

Abstract
ATP-regulated potassium (KATP) channel complexes of inward rectifier potassium channel (Kir) 6.2 and sulfonylurea receptor (SUR) 1 critically regulate pancreatic islet β-cell membrane potential, calcium influx, and insulin secretion, and consequently, represent important drug targets for metabolic disorders of glucose homeostasis. The KATP channel opener diazoxide is used clinically to treat intractable hypoglycemia caused by excessive insulin secretion, but its use is limited by off-target effects due to lack of potency and selectivity. Some progress has been made in developing improved Kir6.2/SUR1 agonists from existing chemical scaffolds and compound screening, but there are surprisingly few distinct chemotypes that are specific for SUR1-containing KATP channels. Here we report the serendipitous discovery in a high-throughput screen of a novel activator of Kir6.2/SUR1: VU0071063 [7-(4-(tert-butyl)benzyl)-1,3-dimethyl-1H-purine-2,6(3H,7H)-dione]. The xanthine derivative rapidly and dose-dependently activates Kir6.2/SUR1 with a half-effective concentration (EC50) of approximately 7 μM, is more efficacious than diazoxide at low micromolar concentrations, directly activates the channel in excised membrane patches, and is selective for SUR1- over SUR2A-containing Kir6.1 or Kir6.2 channels, as well as Kir2.1, Kir2.2, Kir2.3, Kir3.1/3.2, and voltage-gated potassium channel 2.1. Finally, we show that VU0071063 activates native Kir6.2/SUR1 channels, thereby inhibiting glucose-stimulated calcium entry in isolated mouse pancreatic β cells. VU0071063 represents a novel tool/compound for investigating β-cell physiology, KATP channel gating, and a new chemical scaffold for developing improved activators with medicinal chemistry.
Introduction
By integrating cellular metabolism, membrane potential (Vm), and excitability, ATP-sensitive K+ (KATP) channels carry out fundamental roles in nerve, muscle, epithelial, and endocrine tissue physiology (Ashcroft, 1988). KATP channels are octomeric complexes of four pore-forming inward rectifier K+ (Kir) 6.x channel subunits and four regulatory sulfonylurea receptor (SUR) x subunits (Nichols et al., 2006). Kir6.1 (KCNJ8), Kir6.2 (KCNJ11), and SUR1 (ATP binding cassette [ABC] C8) are encoded by different genes; SUR2A and SUR2B (ABCC9) are splice variants of the same gene. The three major channel subtypes created by different subunit combinations exhibit distinctive biophysical, regulatory, and pharmacological properties, as well as cell type–specific expression (Flagg et al., 2010; Hibino et al., 2010; Nichols et al., 2013).
Pancreatic islet β-cell KATP channels are validated drug targets for type 2 diabetes and severe hypoglycemia resulting from excessive insulin secretion (Denton and Jacobson, 2012). Increases in blood glucose induce MgATP-dependent Kir6.2/SUR1 channel inhibition, Vm depolarization, Ca2+ influx through L-type voltage-dependent Ca2+ channels (VDCC), and secretion of insulin, which in turn acts on a myriad of target tissues to promote glucose uptake and utilization (Ashcroft, 2007). Sulfonylurea drugs (e.g., glibenclamide, tolbutamide) that directly inhibit Kir6.2/SUR1 and stimulate insulin secretion are used clinically to help manage glycemic levels in type 2 diabetic patients. In contrast, KATP channel activators (e.g., diazoxide) are used to treat disorders of severe hypoglycemia, such as congenital hyperinsulinism and insulin-producing pancreatic tumors (Ashcroft, 2007; Nichols et al., 2007). The major KATP channel subtype in cardiomyocytes consists of Kir6.2 and SUR2A (Nichols et al., 2013), and potassium channel opener activation of sarcolemmal KATP channels and Vm hyperpolarization afford cardioprotection from subsequent ischemia-reperfusion injury (Grover and Garlid, 2000). Activation of vascular smooth muscle Kir6.1/SUR2B with pinacidil or diazoxide leads to vasodilation and a reduction in blood pressure (Flagg et al., 2010), but can also result in pathologic edema and other cardiovascular pathologies reminiscent of those found in Cantu syndrome, which results from gain-of-function mutations in SUR2 (Nichols et al., 2013).
Given the broad tissue distribution of KATP channels, their important physiologic roles, and therapeutic as well as pathologic potential in various conditions, there is considerable interest in the continued development of pharmacological modulators for targeting specific subtypes of KATP channels (Hansen, 2006). Here, we report the serendipitous discovery of a novel xanthine derivative that directly activates heterologously expressed Kir6.2/SUR1 channels and native pancreatic β-cell KATP channels. The activator, VU0071063 [7-(4-(tert-butyl)benzyl)-1,3-dimethyl-1H-purine-2,6(3H,7H)-dione], is more potent and efficacious than diazoxide and is selective for SUR1-containing KATP channels. VU0071063 represents a new tool compound for interrogating Kir6.2/SUR1 channel physiology and structure-function relationships of KATP channel gating.
Materials and Methods
Expression Vectors.
The following vectors were used in this study: pcDNA5/TO-Kir6.2 (NM_010602), pcDNA5/TO-Kir6.1 (NM_004982), pcDNA3.1-SUR1 (L40623.1), pCMV6c-SUR2A (D83598.1), pcDNA3.1-SUR2B (D86038.2), pcDNA5/TO-Kir2.1 (NM_000891.2), pcDNA5/TO-Kir2.2 (NM_021012), and pcDNA5/TO-Kir2.3 (NM_152868).
Cell Lines and Transfections.
T-REx-human embryonic kidney (HEK) 293 cells were transfected with pcDNA5/TO-Kir6.2 using Lipofectamine 2000 (Life Technologies, Carlsbad, CA) and cultured with blasticidin and hygromycin to select stably transfected cells as previously described (Lewis et al., 2009; Raphemot et al., 2011). After confirming they exhibited tetracycline-inducible Kir6.2 expression by Western blot analysis (Supplemental Fig. 1), the cells were cotransfected with pcDNA3.1-SUR1 and grown in G418-containing medium to select cells carrying stably integrated plasmids for both KATP channel subunits. Monoclonal lines were isolated by limiting dilution, expanded, and tested for tetracycline-inducible thallium (Tl+) flux, as described below. One cell line exhibiting robust Tl+ flux was selected for assay development and small-molecule screening. Monoclonal T-REx-HEK293 cell lines expressing other mammalian Kir channels were generated as described previously (Lewis et al., 2009; Raphemot et al., 2011). Monoclonal metabotropic glutamate receptor 8/G protein–coupled inward rectifier K+ channel/HEK293 cells stably expressing Kir3.1/3.2, the M4 muscarinic receptor, and rat mGlu8a were cultured as described previously (Niswender et al., 2008). For transient transfections, HEK293T cells were transfected with 1 μg of Kir6.x, 2 μg SURx, and 0.5 μg of pcDNA3.1-EGFP (transfection marker) using Lipofectamine LTX Plus according to the manufacturer’s instructions.
Western Blot Analysis.
Western blot analysis of Kir6.2 expression was performed following 24-hour induction with tetracycline essentially as described previously (Lewis et al., 2009). Goat polyclonal Kir6.2 antiserum (SC-11226) and donkey anti-goat horseradish peroxidase–conjugated antiserum (SC-2020) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Tl+ Flux Assays.
Tl+ flux assays were performed essentially as described previously (Raphemot et al., 2013). Briefly, stably transfected T-REx-HEK-293 cells expressing Kir6.2/SUR1 channels were cultured overnight in 384-well plates (20,000 cells/20 μl per well black-walled, clear-bottomed PureCoat amine-coated plates; BD, Bedford, MA) with a platting media containing Dulbecco’s modified Eagle’s medium, 10% dialyzed fetal bovine serum, and 1 μg/ml tetracycline. On the day of the experiment, the cell culture medium was replaced with dye-loading solution containing assay buffer (Hanks’ balanced salt solution with 20 mM HEPES, pH 7.3), 0.01% (w/v) Pluronic F-127 (Life Technologies), and 1.2 μM of the thallium-sensitive dye Thallos-AM (TEFlabs, Austin, TX). Following 1-hour incubation at room temperature, the dye-loading solution was washed from the plates and replaced with 20 μl/well of assay buffer. The plates were transferred to a Hamamatsu Functional Drug Screening System 6000 (FDSS6000; Hamamatsu, Tokyo, Japan) and 20 μl/well of test compounds in assay buffer (as prepared below) was added. After a 20-minute incubation period, a baseline recording was collected at 1 Hz for 10 seconds (excitation 470 ± 20 nm, emission 540 ± 30 nm) followed by addition of the Tl+ stimulus buffer (10 μl/well) and data collection for an additional 4 minutes. The Tl+ stimulus buffer contains in (mM) 125 NaHCO3, 1.8 CaSO4, 1 MgSO4, 5 glucose, 1.8 Tl2SO4, and 10 HEPES, pH 7.4. For the Tl+ flux assay on Kir3.1/3.2-expressing cells, the thallium stimulus buffer contains 12 mM Tl2SO4 and either an EC20 or EC80 of glutamate (Sigma-Aldrich, St. Louis, MO).
Test compounds from the library of Vanderbilt Institute of Chemical Biology were dispensed into in polypropylene 384-well plates (Greiner Bio-One, Monroe, NC) using an Echo555 liquid handler (Labcyte, Sunnyvale, CA) diluted in assay buffer to 2× final concentrations to generate 4- or 11-point 3-fold serial dilution series. The KATP channel inhibitors glibenclamide and tolbutamide were resuspended in assay buffer containing VU0071063 or diazoxide. Tl+ flux assays on Kir2.1, Kir2.2, Kir2.3, and Kir3.1/3.2 were performed as described previously (Raphemot et al., 2011).
Tl+ flux data were analyzed as previously described (Raphemot et al., 2013) using a combination of Excel (Microsoft Corp, Redmond, WA) with XLfit add-in (IDBS, Guildford, Surrey, UK) and OriginPro (OriginLab, Northampton, MA) software. Each data point in a given trace was divided by the first data point from that trace (static ratio) followed by subtraction of data points from control traces generated in presence of vehicle controls. The slope of the fluorescence increase beginning 5 seconds after Tl+ addition and ending 15 seconds after Tl+ addition was calculated. The data were then plotted in Prism software (GraphPad Software, San Diego, CA) to generate concentration-response curves (CRCs). Potencies were calculated from fits to CRC data using a four-parameter logistic equation.
Patch-Clamp Electrophysiology.
Transfected cells were dissociated with trypsin, plated on poly(L-lysine)–coated glass coverslips, and allowed to recover for at least 1 hour before experiments. Coverslips were transferred to a small-volume perfusion chamber and mounted on the stage of an inverted microscope. Patch electrodes were pulled from 1.5-mm outer diameter glass capillaries and had resistances ranging from 3–5 MΩ when filled with the following intracellular solution (in mM): 135 KCl, 2 MgCl2, 1 EGTA, 10 HEPES, and 3 Na2ATP, pH 7.3. The standard bath solution contained (in mM): 135 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 5 glucose, and 10 HEPES, pH 7.4. Whole-cell currents were recorded under voltage-clamp conditions using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA). The cells were voltage-clamped and stepped every 5 seconds from a holding potential of −75 to 120 mV for 200 milliseconds, and then ramped at a rate of 2.4 mV/ms from −75 to 120 mV before returning to −75 mV. Electrophysiologic data were collected at 5 kHz and filtered at 2 kHz. Data acquisition and analysis were performed using pClamp 9.2 software (Molecular Devices).
For excised patch-clamp measurements, COSm6 cells were transiently transfected with Kir6.2, SUR1, and EGFP for 24 hours before patch-clamp analysis. Transfected cells were identified by green fluorescent protein fluorescence and membrane patches were voltage-clamped. The pipette (resistance 1–2.5 MΩ) and bath solutions were (in mM): 140 KCl, 10 HEPES, and 1 EGTA and 0.5 free Mg2+, pH 7.35). After sealing, the membrane patch was excised to the inside-out configuration. Currents were recorded at a membrane potential of −50 mV using pClamp 8.2 software.
Calcium Imaging.
Islet cell intracellular calcium ([Ca2+]i) was measured using Ca2+ sensitive dye fura-2 (Life Technologies) as previously described (Jacobson et al., 2007). Briefly, mouse islets were dissociated in 0.005% trypsin, plated on glass coverslips, and cultured for 16 hours in RPMI 1640 medium supplemented with 10% fetal calf serum, concentrations of glucose specified, 100 IU ml−1 penicillin, and 100 mg ml−1 streptomycin. Cells were dye-loaded for 20 minutes at 37°C with 2 μM fura-2-AM in solution containing (in mM): 119 NaCl, 2.5 CaCl2·[(H2O)6], 4.7 KCl, 10 HEPES, 1.2 MgS04, 1.2 KH2PO4, and 2 glucose, pH 7.35. Fluorescence imaging was performed using a Nikon Eclipse TE2000-U microscope equipped with an epifluorescent illuminator (Sutter instruments, Novato, CA), a CoolSNAP HQ2 camera (Photometrics, Tucson, AZ), and Nikon Elements software (Nikon, Tokyo, Japan). The [Ca2+]i ratios of emitted fluorescence intensities at excitation wavelengths of 340 and 380 nm (F340/F380) were determined every 5 seconds with background subtraction. Cells were perifused at 37°C at a flow of 2 ml/min; the solutions used during the experiments were the loading solution with various glucose concentrations and VU0071063, as indicated.
Measurement of Complex 2 Activity.
Mitochondria were isolated from four mouse hearts using differential centrifugation in sucrose-based buffer as previously described (Wojtovich and Brookes, 2008; Wojtovich et al., 2011). Complex 2 enzymatic activity was measured spectrophotometrically at 600 nm as previously described (Wojtovich and Brookes, 2008; Wojtovich and Brookes, 2009).
Chemicals.
VU0071063 was purchased from AldrichCPR (Sigma-Aldrich, LLC, Milwaukee, WI). Diazoxide, glibenclamide, and tolbutamide were purchased from Sigma-Aldrich. All compounds were dissolved in anhydrous dimethyl sulfoxide (DMSO) (Fisher Scientific, Pittsburgh, PA) and diluted in bath solution before use. The final concentration of DMSO used was less than or equal to 0.3% (v/v).
Results
Serendipitous Discovery of the Kir6.2/SUR1 Activator VU0071063.
A Tl+ flux assay of Kir6.2/SUR1 KATP channels was developed to assess the specificity of inhibitors of a mosquito Kir channel identified in a high-throughput screen (R. Raphemot and J. S. Denton, unpublished data). The assays employ stably transfected T-REx-HEK293 cells expressing Kir6.2 from a tetracycline-inducible promoter and SUR1 constitutively (Supplemental Fig. 1). Kir6.2/SUR1 is inhibited in T-REx-HEK293 cells under control conditions and must be activated by metabolic poisoning (data not shown) or the SUR1-prefering KATP channel opener diazoxide to mediate Tl+ flux. While testing approximately 300 mosquito Kir1 antagonists for selectivity, diazoxide was inadvertently excluded from one plate, revealing a dose-dependent increase in Tl+ flux in wells I3, I4, I5, and I6 containing 30, 10, 3, and 1 μM of a mosquito Kir1 antagonist, respectively (Fig. 1, B and C). The small-molecule added to these wells, which we termed VU0071063, was reordered as a powder, freshly dissolved in DMSO, and characterized in Tl+ flux and electrophysiologic assays.
Fig. 1.
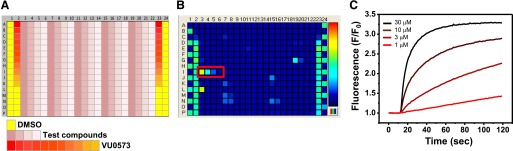
Discovery of VU0071063 in a Tl+ flux assay of Kir6.2/SUR1. (A) Plate map used in CRC analyses. DMSO (solvent) and broad-spectrum Kir channel inhibitor VU0573 (Raphemot et al., 2011) were used as controls. The 4-point test compound CRCs were distributed horizontally, whereas 11-point VU0573 CRCs were distributed vertically. (B) Fluorescence heat map recorded from the assay plate containing four doses of VU0071063 (red box). Fluorescence intensity is indicated by the pseudocolored scale (right), with cooler (blue) to hotter (red) colors corresponding to low high Tl+ flux, respectively. (C) Representative time versus normalized (F/F0) fluorescence intensity in wells containing the indicated concentrations of VU0071063.
Effects of VU0071063 on Pancreatic Kir6.2/SUR1 KATP Channels.
The activity of VU0071063 on Kir6.2/SUR1 was compared with diazoxide and the SUR2-preferring opener pinacidil in 11-point CRCs in Tl+ flux assays. The chemical structures of the three agonists are illustrated in Fig. 2A. Tetracycline-induced T-REx-HEK293-Kir6.2/SUR1 cells were treated with the agonists for 20 minutes prior to Tl+ addition to allow full activation of the channel. As shown in the representative fluorescence traces in Fig. 2B, 30 μM VU0071063 led to a slightly greater steady-state activation of Kir6.2/SUR1-dependent Tl+ flux than did 250 μM diazoxide. VU0071063 and diazoxide led to a dose-dependent activation of Tl+ flux, whereas pinacidil was predictably inactive against Kir6.2/SUR1 (Fig. 2C). Half-maximal effective concentrations (EC50) derived from logistical fits to CRC data for VU0071063 and diazoxide were 10.3 μM (95% confidence intervals [95% CI]: 9.5–11 μM) and greater than 100 μM (EC50 ∼120 μM), respectively (n = 4–6 independent experiments). To confirm that Tl+ flux is dependent on Kir6.2/SUR1 channels and not endogenous Tl+ flux pathways, the dose-dependent effects of KATP channel inhibitors glibenclamide and tolbutamide were evaluated. The cells were pretreated with EC80 doses of VU0071063 (20 μM) or diazoxide (250 μM) and 3-fold dilutions of glibenclamide or tolbutamide ranging from 2 nM to 90 μM. Similar to published half-maximal inhibitory concentration values, the IC50 values for glibenclamide and tolbutamide in VU0071063-treated cells were 5.60 nM (95% CI: 5–6 nM) and 3.07 μM (95% CI: 2–5 μM), respectively. These values are similar to those of diazoxide-treated cells (glibenclamide IC50 = 16.6 nM [95% CI: 15–18 nM]; tolbutamide IC50 = 1.60 μM [95% CI: 1.4–2 μM]).
Fig. 2.
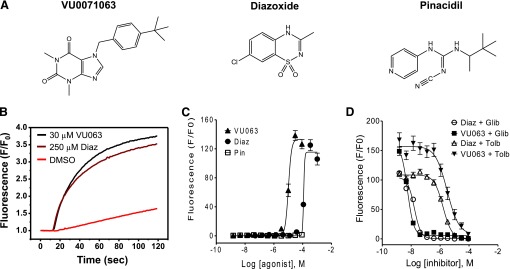
Characterization of VU0071063 activity against Kir6.2/SUR1 in Tl+ flux assays. (A) Chemical structures of VU0071063 (VU063), diazoxide (Diaz), and pinacidil (Pin). (B) Representative Tl+ flux experiment demonstrating activation of Kir6.2/SUR1 by 30 μM VU0071063 or 250 μM diazoxide, but not the vehicle control DMSO (0.3%). Fluorescence data have been normalized (F/F0) to baseline values recorded before Tl+ addition. (C) Dose-dependent activation of Kir6.2/SUR1 by VU0071063 and diazoxide, but not pinacidil (n = 4–6 independent experiments, each performed in triplicate). (D) Dose-dependent inhibition of VU0071063- and diazoxide-dependent Tl+ flux by glibenclamide (Glib) and tolbutamide (Tolb) (n = 3 independent experiments, each performed in triplicate).
Whole-cell patch-clamp electrophysiology was used to further characterize the effects of VU0071063 and diazoxide on Kir6.2/SUR1. Bath application of VU0071063 rapidly (Fig. 3A) and dose-dependently (Fig. 3C) activated Kir6.2/SUR1 currents, with a maximal activation of 1,077 ± 87% at a dose of 50 μM. In contrast, diazoxide activated Kir6.2/SUR1 more slowly (Fig. 3B) and with significantly (t test P = 0.01) lower efficacy (maximal activation 580 ± 105% at 50 μM) than VU0071063. As shown in Fig. 3B, following steady-state activation with 50 μM diazoxide, bath addition of 50 μM VU0071063 led to further Kir6.2/SUR1 activation. These data show that at low micromolar concentrations, VU0071063 is a more potent activator of Kir6.2/SUR1 than diazoxide.
Fig. 3.
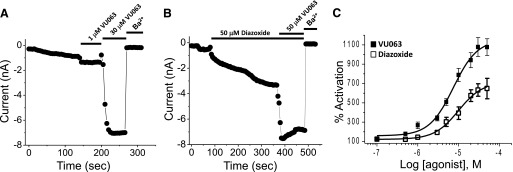
Characterization of VU0071063 activity against Kir6.2/SUR1 with patch-clamp electrophysiology. (A) Transfected cells expressing Kir6.2/SUR1 were voltage-clamped at −75 mV and stepped every 5 seconds to −120 mV to elicit inward current. Minor current run-up was observed following establishment of the whole-cell configuration and dialysis with the pipette solution. Addition of 1 or 30 μM VU0071063 led to rapid activation of inward current. (B) In contrast, addition of 50 mM diazoxide activated Kir6.2/SUR1 slowly. After achieving steady-state activation, addition of 50 μM VU0071063 led to further activation of Kir6.2/SUR1. Inward currents were blocked with 2 mM Ba2+. (C) Mean ± S.E.M. dose-response data fitted with 4-parameter logistic functions to derive EC50 values of 7 and 11 for VU0071063 (▪) and diazoxide (□), respectively (n = 5–10 per concentration). Data are normalized and expressed as percent (%) activation from baseline current in the absence of agonist.
To exclude the possibility that VU0071063 might be activating channels in intact cells by altering cell metabolism, currents were recorded from COSm6 cells expressing Kir6.2 and SUR1, in inside-out membrane patches. In the presence of 0.1 mM MgATP, which inhibits wild-type channels ∼90%, both 10 and 20 μM of VU0071063 markedly increased the patch current (Supplemental Fig. 2).
VU0071063 Is Selective SUR1-Containing KATP Channels.
The pharmacological selectivity of known KATP channel agonists is achieved through interactions with the SUR subunit. To determine if VU0071063 activity is also dependent on the SUR, we tested its effects on Kir6.2 or Kir6.1 channels containing SUR1 or SUR2A using patch-clamp electrophysiology. Diazoxide and pinacidil were used as positive controls for SUR1- and SUR2A-containting channels, respectively. As shown in the representative time-course experiment in Fig. 4A, and summary data (mean ± S.E.M.; n = 7) in Fig. 4C, bath application of 50 μM VU0071063 rapidly and reversibly activated Kir6.2/SUR1 to a greater extent than an equal concentration of diazoxide. Qualitatively similar results were observed in cells transfected with Kir6.1/SUR1 (Fig. 4D). In striking contrast, VU0071063 had no effect on Kir6.2/SUR2A (Fig. 4, B and E) or Kir6.1/SUR2A (Fig. 4F), whereas pinacidil activated both channel subtypes. Dose-response experiments revealed that VU0071063 had no appreciable effects on Kir6.2/SUR2A at concentrations up to 150 μM (Supplemental Fig. 3), which is 15-fold higher than the IC50 for Kir6.2/SUR1.
Fig. 4.
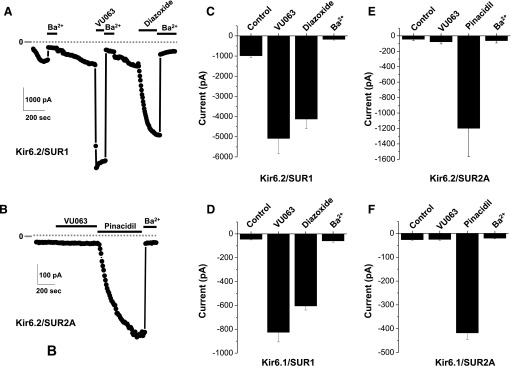
VU0071063 is selective for SUR1-containing KATP channels. (A) Representative whole-cell patch-clamp experiment showing the effects of 50 μM VU0071063, 50 μM diazoxide, and 2 mM Ba2+ on Kir6.2/SUR1 current at −120 mV. Note the differences in the kinetics of activation of VU0071063 and diazoxide. (B) Representative recording showing effects of 50 μM VU0071063, 50 μM pinacidil, and 2 mM Ba2+ on Kir6.2/SUR2A currents. (C–F) Mean ± S.E.M./ current at −120 mV recorded from cells transfected with Kir6.2/SUR1, Kir6.1/SUR1, Kir6.2/SUR2A, or Kir6.1, SUR2A, respectively (n = 4–7).
VU0071063 Inhibits Glucose-Stimulated β-Cell Ca2+ Influx.
Glucose-stimulated closure of β-cell KATP channels results in membrane potential depolarization, activation of voltage-dependent Ca2+ channels (VDCC), Ca2+ influx, and Ca2+-induced insulin secretion. We therefore tested whether VU0071063 activates native Kir6.2/SUR1 channels by measuring the effect of the activator on β-cell Ca2+ influx during glucose-stimulation. Treatment of β cells with high (14 mM) glucose induced a significant rise in Ca2+ as determined by the fluorescent Ca2+ indicator fura-2, which shows an increase in the fluorescent ratio of Ca2+ bound to Ca2+ unbound dye in response to glucose (red cells, Fig. 5). Activation of β-cell KATP channels with VU0071063 in the presence of high (14 mM) glucose resulted in inhibition of β-cell Ca2+ influx and reduction in Ca2+ levels back to those observed in low (2 mM) glucose conditions (green cells, Fig. 5). The reduction in Ca2+ influx mediated via KATP activation is reversible following removal of VU0071063, which results in a return of β-cell Ca2+ levels to high (14 mM) glucose levels (red cells, Fig. 5). These data indicate that VU0071063 activates native β-cell KATP channels and thereby reduces VDCC activation and Ca2+ influx.
Fig. 5.

VU0071063 inhibits glucose-stimulated β-cell calcium entry. (A) Two representative β cells loaded with FURA-2, displayed as a fluorescent ratio (340/380 nM) in response to 2 mM glucose (1), 14 mM glucose (2), 14 mM glucose + 10 μM VU0071063 (3), and 14 mM glucose (4). (B) Relative calcium responses of mouse islet cells following treatment with 2 mM glucose and as indicated by the conditions labeled above (black lines, n = 239 islet cells over 3 days and 13 plates of cells).
Kir6.2/SUR1 Activation by VU0071063 Is Not Mediated by a Phosphodiesterase Inhibitory Pathway.
Vascular smooth muscle KATP channels are activated by cAMP/PKA- and cGMP/PKG-dependent pathways following phosphodiesterase (PDE) inhibition with theophylline (see Discussion). Because VU0071063 contains a theophylline moiety (Supplemental Fig. 4A), we tested whether theophylline could activate Kir6.2/SUR1 in Tl+ flux assays under conditions identical to those used to discover VU0071063. However, as shown in Supplemental Fig. 4B, theophylline at a concentration of 250 μM had no effect on Kir6.2/SUR1-dependent Tl+ flux.
Ancillary Pharmacology.
The selectivity of VU007106 was evaluated in 11-point CRCs in Tl+ flux assays against Kir2.1, Kir2.2, Kir2.3, and Kir3.1/3.2. VU0071063 was inactive against Kir2.1 and Kir2.2 (IC50 > 100 μM) and showed weak inhibitor activity against Kir3.1/3.2 (IC50 = 65 μM) and Kir2.3 (IC50 = 91 μM) (Supplemental Fig. 5). Patch-clamp electrophysiology was used to determine whether VU0071063 acts on the voltage-gated K+ channel (Kv) 2.1, which contributes to action repolarization in pancreatic β cells (Philipson et al., 1994; Roe et al., 1996). Cells were voltage-clamped at a holding potential of −75 mV and stepped to +50 mV every 5 seconds. Bath application of 10 μM VU0071063 led to a 7.8 ± 0.9% (n = 4) reduction in outward Kv2.1 current at 40 mV that was fully reversible (Supplemental Fig. 6).
Discussion
Pancreatic KATP channels are validated drug targets for intractable hypoglycemia due to insulinoma and congenital hyperinsulinism, and therefore considerable efforts have been made to develop specific activators of Kir6.2/SUR1 channels (Hansen, 2006; Pirotte et al., 2010; de Tullio et al., 2011). Diazoxide is the best-known SUR1-preferring opener and has been used clinically for more than 50 years. However, its use has been limited by a lack of potency and selectivity, leading to undesirable side effects, such as low blood pressure, blurred vision, reduced urination, fluid retention, and hirsutism, mimicking the effects of Cantu syndrome, which results from gain-of-function in the cardiovascular SUR2 isoform (Nichols et al., 2013), and reflecting enhanced opening of vascular smooth muscle KATP channels and potentially effects on mitochondrial respiration (Coetzee, 2013). In an effort to develop openers with fewer side effects, several groups have synthesized analogs from existing lead compounds that show improved potency and selectivity toward Kir6.2/SUR1. Structural modifications to the diazoxide scaffold have led to several new series with submicromolar potency and selectivity for pancreatic over smooth muscle KATP channels (Pirotte et al., 2010; de Tullio et al., 2011). One analog, termed NN414 (Dabrowski et al., 2003), shows favorable activity in obese rats (Carr et al., 2003; Alemzadeh et al., 2004), as well as healthy and type 2 diabetes patients (Zdravkovic et al., 2005, 2007). Clinical trials were initiated but later suspended due to drug-induced elevations of key liver enzymes (Hansen, 2006). Analogs of the SUR2-preferring openers cromakalim and pinacidil that exhibit selectivity for pancreatic KATP channels (Khelili et al., 2006, 2008; Sebille et al., 2006, 2008; Florence et al., 2009, 2011) have also been developed, showing that it is possible to switch SUR preference with chemical modifications to the scaffold. To our knowledge, the only unique pancreatic KATP channel activator chemotypes reported in the last 2 decades were identified in screens of small-molecule libraries. These include the 4-sulfamoylphenylbenzamide and nitropyrazole series of KATP activators. A 4-sulfamoylphenylbenzamide derivative was shown to activate heterologously expressed Kir6.2/SUR1 channels and inhibit glucose-stimulated insulin secretion from primary rat islets with submicromolar efficacy; the activity toward SUR2-containing channels was not reported (Nielsen et al., 2004). One nitropyrazole analog exhibits nanomolar-affinity toward Kir6.2/SUR1 and at least 15-fold selectivity over SUR2A and SUR2B-containing channels (Peat et al., 2004). No in vivo efficacy of either series has been published.
To our knowledge, VU0071063 is only the third publicly disclosed SUR1-preferring chemotype identified with compound screening and therefore provides an important starting point for the development of new channel openers. The discovery of VU0071063 underscores the value of mining focused libraries from primary screens for modulators of diverse inward rectifier K+ channels. VU0071063 is slightly more potent than diazoxide, and activates the channel with a faster time-course. The reason for the discrepancy in IC50 values derived from Tl+ flux and patch-clamp experiments is unclear, but likely reflects 1) differences in the behavior of Tl+ and K+ in the KATP channel pore, and 2) the slower kinetics of diazoxide action compared with that of VU0071063 (e.g., Fig. 4). In an effort to avoid the latter issue, T-REx-HEK293-Kir6.2/SUR1 cells were incubated with compounds for 20 minutes before adding Tl+ stimulus buffer; however, we still observed a rightward shift in the EC50 value for diazoxide.
VU0071063 is selective for Kir6.2/SUR1 over Kir6.2/SUR2A and Kir6.1/SUR2A, as well as Kir3.1/3.2, Kir2.1, Kir2.2, Kir2.3, and Kv2.1. Kv2.1 was tested for VU0071063 sensitivity because it plays important roles in the electrophysiology of pancreatic β cells by modulating action potential repolarization, Ca2+ influx through VDCC, and insulin secretion. At the same dose shown to reduce high glucose-induced Ca2+ influx in pancreatic β cells, 10 μM VU0071063 significantly inhibited Kv2.1 currents by approximately 10%. Inhibition of Kv2.1 would be expected to prolong the action potential depolarization and increase, not decrease, Ca2+ influx through VDCC, excluding a potential role of Kv2.1 in the VU0071063 mechanism of action. Specific effects of VU0071063 on the electrical excitability and underlying ion channels in β cells will be examined in future studies. Furthermore, the selectivity of VU0071063 suggests the binding site is located within SUR1, which belongs to the ABC superfamily of transporters. Considering that the ABC transporter family member cystic fibrosis transmembrane conductance regulator is inhibited at high concentrations (IC50 = 250 μM; Sheppard and Welsh, 1992) of diazoxide, future studies should address whether VU0071063 inhibits cystic fibrosis transmembrane conductance regulator or other members of this superfamily. Finally, although VU0071063 does not activate SUR2A-containing channels, potential effects on SUR2B-containing channels and hence vascular smooth muscle tone should be considered before using VU0071063 as an in vivo probe of SUR1-containing KATP channels. Despite several attempts and different experimental conditions, we were unable to measure the activity of SUR2B-containing KATP channels in our expression system and could not determine the effect of VU0071063 on these channels (D. R. Swale and J. S. Denton, unpublished observations).
VU0071063 is structurally related to the xanthine derivative KMUP-1, which induces smooth muscle relaxation and vasodilation through activation of the cGMP and cAMP pathways. KMUP-1 induces the accumulation of cGMP and cAMP in part by inhibiting their degradation by phosphodiesterase enzymes. Pharmacologic agents that prevent cGMP accumulation predictably suppress KMUP-1–induced dilation. Inhibitors of several different families of K+ channels, including tetraethylammonium, 4-aminopyridine, iberiotoxin, charybdotoxin, and glibenclamide, blunt the vasodilatory effects of KMUP-1, indicating an important role of the membrane potential in its mechanism of action (Wu et al., 2001; Lin et al., 2002; Dai et al., 2010). However, this does not appear to involve direct K+ channel activation. For example, (Wu et al., 2004) found that KMUP-1 activates large-conductance Ca2+-activated K+ (BK) currents in cerebral smooth muscle cells, but this was dependent on cGMP generation. Although the effects of KMUP-1 on KATP currents have not been reported, cGMP is known to activate vascular KATP channels (Kubo et al., 1994). The PDE-inhibitory activity of KMUP-1 is likely mediated through the theophylline moiety, since the addition of theophylline, a nonspecific PDE inhibitor, recapitulates the effects of KMUP-1 on cGMP levels and vascular tone (Wu et al., 2004). As noted earlier, VU0071063 also contains a theophylline group, raising the possibility that PDE inhibition and cGMP or cAMP accumulation contribute to Kir6.2/SUR1 activation. However, theophylline at a concentration of 250 μM had no effect on Kir6.2/SUR1-mediated Tl+ flux following 20-minute incubation. This, together with the observation that VU0071063 directly activates Kir6.2/SUR1 in excised membrane patches (Supplemental Fig. 2), suggests that PDE inhibition and cyclic nucleotides are not essential components of its mechanism of action.
There are several important questions regarding VU0071063 and its mechanism of action remaining to be answered. KMUP-1 and VU0071063 differ only in the structure of their side-chains that project off a common theophylline moiety, yet only VU0071063 appears to be a direct SUR1/Kir6.2 channel activator. Determination of VU0071063 structure-activity relationships with medicinal chemistry will inform a deeper understanding of pharmacophore requirements for activation of SUR1- and SUR2-containing KATP channels and may lead to the development of improved xanthine-based activators. Do VU0071063 and diazoxide activate Kir6.2/SUR1 through common molecular mechanisms? For instance, do they share the same receptor binding site in SUR1, and does VU0071063 require ATP hydrolysis for channel activation like diazoxide (Larsson et al., 1993)? It is well established that diazoxide has direct effects on mitochondrial respiration, although the underlying mechanisms are a matter of ongoing debate (Coetzee, 2013). At least some of the effects of diazoxide in cardiac and smooth muscle cells are mediated through inhibition of mitochondrial complex 2 (Grimmsmann and Rustenbeck, 1998; Adebiyi et al., 2008), which has made it difficult to ascribe beneficial and undesirable effects of the drug to KATP channel–mediated effects or other mechanisms. Importantly, VU0071063 (<100 µM) had no effect on complex 2 activity (Supplemental Table 1). It will be important to determine whether VU0071063 action is limited to plasma membrane SUR1-containing channels or also has off-target effects on mitochondrial respiration and potentially other signaling pathways. The activation of KATP channels is linked to signaling pathways that can protect against cellular stress (Wojtovich et al., 2013). While the location of the channel that mediates protection (e.g., canonical surface KATP versus mitochondrial KATP channels) remains elusive (Sato et al., 2000; Suzuki et al., 2002; Wojtovich et al., 2013), VU0071063 may prove to be a valuable tool to investigate the role of KATP channels in stress responses.
In conclusion, VU0071063 is a novel xanthine derivative that directly and selectively activates KATP channels containing SUR1. Despite KATP channels being validated drug targets for numerous diseases, VU0071063 is only the third SUR1-preferring chemotype discovered using small-molecule library screening. We anticipate that the Tl+ flux assay described here will enable the discovery of additional small-molecule modulators of Kir6.2/SUR1 and other KATP channel subtypes.
Supplementary Material
Acknowledgments
The authors thank Emily Days from the Vanderbilt High Throughput Screening Facility for technical assistance in conducting high-throughput screening experiments, David Weaver (VUMC) for supplying the HEK293-Kir3.1/3.2 cell line, and Sujay Kharade (VUMC) for critically reading the manuscript.
Abbreviations
- 95% CI
95% confidence intervals
- ABC
ATP binding cassette
- [Ca2+]I
intracellular calcium concentration
- CRC
concentration-response curve
- DMSO
dimethyl sulfoxide
- HEK
human embryonic kidney
- KATP
ATP-sensitive potassium channel
- Kir
inward rectifier potassium channel
- Kv
voltage-gated potassium channel
- PDE
phosphodiesterase
- SUR
sulfonylurea receptor
- Tl+
thallium
- VDCC
L-type voltage-dependent calcium channels
- Vm
membrane potential
- VU0071063
7-(4-(tert-butyl)benzyl)-1,3-dimethyl-1H-purine-2,6(3H,7H)-dione
Authorship Contributions
Participated in research design: Raphemot, Swale, Jacobson, Cooper, Banerjee, Nichols, Denton.
Conducted experiments: Raphemot, Swale, Dadi, Jacobson, Cooper, Wojtovich, Banerjee.
Performed data analysis: Raphemot, Swale, Jacobson, Cooper, Banerjee, Wojtovich, Nichols, Denton.
Wrote or contributed to the writing of the manuscript: Raphemot, Swale, Jacobson, Cooper, Nichols, Denton.
Footnotes
This work was supported by the Foundation for the National Institutes of Health Vector-Based Transmission Control program of the Grand Challenges in Global Health initiative [Grant PIER11VCTR]; the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grants 1R01-DK082884 and 5R03-DK096122]; the National Institutes of Health National Heart, Lung, and Blood Institute [Grant R01-HL95010]; and the American Heart Association, Founder’s Affiliate Postdoctoral Fellowship [Grant 11POST7290028]. A.P.W. acknowledges support from Paul S. Brookes and Keith Nehrke (University of Rochester); work in whose laboratories is funded by the National Institutes of Health National Institute of General Medical Sciences [Grant R01-GM087483].
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Adebiyi A, McNally EM, Jaggar JH. (2008) Sulfonylurea receptor-dependent and -independent pathways mediate vasodilation induced by ATP-sensitive K+ channel openers. Mol Pharmacol 74:736–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alemzadeh R, Fledelius C, Bodvarsdottir T, Sturis J. (2004) Attenuation of hyperinsulinemia by NN414, a SUR1/Kir6.2 selective K-adenosine triphosphate channel opener, improves glucose tolerance and lipid profile in obese Zucker rats. Metabolism 53:441–447 [DOI] [PubMed] [Google Scholar]
- Ashcroft FM. (1988) Adenosine 5′-triphosphate-sensitive potassium channels. Annu Rev Neurosci 11:97–118 [DOI] [PubMed] [Google Scholar]
- Ashcroft FM. (2007) The Walter B. Cannon Physiology in Perspective Lecture, 2007. ATP-sensitive K+ channels and disease: from molecule to malady. Am J Physiol Endocrinol Metab 293:E880–E889 [DOI] [PubMed] [Google Scholar]
- Carr RD, Brand CL, Bodvarsdottir TB, Hansen JB, Sturis J. (2003) NN414, a SUR1/Kir6.2-selective potassium channel opener, reduces blood glucose and improves glucose tolerance in the VDF Zucker rat. Diabetes 52:2513–2518 [DOI] [PubMed] [Google Scholar]
- Coetzee WA. (2013) Multiplicity of effectors of the cardioprotective agent, diazoxide. Pharmacol Ther 140:167–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowski M, Larsen T, Ashcroft FM, Bondo Hansen J, Wahl P. (2003) Potent and selective activation of the pancreatic beta-cell type K(ATP) channel by two novel diazoxide analogues. Diabetologia 46:1375–1382 [DOI] [PubMed] [Google Scholar]
- Dai ZK, Cheng YJ, Chung HH, Wu JR, Chen IJ, Wu BN. (2010) KMUP-1 ameliorates monocrotaline-induced pulmonary arterial hypertension through the modulation of Ca2+ sensitization and K+-channel. Life Sci 86:747–755 [DOI] [PubMed] [Google Scholar]
- de Tullio P, Servais AC, Fillet M, Gillotin F, Somers F, Chiap P, Lebrun P, Pirotte B. (2011) Hydroxylated analogues of ATP-sensitive potassium channel openers belonging to the group of 6- and/or 7-substituted 3-isopropylamino-4H-1,2,4-benzothiadiazine 1,1-dioxides: toward an improvement in sulfonylurea receptor 1 selectivity and metabolism stability. J Med Chem 54:8353–8361 [DOI] [PubMed] [Google Scholar]
- Denton JS, Jacobson DA. (2012) Channeling dysglycemia: ion-channel variations perturbing glucose homeostasis. Trends Endocrinol Metab 23:41–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagg TP, Enkvetchakul D, Koster JC, Nichols CG. (2010) Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol Rev 90:799–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florence X, Dilly S, de Tullio P, Pirotte B, Lebrun P. (2011) Modulation of the 6-position of benzopyran derivatives and inhibitory effects on the insulin releasing process. Bioorg Med Chem 19:3919–3928 [DOI] [PubMed] [Google Scholar]
- Florence X, Sebille S, Tullio Pd, Lebrun P, Pirotte B. (2009) New R/S-3,4-dihydro-2,2-dimethyl-2H-1-benzopyrans as K(ATP) channel openers: modulation of the 4-position. Bioorg Med Chem 17:7723–7731 [DOI] [PubMed] [Google Scholar]
- Grimmsmann T, Rustenbeck I. (1998) Direct effects of diazoxide on mitochondria in pancreatic B-cells and on isolated liver mitochondria. Br J Pharmacol 123:781–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover GJ, Garlid KD. (2000) ATP-Sensitive potassium channels: a review of their cardioprotective pharmacology. J Mol Cell Cardiol 32:677–695 [DOI] [PubMed] [Google Scholar]
- Hansen JB. (2006) Towards selective Kir6.2/SUR1 potassium channel openers, medicinal chemistry and therapeutic perspectives. Curr Med Chem 13:361–376 [DOI] [PubMed] [Google Scholar]
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. (2010) Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90:291–366 [DOI] [PubMed] [Google Scholar]
- Jacobson DA, Weber CR, Bao S, Turk J, Philipson LH. (2007) Modulation of the pancreatic islet beta-cell-delayed rectifier potassium channel Kv2.1 by the polyunsaturated fatty acid arachidonate. J Biol Chem 282:7442–7449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelili S, Florence X, Bouhadja M, Abdelaziz S, Mechouch N, Mohamed Y, de Tullio P, Lebrun P, Pirotte B. (2008) Synthesis and activity on rat aorta rings and rat pancreatic beta-cells of ring-opened analogues of benzopyran-type potassium channel activators. Bioorg Med Chem 16:6124–6130 [DOI] [PubMed] [Google Scholar]
- Khelili S, Lebrun P, de Tullio P, Pirotte B. (2006) Synthesis and pharmacological evaluation of some N-arylsulfonyl-N-methyl-N’-(2,2-dimethyl-2H-1-benzopyran-4-yl)ureas structurally related to cromakalim. Bioorg Med Chem 14:3530–3534 [DOI] [PubMed] [Google Scholar]
- Kubo M, Nakaya Y, Matsuoka S, Saito K, Kuroda Y. (1994) Atrial natriuretic factor and isosorbide dinitrate modulate the gating of ATP-sensitive K+ channels in cultured vascular smooth muscle cells. Circ Res 74:471–476 [DOI] [PubMed] [Google Scholar]
- Larsson O, Ammälä C, Bokvist K, Fredholm B, Rorsman P. (1993) Stimulation of the KATP channel by ADP and diazoxide requires nucleotide hydrolysis in mouse pancreatic beta-cells. J Physiol 463:349–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis LM, Bhave G, Chauder BA, Banerjee S, Lornsen KA, Redha R, Fallen K, Lindsley CW, Weaver CD, Denton JS. (2009) High-throughput screening reveals a small-molecule inhibitor of the renal outer medullary potassium channel and Kir7.1. Mol Pharmacol 76:1094–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RJ, Wu BN, Lo YC, Shen KP, Lin YT, Huang CH, Chen IJ. (2002) KMUP-1 relaxes rabbit corpus cavernosum smooth muscle in vitro and in vivo: involvement of cyclic GMP and K(+) channels. Br J Pharmacol 135:1159–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols CG, Enkvetchakul D, Flagg TP. (2006) KATP channels: From structure to disease. Biological Membranes 23:101–110 [Google Scholar]
- Nichols CG, Koster JC, Remedi MS. (2007) beta-cell hyperexcitability: from hyperinsulinism to diabetes. Diabetes Obes Metab 9 (Suppl 2):81–88 [DOI] [PubMed] [Google Scholar]
- Nichols CG, Singh GK, Grange DK. (2013) KATP channels and cardiovascular disease: suddenly a syndrome. Circ Res 112:1059–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen FE, Jacobsen P, Worsaae A, Arkhammar PO, Wahl P, Bondo Hansen J. (2004) 2-(4-Methoxyphenoxy)-5-nitro-N-(4-sulfamoylphenyl)benzamide activates Kir6.2/SUR1 K(ATP) channels. Bioorg Med Chem Lett 14:5727–5730 [DOI] [PubMed] [Google Scholar]
- Niswender CM, Johnson KA, Luo Q, Ayala JE, Kim C, Conn PJ, Weaver CD. (2008) A novel assay of Gi/o-linked G protein-coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Mol Pharmacol 73:1213–1224 [DOI] [PubMed] [Google Scholar]
- Peat AJ, Townsend C, Craig McKay M, Garrido D, Terry CM, Wilson JL, Thomson SA. (2004) 3-trifluoromethyl-4-nitro-5-arylpyrazoles are novel K(ATP) channel agonists. Bioorg Med Chem Lett 14:813–816 [DOI] [PubMed] [Google Scholar]
- Philipson LH, Rosenberg MP, Kuznetsov A, Lancaster ME, Worley JF, 3rd, Roe MW, Dukes ID. (1994) Delayed rectifier K+ channel overexpression in transgenic islets and beta-cells associated with impaired glucose responsiveness. J Biol Chem 269:27787–27790 [PubMed] [Google Scholar]
- Pirotte B, de Tullio P, Nguyen QA, Somers F, Fraikin P, Florence X, Wahl P, Hansen JB, Lebrun P. (2010) Chloro-substituted 3-alkylamino-4H-1,2,4-benzothiadiazine 1,1-dioxides as ATP-sensitive potassium channel activators: impact of the position of the chlorine atom on the aromatic ring on activity and tissue selectivity. J Med Chem 53:147–154 [DOI] [PubMed] [Google Scholar]
- Raphemot R, Lonergan DF, Nguyen TT, Utley T, Lewis LM, Kadakia R, Weaver CD, Gogliotti R, Hopkins C, Lindsley CW, et al. (2011) Discovery, characterization, and structure-activity relationships of an inhibitor of inward rectifier potassium (Kir) channels with preference for Kir2.3, Kir3.x, and Kir7.1. Front Pharmacol 2:1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphemot R, Weaver CD, Denton JS. (2013) High-throughput screening for small-molecule modulators of inward rectifier potassium channels. J Vis Exp:71:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe MW, Worley JF, 3rd, Mittal AA, Kuznetsov A, DasGupta S, Mertz RJ, Witherspoon SM, 3rd, Blair N, Lancaster ME, McIntyre MS, et al. (1996) Expression and function of pancreatic beta-cell delayed rectifier K+ channels. Role in stimulus-secretion coupling. J Biol Chem 271:32241–32246 [DOI] [PubMed] [Google Scholar]
- Sato T, Sasaki N, Seharaseyon J, O’Rourke B, Marbán E. (2000) Selective pharmacological agents implicate mitochondrial but not sarcolemmal K(ATP) channels in ischemic cardioprotection. Circulation 101:2418–2423 [DOI] [PubMed] [Google Scholar]
- Sebille S, de Tullio P, Florence X, Becker B, Antoine MH, Michaux C, Wouters J, Pirotte B, Lebrun P. (2008) New R/S-3,4-dihydro-2,2-dimethyl-6-halo-4-(phenylaminothiocarbonylamino)-2H-1-benzopyrans structurally related to (+/-)-cromakalim as tissue-selective pancreatic beta-cell K(ATP) channel openers. Bioorg Med Chem 16:5704–5719 [DOI] [PubMed] [Google Scholar]
- Sebille S, Gall D, de Tullio P, Florence X, Lebrun P, Pirotte B. (2006) Design, synthesis, and pharmacological evaluation of R/S-3,4-dihydro-2,2-dimethyl- 6-halo-4-(phenylaminocarbonylamino)-2H-1-benzopyrans: toward tissue-selective pancreatic beta-cell KATP channel openers structurally related to (+/-)-cromakalim. J Med Chem 49:4690–4697 [DOI] [PubMed] [Google Scholar]
- Sheppard DN, Welsh MJ. (1992) Effect of ATP-sensitive K+ channel regulators on cystic fibrosis transmembrane conductance regulator chloride currents. J Gen Physiol 100:573–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Sasaki N, Miki T, Sakamoto N, Ohmoto-Sekine Y, Tamagawa M, Seino S, Marbán E, Nakaya H. (2002) Role of sarcolemmal K(ATP) channels in cardioprotection against ischemia/reperfusion injury in mice. J Clin Invest 109:509–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtovich AP, Brookes PS. (2008) The endogenous mitochondrial complex II inhibitor malonate regulates mitochondrial ATP-sensitive potassium channels: implications for ischemic preconditioning. Biochim Biophys Acta 1777:882–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtovich AP, Brookes PS. (2009) The complex II inhibitor atpenin A5 protects against cardiac ischemia-reperfusion injury via activation of mitochondrial KATP channels. Basic Res Cardiol 104:121–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtovich AP, Sherman TA, Nadtochiy SM, Urciuoli WR, Brookes PS, Nehrke K. (2011) SLO-2 is cytoprotective and contributes to mitochondrial potassium transport. PLoS ONE 6:e28287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtovich AP, Urciuoli WR, Chatterjee S, Fisher AB, Nehrke K, Brookes PS. (2013) Kir6.2 is not the mitochondrial KATP channel but is required for cardioprotection by ischemic preconditioning. Am J Physiol Heart Circ Physiol 304:H1439–H1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu BN, Lin RJ, Lin CY, Shen KP, Chiang LC, Chen IJ. (2001) A xanthine-based KMUP-1 with cyclic GMP enhancing and K(+) channels opening activities in rat aortic smooth muscle. Br J Pharmacol 134:265–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu BN, Lin RJ, Lo YC, Shen KP, Wang CC, Lin YT, Chen IJ. (2004) KMUP-1, a xanthine derivative, induces relaxation of guinea-pig isolated trachea: the role of the epithelium, cyclic nucleotides and K+ channels. Br J Pharmacol 142:1105–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdravkovic M, Kruse M, Rost KL, Møss J, Kecskes A. (2007) The effects of NN414, a SUR1/Kir6.2 selective potassium channel opener in subjects with type 2 diabetes. Exp Clin Endocrinol Diabetes 115:405–406 [DOI] [PubMed] [Google Scholar]
- Zdravkovic M, Kruse M, Rost KL, Møss J, Kecskes A, Dyrberg T. (2005) The effects of NN414, a SUR1/Kir6.2 selective potassium channel opener, in healthy male subjects. J Clin Pharmacol 45:763–772 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.