entry into mitosis and coincides with Cyclin A destruction [15]. Moreover, Shirayama et al. found that APC/CCdc20 promotes the exit form mitosis in part via mediating the proteolysis of the anaphase inhibitor Pds1 (yeast homolog of mammalian Securin) and the S phase cyclin Clb5 [12]. These results demonstrate that APC/CCdc20 drives the exit from mitosis in part through degradation of Securin and mitotic cyclins, leading to the necessary inactivation of mitotic Cdk. Recently, Harley et al. showed that APC/CCdc20-mediated destruction of Mcl-1, which was phosphorylated by CDK1-Cyclin B1 initiates apoptosis during mitotic arrest [17]. One elegant study from Pagano’s lab demonstrated that APC/CCdc20 controls the ubiquitin-mediated degradation of p21, a well-characterized CDK inhibitor, leading to the full activation of CDK1 necessary for mitotic events [16]. It is well characterized that p21 functions as a master effector of multiple tumor suppressor pathways for promoting anti-proliferative activities, in part by inactivating Cdk-dependent kinase activities [18]. The down-regulation of p21 expression has been observed in a variety of human cancers. Therefore, Cdc20 might exert its anti-cancer activity partly though degradation of p21 tumor suppressor protein.
Interestingly, Cdc20 has also been suggested to possess functions independently of the APC/C [19]. Clarke et al. found that in budding yeasts, Mec1p and Rad53p repress the accumulation of Cdc20 in S phase, thus avoiding premature entry into mitosis during the S phase [19], suggesting that S-phase checkpoint governs mitosis through an APC-independent Cdc20 function. Additionally, the Reed group has also found an APC-independent pool of Cdc20 that are actively involved in regulating gene transcription [19]. However, further studies are required to investigate the detailed molecular mechanisms for APC/C-independent function of Cdc20.
3. Upstream regulators of Cdc20
In recent years, most studies have focused on Cdc20 function and its downstream ubiquitin targets; however, the upstream regulators of Cdc20 in human cancer progression are largely undisclosed. Until recently, some studies began to reveal the identities of the upstream Cdc20 regulators. To this end, several groups have found that multiple genes can regulate Cdc20 expression. For example, Kidokoro et al. found that Cdc20 expression was suppressed by ectopic over-expression of p53, whereas siRNA-mediated silencing of p53 induced Cdc20 [20], suggesting that Cdc20 is negatively regulated by p53. Furthermore, inhibition of Cdc20 by siRNA induced G2/M cell cycle arrest and suppressed cell growth. Moreover, this group observed that p53 inhibits tumor cell growth through regulation of Cdc20 [20].
More importantly, multiple spindle checkpoint regulators have been discovered to inhibit APCCdc20 activity. There are accumulated data indicating the central role of suppressing APCCdc20 in the establishment and maintenance of the SAC that governs genomic stability. For example, the spindle checkpoint protein Mad2 inhibited APCCdc20 activity by forming a Mad2-Cdc20-APC complex to control anaphase initiation [21]. Later on, Luo et al. confirmed this finding by investigating the solution structure of human Mad2 and its interaction with Cdc20, which provides the structural insights into molecular mechanisms of the SAC [22]. Additionally, Reimann et al. demonstrated that Emi1 (early mitotic inhibitor 1), a conserved F-box protein, governs mitosis through inhibition of the APC [23; 24]. The elegant study from the Jackson group revealed that Emi1 could bind to Cdc20 as a pseudo-Cdc20-substrate, subsequently rescues an Emi1-induced block to Cyclin B degradation [23]. Furthermore, an independent study has shown that budding yeast Bub 1 and Bub3 binds to unattached kinetochores and blocks progression through mitosis by inhibition of APCCdc20-mediated degradation of Pds1 and Cyclin B [25]. Interestingly, a groundbreaking study from Dr. Elledge’s group identified that the deubiquitinating enzyme USP44 (ubiquitin-specific protease 44) prevents the premature activation of the APC through stabilization of the APC-inhibitory Mad2-Cdc20 complex [26]. USP44 deubiquitinates the Cdc20 and prevents the APC-driven disassembly of Mad2-Cdc20 complex [26]. This work demonstrated the dynamic balance between ubiquitination by the APC and deubiquitination by USP44 to control cells entry into mitosis.
In addition to the above described mechanisms, RASSF1A has been found to control APCCdc20 activity as well [27]. It is known that RASSF1A is a tumor suppressor gene and is frequently silenced due to its CpG hypermethylation within its promoter in a variety of human cancers [28; 29]. In addition, RASSF1A can bind to NORE1 (novel Ras effector 1) and MST1 (mammalian sterile 20-like 1), and subsequently contributes to cellular apoptosis [30]. More importantly, RASSF1A is necessary for normal mitotic progression as it can inhibit APC activity during mitosis [27]. Moreover, RASSF1A inhibits APCCdc20 and prevent degradation of Cyclins A and B at the spindle poles [27]. As a possible feedback mechanism, it has been recently reported that Aurora A controls prometaphase progression through inhibition of the ability of RASSF1A to regulate APCCdc20 activity [31].
Notably, it has been discovered that the APC/C subunit Apc15 caused Cdc20 auto-ubiquitination and promoted SAC inactivation [32]. Moreover, APC15 drives the turnover of Cdc20 and MCCs (mitotic checkpoint proteins conplexes) on the APC/C to allow the SAC to respond to the kinetochore attachment [33]. More importantly, in addition to ubiqutination-dependent regulatory mechanism, Kim et al. recently found that Sirt2 (silence information regulator 2) regulates APCCdc20 and APCCdh1 activities to maintain genome integrity and subsequently suppress tumorigenesis [34], indicating a critical role of acetylation-dependent regulation in modulating Cdc20 activities. Strikingly, it has been found that both phosphorylation by CDK (cyclin-dependent kinase) and dephosphorylation by protein phosphatase 2A are critical for the activation of the APCCdc20 [35]. These recent findings altogether suggest that Cdc20 activity is subjected to multiple layers of tight regulation in order to precisely control the SAC establishment and the ordered mitotsis progression scheme.
Notably, two recent studies have indicated that microRNAs (miRNAs) could regulate Cdc20 expression [36; 37]. It is well known that miRNAs are small non-coding RNA molecules with 21–25 nucleotides in length [38]. To exert their functions, miRNAs typically bind to the 3′UTR (3′ untranslated region) of target mRNA, thus resulting in down-regulation of gene expression via various mechanisms including translational repression, mRNA cleavage and deadenylation [38]. Interestingly, some miRNAs have oncogenic activities, whereas other miRNAs play anti-tumor activities in human cancers [39], depending on the specific target mRNAs they suppress. In this regard, one miRNA could regulate the expression of many genes, and one single gene could be controlled by multiple miRNAs [39]. Recently, Lize et al. found that miR-449a could govern the expression of Cdc20B (a paralog of CDC20) that contains the miR-449a encoding region within an intron [36]. In addition, the expression of Cdc20B was found to be up-regulated when miR-449a levels sharply increased [36]. In addition to miR-449a, miRNA-494 could also regulate Cdc20 expression in human cholangiocarcinoma [37]. Given the central role of miRNA in regulating various cellular functions including tumorigenesis, we believe that more miRNAs that may regulate Cdc20 will be discovered in the near future.
4. Cdc20-conditional knockout mice
Since Cdc20 RNAi failed to induce mitotic arrest in many studies [40], it is better to use the transgenic mouse model to investigate the requirements for mitotic exit. The study by the Zhang’s group has shown that lack of Cdc20 results in embryonic lethality at the two-cell stage and display condensed chromosomes without separation of sister chromatids [41]. To overcome this pitfall, Dr. Malumbres’s group created the Cdc20-conditional mutant mice [42]. They observed that Cdc20 (+/−) mice are viable and fertile. In Cdc20-conditional KO (knock out) mice, a tamoxifen-inducible form of the Cre recombinase (Cre-ERT2) is expressed from the 3′-UTR of the RNA polymerase II gene (RERT allele), which facilitates Cdc20 KO in adult animals upon administration of 4-hydroxytamoxifen (4-OHT) [42]. Moreover, this study has also shown that skin tumors were induced using two-stage carcinogenesis protocol in Cdc20 (−/lox) mice expressing the inducible Cre recombinase [42]. Tumors continue to grow in Cdc20 (+/lox); RERT(+/+) and Cdc20 (+/Δ); RERT(+/Cre). However, tumors were arrested completely in Cdc20 (−/Δ); RERT (+/Cre) mice soon after 4-OHT treatment. Moreover, in most cases these tumor cells contain abundant apoptotic figures. In addition, depletion of Cdc20 led to a massive arrest of tumor cells in metaphase-like stages [42].
Furthermore, to investigate the role of Cdc20 in aggressive fibrosarcomas, Manchado et al. transfected RasV12 and E1A-expressing vectors to primary MEF (mouse embryonic fibroblasts) and got the stable clones [42]. These cells after 4-OHT treatments had a mitotic arrest at 24 hours, and most cells had apoptosis or stayed at metaphase at 48 hours. However, 4-OHT treatments do not have any effect in Cdc20 (+/lox); RERT (+/Cre) cells [42]. More importantly, Cdc20 (lox/lox); RERT (+/Cre) cells grew rapid and developed fibrosarcomas in SCID mice within 10 days. 4-OHT treatments caused slightly increased in tumor size. However, Cdc20 (Δ/Δ); RERT (+/Cre) tumors stopped growing and even disappeared after 4-OHT treatment for 16 days [42]. These results coherently suggest that elimination of Cdc20 led to complete regression of aggressive tumors in vivo, thereby indicating Cdc20 as an attractive anti-cancer drug target.
5. Cdc20 is frequently over-expressed in human cancers
Consistent with the notion that Cdc20 may function as an oncoprotein, recent studies have shown that Cdc20 is highly expressed in various types of human tumors. Jiang et al reported that Cdc20 is over-expressed in breast cancer cells compared to normal mammary epithelial cells [43]. Moreover, higher expression of Cdc20 was observed in breast tumors when compared to the tissue surrounding the tumors in human breast cancer patient specimens [43]. Cdc20 gene was also over-expressed in cervical cancer and in CIN3/CLS (cervical intraepithelial neoplasia/carcinoma in situ) compared to both normal and CIN1/CIN2 [44]. Similarly, Cdc20 was up-regulated in glioblastomas and down-regulated in low-grade gliomas [45]. Moreover, over-expression of Cdc20 was found to be highly corelated with the development of glioblastomas, suggesting that Cdc20 could be a useful marker for the identification of glioblastomas in biopsy [45]. In line with these results, high expression of Cdc20 was tightly associated with a poor prognosis in epithelial ovarian tumors, indicating that Cdc20 could be a biomarker for prognosis in ovarian cancer [46]. In accordance with these findings, it has also been observed that the over-expression of Cdc20 is a common features of the urinary bladder cancer as demonstrated by whole genome microarrays [47]. Kim et al. reported that Cdc20 was over-expressed in more than 70% of gastric cancer tissues, arguing that Cdc20 could represent a potential biomarker of human gastric cancer [48]. Taken together, Cdc20 is often over-expressed in a majority of human cancers, supporting its oncogenic role in promoting tumorigenesis, and thus Cdc20 is a legitimate target of drug development for the treatment of human malignancies.
6. Over-expression of Cdc20 is correlated with clinicopathological parameters
Recently, some studies have shown that increased Cdc20 expression is associated with clinical progression in human cancers. For example, high expression of Cdc20 was observed in primary Non-small cell lung cancer (NSCLC) patients [49]. Moreover, Cdc20 expression was positively corelated with clinicopathological parameters. Specifically, higher expression of Cdc20 was associated with male sex, pleural invasion, and pathological tumor status [49]. Interestingly, Cdc20 has been shown to be an independent prognostic factor in lung adenocarcinoma patients, but not in squamous cell carcinoma patients. More importantly, NSCLC patients with higher expression of Cdc20 have significantly shorter five-year survival, suggesting that Cdc20 could be a biomarker for prognosis in NSCLC patients [49]. It has also been shown that the expression of Cdc20 was associated with the clinical stage of epithelial ovarian tumor [46]. Cdc20 expression is more tightly associated with a poor prognosis, suggesting that Cdc20 could be a biomarker in the prognosis for patients with epithelial ovarian tumors [46]. In line with these reports, higher expression of Cdc20 in pancreatic ductal adenocarcinoma (PDAC) tissues compared with normal pancreas and chronic pancreatic tissues using microarray analysis and qRT-PCR analysis [50]. Surprisingly, it has no any significant association between higher expression of Cdc20 and tumor size, tumor stage, and tumor metastasis [50]. However, this group showed that high expression of Cdc20 was associated with poor differentiation and poor recurrence-free survival rates in PDAC patients [50], indicating that Cdc20 could be useful as a prognostic marker and therapeutic target in PDAC.
7. Cdc20 inhibitors
Since Cdc20 is over-expressed in human tumors and play an oncogenic role in tumorigenesis, thus targeting Cdc20 by its inhibitors could be a novel strategy for the treatment of human cancers. Although proteasome inhibitors can block APC-dependent proteolysis, they simultaneously inhibit the destruction of many other substrates of the UPS in a non-specific fashion. Therefore, it might be better to use Cdc20 specific inhibitors instead to achieve targeted inhibition of APCCdc20 as an efficient anti-cancer therapeutic approach. Along this line, multiple recent studies have reported several inhibitors that suppress the expression of Cdc20. However, it is important to note that there are no specific Cdc20 inhibitors available so far although a promising pan-APC inhibitor has been recently discovered as discussed below.
Zeng et al. have reported a small molecule inhibitor of APC, TAME (tosyl-L-arginine methyl ester), which can bind to the APC and prevent its activation by Cdc20 and Cdh1 [51]. TAME is a mimetic of the IR motif that is involved in interaction with the APC core complex, thereby it functions as a Cdc20 (and Cdh1) competitor to disrupt the IR-tail dependent interaction between Cdc20 and the APC core complex [51]. Since TAME is not cell permeable, Zeng et al. synthesized a TAME prodrug that called proTAME, which can be processed by intracellular esterases to yield the parent compound inside the cell. The evidence from this group further suggests that ProTAME induces mitotic arrest in the absence of spindle damage in SAC dependent manner [51].
Recently, it has been independently found that NAHA, a novel hydroxamic acid-derivative, could down-regulate the expression of Cdc20 in breast cancer cells, leading to the inhibition of cell proliferation and colony formation [52]. Emerging evidence showed that NAHA inhibited cell growth, adhesion, migration, and invasion in breast cancer cells. Moreover, NAHA suppressed tumor volume and tumor weight in vivo [52]. For molecular mechanism underlying NAHA-mediated anti-tumor activity, Jiang et al. observed that NAHA down-regulated the expression of several genes including Cdc20, CDK2, uPA (urokinase-type plasminogen activator), VEGF (vascular endothelial growth factor) and CD31 [52]. Using cDNA microarray analysis, Jiang et al. further revealed that novel medicinal mushroom blend inhibited breast cancer cell growth via suppression of multiple gene expressions including Cdc20 [53]. Additionally, the same group also demonstrated that ganodermanontriol (GDNT), a ganoderma alcohol from medicinal mushroom, inhibited cell growth through down-regulation of Cdc20 in breast cancer cells [43]. Strikingly, GDNT inhibited cell proliferation and colony formation, and also inhibited invasive behavior of highly invasive breast cancer cells. Moreover, GDNT exerts these functions through down-regulation of Cdc20, uPA and uPAR (urokinase-type plasminogen activator receptor) expression [43]. Without a doubt, due to the key oncogenic roles of Cdc20 in tumorigenesis, discovery and development of more specific Cdc20 inhibitors are important which will benefit human caner patients in the near future.
8. Conclusion
In conclusion, Cdc20 may play a critical oncogenic role in the development and progression of human cancers. More importantly, accumulating evidence showed that anti-mitotic reagents including Taxol and Nocodazole, which have long been utilized as anti-cancer therapies, could activate SAC presumably through inhibiting APCCdc20 [54]. In line with this argument, it has been recently shown that Cdc20 knockdown caused a checkpoint-independent mitotic arrest that subsequently kills cancer cells more effectively than spindle-perturbing drugs, suggesting that targeting mitotic exit is a promising therapeutic option [54]. In this sense, targeting the Cdc20 oncoprotein may be a better anti-cancer therapeutic strategy than perturbing the spindle assembly by those standard spindle-perturbing drugs, in part due to that fact that it can reduce the off-target and side effects. Therefore, development of inhibitors that will specifically target Cdc20 could be a novel strategy for the treatment of a broad spectrum of human cancers. One alternative strategy may be to target several upstream genes that control Cdc20 expression, such as p53, RASSF1A, EMI1 and USP44. Furthermore, we have discussed that TAME could function as APC/CCdc20 inhibitor to retard tumor cell growth, indicating that Cdc20 could represent a novel therapeutic target. Interestingly, natural agents from mushroom were also found to inhibit Cdc20 expression in breast cancers and because of its non-toxic nature, targeting Cdc20 by natural agents alone or combined with conventional chemotherapeutics could be a promising approach for the treatment of human malignancies. We sincerely hope that this review article would attract significant attention by investigators in cancer research, and therefore stimulate further work to explore the molecular mechanism(s) by which Cdc20 is involved in tumor development and progression. We believe that these experimental evidences will also provide the rationale for designing Cdc20 specific inhibitors as novel approaches to achieve better clinical benefit in treating various types of human cancer patients.
Figure 1. Illustrates pathways for APC/Ccdc20-mediated degradation of its major downstream targets as well as the identified Cdc20 upstream regulators.
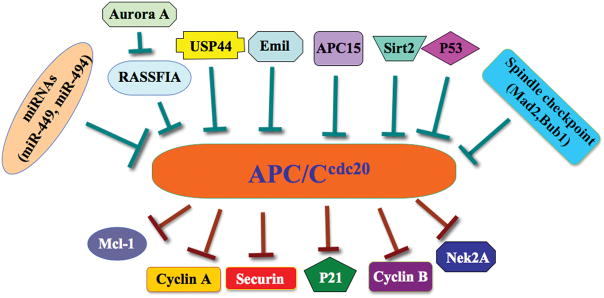
The specific substrates of APC/Ccdc20 include Mcl-1, Cyclin A, Securin, p21, Cyclin B, and Nek2A. Several upstream proteins such as p53, Aurora A, RASSF1A, Emi1, Sirt2, APC15, and USP44, as well as various microRNAs including miR-449 and miR-494, have been recently found to regulate the expression of Cdc20.
Acknowledgments
In this review article, we apologize to those authors whose work was not cited because of space limitation. This work was supported by grants from the National Institute of General Medicine, NIH (GM089763, GM094777) to W.W, and W.W is an American Cancer Society Scholar. Z. W is supported by NIH NRSA fellowship.
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.Hoeller D, Hecker CM, Dikic I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat Rev Cancer. 2006;6:776–88. doi: 10.1038/nrc1994. [DOI] [PubMed] [Google Scholar]
- 2.Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008;8:438–49. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6:369–81. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 4.McLean JR, Chaix D, Ohi MD, Gould KL. State of the APC/C: organization, function, and structure. Crit Rev Biochem Mol Biol. 2011;46:118–36. doi: 10.3109/10409238.2010.541420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qiao X, Zhang L, Gamper AM, Fujita T, Wan Y. APC/C-Cdh1: from cell cycle to cellular differentiation and genomic integrity. Cell Cycle. 2010;9:3904–12. doi: 10.4161/cc.9.19.13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eguren M, Manchado E, Malumbres M. Non-mitotic functions of the Anaphase-Promoting Complex. Semin Cell Dev Biol. 2011;22:572–8. doi: 10.1016/j.semcdb.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 7.Smolders L, Teodoro JG. Targeting the anaphase promoting complex: common pathways for viral infection and cancer therapy. Expert Opin Ther Targets. 2011;15:767–80. doi: 10.1517/14728222.2011.558008. [DOI] [PubMed] [Google Scholar]
- 8.Hartwell LH, Culotti J, Reid B. Genetic control of the cell-division cycle in yeast. I. Detection of mutants. Proc Natl Acad Sci U S A. 1970;66:352–9. doi: 10.1073/pnas.66.2.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hartwell LH, Mortimer RK, Culotti J, Culotti M. Genetic Control of the Cell Division Cycle in Yeast: V. Genetic Analysis of cdc Mutants. Genetics. 1973;74:267–86. doi: 10.1093/genetics/74.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zur A, Brandeis M. Securin degradation is mediated by fzy and fzr, and is required for complete chromatid separation but not for cytokinesis. EMBO J. 2001;20:792–801. doi: 10.1093/emboj/20.4.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lim HH, Goh PY, Surana U. Cdc20 is essential for the cyclosome-mediated proteolysis of both Pds1 and Clb2 during M phase in budding yeast. Curr Biol. 1998;8:231–4. doi: 10.1016/s0960-9822(98)70088-0. [DOI] [PubMed] [Google Scholar]
- 12.Shirayama M, Toth A, Galova M, Nasmyth K. APC(Cdc20) promotes exit from mitosis by destroying the anaphase inhibitor Pds1 and cyclin Clb5. Nature. 1999;402:203–7. doi: 10.1038/46080. [DOI] [PubMed] [Google Scholar]
- 13.Geley S, Kramer E, Gieffers C, et al. Anaphase-promoting complex/cyclosome-dependent proteolysis of human cyclin A starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J Cell Biol. 2001;153:137–48. doi: 10.1083/jcb.153.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohtoshi A, Maeda T, Higashi H, Ashizawa S, Hatakeyama M. Human p55(CDC)/Cdc20 associates with cyclin A and is phosphorylated by the cyclin A-Cdk2 complex. Biochem Biophys Res Commun. 2000;268:530–4. doi: 10.1006/bbrc.2000.2167. [DOI] [PubMed] [Google Scholar]
- 15.Hames RS, Wattam SL, Yamano H, Bacchieri R, Fry AM. APC/C-mediated destruction of the centrosomal kinase Nek2A occurs in early mitosis and depends upon a cyclin A-type D-box. EMBO J. 2001;20:7117–27. doi: 10.1093/emboj/20.24.7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amador V, Ge S, Santamaria PG, Guardavaccaro D, Pagano M. APC/C(Cdc20) controls the ubiquitin-mediated degradation of p21 in prometaphase. Mol Cell. 2007;27:462–73. doi: 10.1016/j.molcel.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harley ME, Allan LA, Sanderson HS, Clarke PR. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 2010;29:2407–20. doi: 10.1038/emboj.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–14. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clarke DJ, Segal M, Andrews CA, et al. S-phase checkpoint controls mitosis via an APC-independent Cdc20p function. Nat Cell Biol. 2003;5:928–35. doi: 10.1038/ncb1046. [DOI] [PubMed] [Google Scholar]
- 20.Kidokoro T, Tanikawa C, Furukawa Y, et al. CDC20, a potential cancer therapeutic target, is negatively regulated by p53. Oncogene. 2008;27:1562–71. doi: 10.1038/sj.onc.1210799. [DOI] [PubMed] [Google Scholar]
- 21.Fang G, Yu H, Kirschner MW. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev. 1998;12:1871–83. doi: 10.1101/gad.12.12.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo X, Fang G, Coldiron M, et al. Structure of the Mad2 spindle assembly checkpoint protein and its interaction with Cdc20. Nat Struct Biol. 2000;7:224–9. doi: 10.1038/73338. [DOI] [PubMed] [Google Scholar]
- 23.Reimann JD, Freed E, Hsu JY, et al. Emi1 is a mitotic regulator that interacts with Cdc20 and inhibits the anaphase promoting complex. Cell. 2001;105:645–55. doi: 10.1016/s0092-8674(01)00361-0. [DOI] [PubMed] [Google Scholar]
- 24.Reimann JD, Gardner BE, Margottin-Goguet F, Jackson PK. Emi1 regulates the anaphase-promoting complex by a different mechanism than Mad2 proteins. Genes Dev. 2001;15:3278–85. doi: 10.1101/gad.945701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fraschini R, Formenti E, Lucchini G, Piatti S. Budding yeast Bub2 is localized at spindle pole bodies and activates the mitotic checkpoint via a different pathway from Mad2. J Cell Biol. 1999;145:979–91. doi: 10.1083/jcb.145.5.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stegmeier F, Rape M, Draviam VM, et al. Anaphase initiation is regulated by antagonistic ubiquitination and deubiquitination activities. Nature. 2007;446:876–81. doi: 10.1038/nature05694. [DOI] [PubMed] [Google Scholar]
- 27.Song MS, Song SJ, Ayad NG, et al. The tumour suppressor RASSF1A regulates mitosis by inhibiting the APC-Cdc20 complex. Nat Cell Biol. 2004;6:129–37. doi: 10.1038/ncb1091. [DOI] [PubMed] [Google Scholar]
- 28.Amin KS, Banerjee PP. The cellular functions of RASSF1A and its inactivation in prostate cancer. J Carcinog. 2012;11:3. doi: 10.4103/1477-3163.93000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Wang B, Chen X, Bi J. The prognostic value of RASSF1A promoter hypermethylation in non-small cell lung carcinoma: a systematic review and meta-analysis. Carcinogenesis. 2011;32:411–6. doi: 10.1093/carcin/bgq266. [DOI] [PubMed] [Google Scholar]
- 30.Avruch J, Praskova M, Ortiz-Vega S, Liu M, Zhang XF. Nore1 and RASSF1 regulation of cell proliferation and of the MST1/2 kinases. Methods Enzymol. 2006;407:290–310. doi: 10.1016/S0076-6879(05)07025-4. [DOI] [PubMed] [Google Scholar]
- 31.Song SJ, Song MS, Kim SJ, et al. Aurora A regulates prometaphase progression by inhibiting the ability of RASSF1A to suppress APC-Cdc20 activity. Cancer Res. 2009;69:2314–23. doi: 10.1158/0008-5472.CAN-08-3984. [DOI] [PubMed] [Google Scholar]
- 32.Foster SA, Morgan DO. The APC/C Subunit Mnd2/Apc15 Promotes Cdc20 Autoubiquitination and Spindle Assembly Checkpoint Inactivation. Mol Cell. 2012 doi: 10.1016/j.molcel.2012.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mansfeld J, Collin P, Collins MO, Choudhary JS, Pines J. APC15 drives the turnover of MCC-CDC20 to make the spindle assembly checkpoint responsive to kinetochore attachment. Nat Cell Biol. 2011;13:1234–43. doi: 10.1038/ncb2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim HS, Vassilopoulos A, Wang RH, et al. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell. 2011;20:487–99. doi: 10.1016/j.ccr.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Labit H, Fujimitsu K, Bayin NS, et al. Dephosphorylation of Cdc20 is required for its C-box-dependent activation of the APC/C. EMBO J. 2012;31:3351–62. doi: 10.1038/emboj.2012.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lize M, Herr C, Klimke A, Bals R, Dobbelstein M. MicroRNA-449a levels increase by several orders of magnitude during mucociliary differentiation of airway epithelia. Cell Cycle. 2010;9:4579–83. doi: 10.4161/cc.9.22.13870. [DOI] [PubMed] [Google Scholar]
- 37.Yamanaka S, Campbell NR, An F, et al. Coordinated effects of microRNA-494 induce G 2/M arrest in human cholangiocarcinoma. Cell Cycle. 2012:11. doi: 10.4161/cc.21105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Kouwenhove M, Kedde M, Agami R. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat Rev Cancer. 2011;11:644–56. doi: 10.1038/nrc3107. [DOI] [PubMed] [Google Scholar]
- 39.Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–69. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 40.Baumgarten AJ, Felthaus J, Wasch R. Strong inducible knockdown of APC/CCdc20 does not cause mitotic arrest in human somatic cells. Cell Cycle. 2009;8:643–6. doi: 10.4161/cc.8.4.7810. [DOI] [PubMed] [Google Scholar]
- 41.Li M, York JP, Zhang P. Loss of Cdc20 causes a securin-dependent metaphase arrest in two-cell mouse embryos. Mol Cell Biol. 2007;27:3481–8. doi: 10.1128/MCB.02088-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manchado E, Guillamot M, de Carcer G, et al. Targeting mitotic exit leads to tumor regression in vivo: Modulation by Cdk1, Mastl, and the PP2A/B55alpha,delta phosphatase. Cancer Cell. 2010;18:641–54. doi: 10.1016/j.ccr.2010.10.028. [DOI] [PubMed] [Google Scholar]
- 43.Jiang J, Jedinak A, Sliva D. Ganodermanontriol (GDNT) exerts its effect on growth and invasiveness of breast cancer cells through the down-regulation of CDC20 and uPA. Biochem Biophys Res Commun. 2011;415:325–9. doi: 10.1016/j.bbrc.2011.10.055. [DOI] [PubMed] [Google Scholar]
- 44.Rajkumar T, Sabitha K, Vijayalakshmi N, et al. Identification and validation of genes involved in cervical tumourigenesis. BMC Cancer. 2011;11:80. doi: 10.1186/1471-2407-11-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marucci G, Morandi L, Magrini E, et al. Gene expression profiling in glioblastoma and immunohistochemical evaluation of IGFBP-2 and CDC20. Virchows Arch. 2008;453:599–609. doi: 10.1007/s00428-008-0685-7. [DOI] [PubMed] [Google Scholar]
- 46.Ouellet V, Guyot MC, Le Page C, et al. Tissue array analysis of expression microarray candidates identifies markers associated with tumor grade and outcome in serous epithelial ovarian cancer. Int J Cancer. 2006;119:599–607. doi: 10.1002/ijc.21902. [DOI] [PubMed] [Google Scholar]
- 47.Zaravinos A, Lambrou GI, Boulalas I, Delakas D, Spandidos DA. Identification of common differentially expressed genes in urinary bladder cancer. PLoS One. 2011;6:e18135. doi: 10.1371/journal.pone.0018135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim JM, Sohn HY, Yoon SY, et al. Identification of gastric cancer-related genes using a cDNA microarray containing novel expressed sequence tags expressed in gastric cancer cells. Clin Cancer Res. 2005;11:473–82. [PubMed] [Google Scholar]
- 49.Kato T, Daigo Y, Aragaki M, et al. Overexpression of CDC20 predicts poor prognosis in primary non-small cell lung cancer patients. J Surg Oncol. 2012 doi: 10.1002/jso.23109. [DOI] [PubMed] [Google Scholar]
- 50.Chang DZ, Ma Y, Ji B, et al. Increased CDC20 expression is associated with pancreatic ductal adenocarcinoma differentiation and progression. J Hematol Oncol. 2012;5:15. doi: 10.1186/1756-8722-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zeng X, Sigoillot F, Gaur S, et al. Pharmacologic inhibition of the anaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage. Cancer Cell. 2010;18:382–95. doi: 10.1016/j.ccr.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang J, Thyagarajan-Sahu A, Krchnak V, et al. NAHA, a novel hydroxamic acid-derivative, inhibits growth and angiogenesis of breast cancer in vitro and in vivo. PLoS One. 2012;7:e34283. doi: 10.1371/journal.pone.0034283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jiang J, Sliva D. Novel medicinal mushroom blend suppresses growth and invasiveness of human breast cancer cells. Int J Oncol. 2010;37:1529–36. doi: 10.3892/ijo_00000806. [DOI] [PubMed] [Google Scholar]
- 54.Huang HC, Shi J, Orth JD, Mitchison TJ. Evidence that mitotic exit is a better cancer therapeutic target than spindle assembly. Cancer Cell. 2009;16:347–58. doi: 10.1016/j.ccr.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]