

Abstract
Head and neck cancer (HNC) is the sixth most common human malignancy worldwide. The main forms of treatment for HNC are surgery, radiotherapy (RT) and chemotherapy (CT). However, the choice of therapy depends on the tumor staging and approaches, which are aimed at organ preservation. Because of systemic RT and CT genotoxicity, one of the important side effects is a secondary cancer that can result from the activity of radiation and antineoplastic drugs on healthy cells. Ionizing radiation can affect the DNA, causing single and double-strand breaks, DNA-protein crosslinks and oxidative damage. The severity of radiotoxicity can be directly associated with the radiation dosimetry and the dose-volume differences. Regarding CT, cisplatin is still the standard protocol for the treatment of squamous cell carcinoma, the most common cancer located in the oral cavity. However, simultaneous treatment with cisplatin, bleomycin and 5-fluorouracil or treatment with paclitaxel and cisplatin are also used. These drugs can interact with the DNA, causing DNA crosslinks, double and single-strand breaks and changes in gene expression. Currently, the late effects of therapy have become a recurring problem, mainly due to the increased survival of HNC patients. Herein, we present an update of the systemic activity of RT and CT for HNC, with a focus on their toxicogenetic and toxicogenomic effects.
Keywords: Chemotherapy, Head and neck cancer, Radiotherapy, Toxicogenetic, Toxicogenomic
Core tip: The main therapies for head and neck cancer (HNC) are surgery, radiotherapy (RT) and chemotherapy. Considering that both RT and chemotherapeutical drugs can interact with the DNA, one of the important, late-occurring complications is a therapy-related secondary tumor resulting from the genotoxic effects of the therapy on the healthy cells. This review presents an update of the toxicogenetic and toxicogenomic effects of HNC treatments, highlighting the main mechanisms evolved in the secondary damage caused by RT and chemotherapies.
INTRODUCTION
Head and neck cancer (HNC) is the sixth most common human malignancy[1], representing 3% of all types of malignant tumors. The head and neck are anatomically complex regions where a wide variety of cancers with different phenotypes, histologies and invasiveness may occur[2]. Approximately 48% of the cases are located in the oral cavity, and 90% of these cases are squamous cell carcinoma (SCC), which affects the lips, mouth, tongue, nasopharynx, oropharynx, hypopharynx, larynx and paranasal sinuses[2,3]. Annually, more than 500000 new cases of SCC are diagnosed[4]. High rates of morbidity and mortality are observed[5], mainly because of the advanced clinical stage at the time of diagnosis[6]. However, the use of concurrent chemotherapy (CT) and radiation demonstrates that survival has substantially improved over the past decades for patients with most of the forms of HNC[7]. Data from some studies have suggested that SCC develops through two mechanisms: directly from the normal mucosa, called “de novo”, and following the sequence “dysplasia-carcinoma”[8-10].
Tobacco, alcohol consumption and human papillomavirus (subtypes 16, 18 and 33) are responsible for, at least, 75% of HNC[11]. However, other factors, such as diet[12], mechanical trauma[13], occupational factors, oral hygiene, inflammation[14,15] and some gene polymorphisms, are also associated[12,16]. Little is known about the molecular mechanism of HNC. Some studies have shown that alterations in the PI3K pathways, such as mutations in the PIK3A gene, have been reported in HNC[17,18]. The PI3K/AKT/mTOR pathway is activated in 57%-81% of SCC patients, and AKT is usually upregulated[19]. Moreover, it has been reported that TP53 mutations are frequently detected in the tissues of young adult SCC patients[20], and NOTCH mutations have also been found in head and neck carcinomas[21]. Furthermore, EGFR expression has been described in up to 90% of cases, and it is associated with poor prognosis[22].
Several therapies and treatment protocols are used for HNC, including surgery, CT[2] and radiotherapy (RT) and more recently, immunotherapy[23], gene therapy[24] and photodynamic therapy[25]. While treatment for loco-regionally recurrent HNC may include surgery or RT, systemic CT is used in locally advanced tumor, recurrence or metastasis[26]. CT may, or may not, be associated with surgery and/or RT[27]. The choice of HNC therapy depends on the tumor staging and the approaches for organ preservation[28]. Currently, the late effects of therapies have become a recurring problem, mainly due to the increased survival of HNC patients.
In the following paragraphs, we highlight some toxicogenetic (stable and heritable alterations in the genome that are able to influence the relative susceptibility of an individual to the adverse effects that may result from exposure to an exogenous material) and toxicogenomic (relationship between the genome and the biological response of the body after exposure to toxic agents or stressors) effects of HNC therapies.
RT
In recent years, RT has technically and biologically improved, aiming at including only the target, with minimal unnecessary irradiation to normal tissue[29,30]. Actually, RT fractionation schedules includes more fractions per day in order to reduce the overall treatment time (accelerated fractionation) and/or the use of multiple small fraction doses (hyperfractionation), which allows a higher total dose to be given without enhancing the risk of morbidity induced by radiation[31].
Radioresistance is one of the main determinants of treatment outcome in HNC patients, and it can be related to tumor hypoxia and changes in gene expression[32,33]. A meta-analysis study showed that when hypoxic modification is given in conjunction with curative intended RT result in a significant improvement in loco-regional control, disease specific control and overall survival[34]. However, another study showed that RT (5 Gy radiation) had no effect on hypoxia-inducible factor-1α (HIF-1α) gene expression in human oral SCC cell lines (SAS, Ca9-22, TT, BSC-OF and IS-FOM). Conversely, SCC cells expressing high levels of HIF-1α were resistant to radiation[35].
Several genes, such as sensors, transducers and effectors of DNA damage, have been associated with the ionizing radiation-induced cellular response[36]. One of the main molecules is the tumor-suppressor protein p53, which acts through the transcriptional control of target genes, influences multiple response pathways and leads to a diverse response to ionizing radiation in mammalian cells[37]. Additionally, one study demonstrated that the radiation-resistant capacity of nasopharyngeal tumors was mostly due to changes in the expression of genes related to cell Ca2+ homeostasis. This same study also showed that cell proliferation induced by extracellular and intracellular factors may maintain the tumor size during RT, leading to recurrence after treatment[38]. Bøhn et al[39] showed that RT induces a systemic stress response, as revealed by the induction of stress relevant gene expression in blood cells. However, the authors discuss that further studies are still needed to confirm that these changes reflect a systemic effect or are biomarkers of the tumor microenvironment. Moreover, a genomic (transcriptomic and proteomic) study of the response to radioresistance showed 265 up-regulated and 268 down-regulated genes, 30 of which were cancer-related genes. The proteomic analysis identified 51 proteins with altered expression in the radioresistant cell lines, 18 of which were cancer-related proteins. In both methodologies, the over-expression of NM23-H1 and PA2G4 were identified[33]. The influence of GP96 protein in radioresistance has been also described[40-43]. GP96 is a multifunctional protein[44] that acts as a chaperonin for peptides and modulates the innate and adaptive immune responses that are mediated by antigen-presenting cells and other immune cells[40-43]. Furthermore, GP96 is important for protein maturation and protein homeostasis[45]. In patients undergoing RT, it was shown that GP96 may serve as a novel prognostic marker of RT and may play important role in radioresistance, favoring tumor invasiveness[46].
In addition to gene expression changes, it is well known that ionizing radiation damages the DNA, causing single (SSB) and double-strand breaksdue to direct action or by the generation of free radicals[47], resulting in oxidative stress[48], base damage and DNA-protein crosslinks[49-51]. The oxidative DNA damage arising from RT can be responsible for both therapeutic and adverse effects[52]. In other words, RT may kill the tumor cells, but it can also damage normal tissues[53]. A second tumor may develop immediately or years after the primary tumor treatment[54]. Therefore, the most important dose-limiting factor is the tolerance of the adjacent normal tissue, which depends on the stage and location of the primary tumor[55]. Quantification of chromosome aberrations in circulating lymphocytes is used to estimate the effects of the RT dose[56].
RT mutagenicity on tumor and healthy tissues depends on the DNA repair capacity[57]. Genomic integrity following irradiation is maintained by specific DNA repair pathways that are initiated based on the type of DNA damage[58]. Double-strand DNA breaks are repaired via homologous recombination repair and/or non-homologous end joining repair[58]. In HNC, variants of the XRCC2, XRCC3 and RAD-5[59] genes were found to be associated with acute mucositis[60,61]. The alkaline comet assay has become a popular technique for detecting a range of DNA damage types during the last decade[62], including the effects of RT. In this sense, DNA damage and repair efficiency were evaluated by comparing the peripheral blood lymphocytes, which were isolated from tissue biopsies and from metastases biopsies of SCC patients after treatment with gamma radiation (Cobalt 60). It was observed that the repair mechanisms were less effective in patients with metastasis than in the healthy controls. Thus, the differences in radiation sensitivity of the cancer and control cells suggests that DNA repair might be critical for the treatment of SCC[57]. On the other hand, the 8-oxoGua is one of the most common DNA lesions that results from reactive oxygen species (ROS)[63], and this lesion can lead to mismatched pairing[64]. The repair hOGG1 glycosylase removes 8-oxoGua from the cellular DNA and repairs the base excision[52,65,66]. Thus, Cooke et al[65] showed that urinary excretion of 8-oxoGua and 8-oxodG can be used to measure the activity of the enzymes that are involved in the removal of oxidative DNA damage. In this sense, Roszkowski et al[52] demonstrated that fractionated RT of HNC patients resulted in elevated urinary excretion of 8-oxodG and a significant reduction of uric acid in the blood. The authors suggested that the RT is responsible for oxidative stress/oxidative DNA damage throughout the whole body and, therefore, may be responsible for a significant increase in the level of 8-oxodG in a distinct subpopulation of HNC patients[52]. The lack of increase of urinary 8-oxoGua in irradiated patients may reflect a reduction in the activity of hOGG1. Recently, it has been observed that HNC patients have a significantly reduced ability to repair the 8-oxoGua lesion that is generated by oxidative stress[67]. However, the decreased activity of the main enzyme responsible for 8-oxoGua removal should result in the accumulation of the lesion in cellular DNA[52]. Some authors have also shown different inter-individual responses in patients after RT. Kadam et al[68] studied gamma radiation-induced SSBs in peripheral blood leukocytes of SCC HNC patients with different lifestyles, both during and after RT. The results indicated that gamma radiation caused considerable DNA damage in all of the dose intervals of the treatment. However, a comparison between the smokers and non-smokers revealed the significantly greater DNA damage in the smoking patients at the pre-therapy level (10 Gy and 60 Gy of irradiation), indicating a higher sensitivity of the smokers to gamma radiation at these doses. At doses of 20-50 Gy, gamma irradiation failed to cause increased DNA damage in smokers, indicating the radio-protective or shielding effect of the tobacco (nicotine) that is possibly related to the antiapoptotic property of nicotine in the targeted/non-targeted cells. This may have important implications for RT that could indicate less effective treatment in smokers. Prolonged exposure to gamma radiation (40-60 Gy) led to a gradual decline in the intensity of the DNA damage, suggesting saturation of DNA damage in the peripheral blood leukocytes[68].
One of the cytogenetic biomarkers that is widely used to predict cancer risk in humans is the presence of micronuclei (MN)[69,70]. MN are chromosome fragments or whole chromosomes that are not incorporated into the main nucleus during mitosis. Therefore, they only appear in cells that have undergone a nuclear division[71]. The MN test is being applied to cytokinesis-blocked peripheral blood lymphocytes and exfoliated cells to monitor human exposure to mutagens[72-74]. The evaluation of cytogenetic damage by measuring the frequency of micronucleated cells (MNC) in peripheral blood and buccal mucosa of HNC patients undergoing RT showed a significant increase in the number of MNC during RT. The number of micronucleated lymphocytes remained high 30 to 140 d after the end of treatment. These data confirmed the clastogenic potential of RT in the circulating lymphocytes and buccal cells of HNC patients[75] (Figures 1 and 2).
Figure 1.
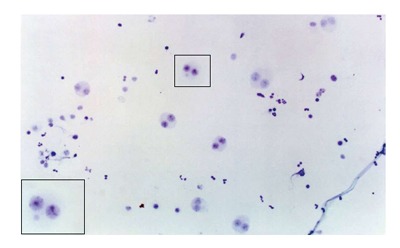
Micronucleated lymphocytes. Stained with 5% Giemsa (increased × 400).
Figure 2.
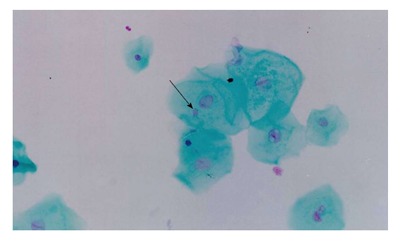
Micronucleated buccal mucosa cells. Stained with feulgen/fast-green (increased × 400).
The severity of radiotoxicity can be directly associated with the radiation dosimetry and the dose-volume differences[76]. While epidemiological studies have demonstrated the correlation between the formation of a secondary tumor with exposure to moderate-to-high doses of ionizing radiation, a statistically significant increase has hardly been described with low doses of radiation[77]. The principal RT effects in normal tissue are acute radiotoxicity (mucositis, dysphagia and dermatitis) that occurs in tissues with rapid turnover rates and late radiotoxicity [subcutaneous skin fibrosis and osteoradionecrosis (ORN)] in tissues with slower turnover rates; these effects may become evident months or years after therapy[78]. There are variable normal tissue responses to RT[79], and this may be due to the stochastic or deterministic variation effects in radioresponsiveness[76]. Mucositis, for instance, is characterized by mucosal ulceration in the oro-esophageal and gastrointestinal tract, resulting in pain, dysphagia and diarrhea, depending on the dysfunction of the affected tissue[80]. Some authors have demonstrated that intestinal mucositis is the consequence of a complex cascade of biological events, rather than solely due to direct clonogenic cell death of the epithelial cells[81]. However, evidence suggests that EGF and its receptor do not have a critical role in prevention or repair of fluorouracil CT-induced intestinal damage[82]. In addition to cell death events that are associated with the pathogenesis of mucositis, the activation of a wide variety of transcription factors, the production of proinflammatory cytokines, matrix metalloproteinases, leukotrienes, and ceramide[81], caspase activation and the generation of oxidative stress and ROS by chemotherapeutic agents or radiation appear to be primary events in most pathways leading to mucositis. ROS causes DNA damage and subsequent clonogenic cell death in the epithelial layer[81]. Of the transcriptional factors that may be significant, nuclear factor κB (NF-κB), which is activated by either RT or CT, has many of the characteristics that suggest it may be a key element in the genesis of mucositis. Once activated, NF-κB leads to the up-regulation of many genes, including those that result in the production of the pro-inflammatory cytokines TNF-α, IL-1β and IL-6. This leads to tissue injury and apoptosis[81].
Moreover, evidence of genetic mutations and the role of single nucleotide polymorphisms (SNPs) have been shown to underlie the inter-individual differences in the adverse responses of normal tissues to radiation[83-85]. Interindividual variations in radiotoxicity responses exist despite the uniform treatment protocols. It is speculated that normal genetic variations, particularly SNPs, may influence normal head and neck tissue radiotoxicity[78]. The first systematic review of the association of SNPs with the occurrence of HNC radiotoxicity evaluated the association of 11 polymorphisms in 8 genes with acute radiotoxicity and 6 polymorphisms in 4 genes for late radiotoxicity[78]. The risk of severe acute mucositis was associated with the G allele of XRCC1 (1196 A > G) in patients treated with RT alone or CT. Severe acute dysphagia was associated with the T allele of XRCC3 (722 C > T) and the G allele of XRCC6 (1310 C > G), and severe acute dermatitis was associated with the T allele of RAD51 (3392 G > T). The G allele of XRCC1 (1196 A > G) was associated with a lower grade of subcutaneous fibrosis, suggesting that the wild-type alleles were the risk alleles[86]. ORN was found to be associated with the T allele of the TGFb1 (509 C > T) polymorphism, while the CC genotype was significantly associated with post-extraction related ORN[78] (Figure 3).
Figure 3.
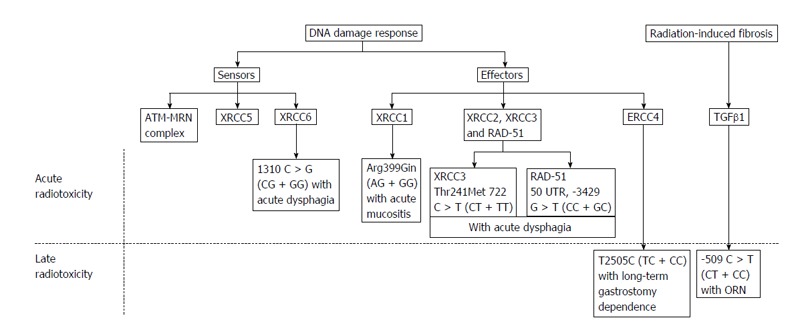
Diagram summarizing the genetic variants associated with radiotoxicity. Extracted from Ghazali et al[78].
In addition to gene expression changes and DNA damage, microRNAs have been associated with the RT response. In oral SCC cells, it was observed that ICAM2 gene inhibition by miR-125b expression downregulation induces radiosensitization, suggesting that this miRNA was associated with proliferation and radioresistance mechanisms. Therefore, the control of the expression or activity of miR-125b might contribute to the suppression of proliferation and overcoming radioresistance in OSCC[87].
CT
Cisplatin-based CT is still the standard protocol for treating SCC[26,88]. However, simultaneous treatment with cisplatin, bleomycin (radiomimetic antitumoral drug) and 5-fluorouracil (anti-metabolite drug) has also been used[89]. Other protocols have added paclitaxel to the traditional cisplatin regimen[90,91]. Combinations with methotrexate and docetaxel may be also used. The administration of the combination therapies can be curative or palliative[92].
Considering that these drugs can induce DNA damage, one of the important late-occurring complications from treatment of the primary tumor is therapy-related secondary cancer that can result from the genotoxic activity of the drugs on healthy cells[89,93]. Desai et al[94] showed that head and neck SCC patients were predisposed to chromosomal rearrangements by anticancer drug treatment, which may promote secondary tumorigenesis. Furthermore, Minicucci et al[95] observed higher frequencies of micronucleated lymphocytes in children with malignant tumors before therapy than in healthy children. The authors suggest that the presence of malignant tumors may increase the frequency of DNA damage in circulating lymphocytes. Other authors have shown that adjacent tumor tissues shared common genetic changes, and it appears that multiple tumors can arise from a single transforming event that spreads throughout the mucosa surface. On the other hand, some authors have demonstrated the presence of a second tumor that is not clonally-related to the first, which supports the hypothesis of widespread genetic changes after exposure to carcinogen[96]. In this sense, Ronchetti et al[97] studied patients with HNC and found that the pattern of microsatellite changes observed in the primary cancer exhibited a completely different genetic arrangement than the second tumor, indicating an independent origin in about three-quarters of the cases.
Cisplatin activity is based on the formation of DNA adducts that block DNA replication and transcription[98]. These crosslinks represent about 90% of the total DNA damage induced by this drug and are the major contributing factor to its cytotoxic effects[99]. Carboplatin is another CT drug used for HNC. It belongs to the same group as cisplatin; thus, it is a platinum-based antineoplastic agent. However, when compared to cisplatin, a higher concentration of carboplatin is required to reach equivalent DNA binding because it forms intrastrand DNA crosslinks at a slower rate, and the elimination of free platinum is 10-fold lower than cisplatin[100]. In relation to treatment with cisplatin, bleomycin and 5-fluorouracil, some authors using the wing somatic mutation and recombination test (Smart) in Drosophila melanogaster (D. melanogaster) have shown that the combination of these drugs produced synergistic and antagonistic genotoxic effects depending on the concentrations used, and these studies suggest that secondary effects associated with their genotoxic effects could exist, emphasizing the importance of long-term monitoring in these patients[101]. The anti-metabolite 5-fluorouracil was developed by Heidelberger et al[102] and is based on the observation that, during DNA synthesis, the uracil base was used more effectively by tumor cells than normal cells. 5-fluoracil is considered to be an S-phase active chemotherapeutic agent and causes DNA damage, such as double and single-strand breaks[103]. The genotoxicity of treatment with paclitaxel and cisplatin was also studied, as their effects are not restricted to malignant cells. Using the wing Smart method in D. melanogaster, the authors suggested that the combination of paclitaxel and cisplatin did not appear to increase the risk of secondary cancer development; however, the aneugenic activity of paclitaxel could be responsible for the reduced genotoxicity of cisplatin[104]. Methotrexate blocks the formation of tetrahydrofolic acid because of its high affinity for dihydrofolic acid reductase and suppresses protein synthesis in the G1 phase. Thus, high dose methotrexate could be possible in combination with leucovorin rescue, which prevents normal cells from being affected by the methotrexate-induced folic acid deficiency[92].
Effects of the genotype are also observed. In patients with prior HNC, 13-cis-retinoic acid (13-cRA) has been shown to prevent second primary tumors (SPT)[105]. However, other authors demonstrated that low dose 13-cRA treatment did not significantly reduce the occurrence of SPT. The results seem to be influenced by the genotype of the patient. The GST-M1 genotype is an influential risk factor for the development of SPTs in patients who were successfully treated for HNC, and the absence of the GST-T1 enzyme demonstrated a protective effect against SPT[106].
To avoid late effects of systemic CT, targeted therapy has been also used for treating HNC patients. Bevacizumab, an antiangiogenic monoclonal antibody that targets vascular endothelial growth factor[107]; transtuzumab, a monoclonal antibody that targets human epidermal growth factor receptor 2 (HER-2)[108]; lapatinib, a dual EGFR and HER-2 receptor tyrosine kinase inhibitor[109]; rapamycin, an mTOR inhibitor with antiproliferative effects[110]; sorafenib, an ERCC1 protein expression DNA repair inhibitor[111] are some of the drugs currently used in target therapy.
Among the chemoradiation-induced toxicities, mucosal barrier injury has been well studied. In this sense, peripheral blood cells of patients treated with carboplatinum, paclitaxel and radiation demonstrated the potential impact of the chemoradiation on healthy tissues. Of potential relevance to the development of mucosal injury, it has been observed that Dkk-1, a specific Wnt inhibitor, is upregulated in the tumors[112]. Some authors suggest that the presence of the Wnt inhibitor provides a mechanism for a reduction in epithelial proliferation, as studies have demonstrated that the presence of Dkk-1 is associated with crypt loss in mice[113].
CONCLUSION
Nowadays, due to advances in cancer therapies, a significant increase of survival has been observed for cancer patients. Reduction of the toxicogenetic and toxicogenomic side effects has been one of the major goals in the search for new anticancer drugs and therapy protocols.
Rapid innovations in RT have resulted in an urgent need for methods to predict cancer risks from RT, as direct observation of the late effects of newer treatments will require patient follow-up for a decade or more[114]. Cancer RT involves the eradication of the cancer cells while sparing the surrounding normal tissues. Currently, global tissue responses to RT appear to be directed towards limiting the damage, inducing repair processes and restoring tissue homeostasis[115]. These newer treatments aim to reduce the amount of healthy tissue exposed to high doses of radiation, but this may occur by increasing the amount of normal tissue exposed to lower doses of radiation[114]. Combined chemotherapeutic protocols have also been used aiming synergistic effects and decreased toxicity. Furthermore, the concurrent use of chemo and RT has shown a substantially improvement of survival over the past decades[7]. Alternative therapies, as well as target therapy have also been developed for treating HNC patients. Prevention of second primary tumors is also a field of research that has increased considerably, because of its impact on long-term survival.
Footnotes
P- Reviewers: Kawamata H, Sanabria A S- Editor: Zhai HH L- Editor: A E- Editor: Liu SQ
References
- 1.Mitra D, Malkoski SP, Wang XJ. Cancer stem cells in head and neck cancer. Cancers (Basel) 2011;3:415–427. doi: 10.3390/cancers3010415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Argiris A, Karamouzis MV, Raben D, Ferris RL. Head and neck cancer. Lancet. 2008;371:1695–1709. doi: 10.1016/S0140-6736(08)60728-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jemal A, Center MM, Ward E, Thun MJ. Cancer occurrence. Methods Mol Biol. 2009;471:3–29. doi: 10.1007/978-1-59745-416-2_1. [DOI] [PubMed] [Google Scholar]
- 4.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 5.Prince ME, Ailles LE. Cancer stem cells in head and neck squamous cell cancer. J Clin Oncol. 2008;26:2871–2875. doi: 10.1200/JCO.2007.15.1613. [DOI] [PubMed] [Google Scholar]
- 6.Tanaka T, Tanaka M, Tanaka T. Oral carcinogenesis and oral cancer chemoprevention: a review. Patholog Res Int. 2011;2011:431246. doi: 10.4061/2011/431246. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Pulte D, Brenner H. Changes in survival in head and neck cancers in the late 20th and early 21st century: a period analysis. Oncologist. 2010;15:994–1001. doi: 10.1634/theoncologist.2009-0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lippman SM, Sudbø J, Hong WK. Oral cancer prevention and the evolution of molecular-targeted drug development. J Clin Oncol. 2005;23:346–356. doi: 10.1200/JCO.2005.09.128. [DOI] [PubMed] [Google Scholar]
- 9.Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138:822–829. doi: 10.1016/j.cell.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 10.Okazaki Y, Tanaka Y, Tonogi M, Yamane G. Investigation of environmental factors for diagnosing malignant potential in oral epithelial dysplasia. Oral Oncol. 2002;38:562–573. doi: 10.1016/s1368-8375(01)00119-1. [DOI] [PubMed] [Google Scholar]
- 11.Smith EM, Rubenstein LM, Haugen TH, Pawlita M, Turek LP. Complex etiology underlies risk and survival in head and neck cancer human papillomavirus, tobacco, and alcohol: a case for multifactor disease. J Oncol. 2012;2012:571862. doi: 10.1155/2012/571862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serefoglou Z, Yapijakis C, Nkenke E, Vairaktaris E. Genetic association of cytokine DNA polymorphisms with head and neck cancer. Oral Oncol. 2008;44:1093–1099. doi: 10.1016/j.oraloncology.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 13.Brown LM, Gridley G, Diehl SR, Winn DM, Harty LC, Otero EB, Fraumeni JF, Hayes RB. Family cancer history and susceptibility to oral carcinoma in Puerto Rico. Cancer. 2001;92:2102–2108. doi: 10.1002/1097-0142(20011015)92:8<2102::aid-cncr1551>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 14.Tsantoulis PK, Kastrinakis NG, Tourvas AD, Laskaris G, Gorgoulis VG. Advances in the biology of oral cancer. Oral Oncol. 2007;43:523–534. doi: 10.1016/j.oraloncology.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 15.Taghavi N, Yazdi I. Type of food and risk of oral cancer. Arch Iran Med. 2007;10:227–232. [PubMed] [Google Scholar]
- 16.Eliot MN, Michaud DS, Langevin SM, McClean MD, Kelsey KT. Periodontal disease and mouthwash use are risk factors for head and neck squamous cell carcinoma. Cancer Causes Control. 2013;24:1315–1322. doi: 10.1007/s10552-013-0209-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mandal M, Younes M, Swan EA, Jasser SA, Doan D, Yigitbasi O, McMurphey A, Ludwick J, El-Naggar AK, Bucana C, et al. The Akt inhibitor KP372-1 inhibits proliferation and induces apoptosis and anoikis in squamous cell carcinoma of the head and neck. Oral Oncol. 2006;42:430–439. doi: 10.1016/j.oraloncology.2005.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Braakhuis B, Rietbergen M, Buijze M, Snijders P, Bloemena E, Brakenhoff R, Leemans C. TP53 mutation and human papilloma virus status of oral squamous cell carcinomas in young adult patients. Oral Dis. 2013:Epub ahead of print. doi: 10.1111/odi.12178. [DOI] [PubMed] [Google Scholar]
- 21.Wang NJ, Sanborn Z, Arnett KL, Bayston LJ, Liao W, Proby CM, Leigh IM, Collisson EA, Gordon PB, Jakkula L, et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc Natl Acad Sci USA. 2011;108:17761–17766. doi: 10.1073/pnas.1114669108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohen EE. Role of epidermal growth factor receptor pathway-targeted therapy in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck. J Clin Oncol. 2006;24:2659–2665. doi: 10.1200/JCO.2005.05.4577. [DOI] [PubMed] [Google Scholar]
- 23.Agada FO, Alhamarneh O, Stafford ND, Greenman J. Immunotherapy in head and neck cancer: current practice and future possibilities. J Laryngol Otol. 2009;123:19–28. doi: 10.1017/S0022215108003356. [DOI] [PubMed] [Google Scholar]
- 24.Ayllón Barbellido S, Campo Trapero J, Cano Sánchez J, Perea García MA, Escudero Castaño N, Bascones Martínez A. Gene therapy in the management of oral cancer: review of the literature. Med Oral Patol Oral Cir Bucal. 2008;13:E15–E21. [PubMed] [Google Scholar]
- 25.Corti L, Toniolo L, Boso C, Colaut F, Fiore D, Muzzio PC, Koukourakis MI, Mazzarotto R, Pignataro M, Loreggian L, et al. Long-term survival of patients treated with photodynamic therapy for carcinoma in situ and early non-small-cell lung carcinoma. Lasers Surg Med. 2007;39:394–402. doi: 10.1002/lsm.20513. [DOI] [PubMed] [Google Scholar]
- 26.Pendleton KP, Grandis JR. Cisplatin-Based Chemotherapy Options for Recurrent and/or Metastatic Squamous Cell Cancer of the Head and Neck. Clin Med Insights Ther. 2013:2013. doi: 10.4137/CMT.S10409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Epstein JB, Thariat J, Bensadoun RJ, Barasch A, Murphy BA, Kolnick L, Popplewell L, Maghami E. Oral complications of cancer and cancer therapy: from cancer treatment to survivorship. CA Cancer J Clin. 2012;62:400–422. doi: 10.3322/caac.21157. [DOI] [PubMed] [Google Scholar]
- 28.Lung T, Tăşcău OC, Almăşan HA, Mureşan O. Head and neck cancer, treatment, evolution and post therapeutic survival - Part 2: a decade’s results 1993-2002. J Craniomaxillofac Surg. 2007;35:126–131. doi: 10.1016/j.jcms.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 29.Corvò R. Evidence-based radiation oncology in head and neck squamous cell carcinoma. Radiother Oncol. 2007;85:156–170. doi: 10.1016/j.radonc.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 30.Lee NY, Le QT. New developments in radiation therapy for head and neck cancer: intensity-modulated radiation therapy and hypoxia targeting. Semin Oncol. 2008;35:236–250. doi: 10.1053/j.seminoncol.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Withers HR, Taylor JM, Maciejewski B. The hazard of accelerated tumor clonogen repopulation during radiotherapy. Acta Oncol. 1988;27:131–146. doi: 10.3109/02841868809090333. [DOI] [PubMed] [Google Scholar]
- 32.Overgaard J. Hypoxic radiosensitization: adored and ignored. J Clin Oncol. 2007;25:4066–4074. doi: 10.1200/JCO.2007.12.7878. [DOI] [PubMed] [Google Scholar]
- 33.Lee SY, Park HR, Cho NH, Choi YP, Rha SY, Park SW, Kim SH. Identifying genes related to radiation resistance in oral squamous cell carcinoma cell lines. Int J Oral Maxillofac Surg. 2013;42:169–176. doi: 10.1016/j.ijom.2012.10.022. [DOI] [PubMed] [Google Scholar]
- 34.Overgaard J. Hypoxic modification of radiotherapy in squamous cell carcinoma of the head and neck--a systematic review and meta-analysis. Radiother Oncol. 2011;100:22–32. doi: 10.1016/j.radonc.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 35.Hosokawa Y, Okumura K, Terashima S, Sakakura Y. Radiation protective effect of hypoxia-inducible factor-1α (HIF-1α) on human oral squamous cell carcinoma cell lines. Radiat Prot Dosimetry. 2012;152:159–163. doi: 10.1093/rpd/ncs215. [DOI] [PubMed] [Google Scholar]
- 36.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 37.Gudkov AV, Komarova EA. The role of p53 in determining sensitivity to radiotherapy. Nat Rev Cancer. 2003;3:117–129. doi: 10.1038/nrc992. [DOI] [PubMed] [Google Scholar]
- 38.Yang S, Chen J, Guo Y, Lin H, Zhang Z, Feng G, Hao Y, Cheng J, Liang P, Chen K, et al. Identification of prognostic biomarkers for response to radiotherapy by DNA microarray in nasopharyngeal carcinoma patients. Int J Oncol. 2012;40:1590–1600. doi: 10.3892/ijo.2012.1341. [DOI] [PubMed] [Google Scholar]
- 39.Bøhn SK, Russnes KM, Sakhi AK, Thoresen M, Holden M, Moskaug JØ, Myhrstad MC, Olstad OK, Smeland S, Blomhoff R. Stress associated gene expression in blood cells is related to outcome in radiotherapy treated head and neck cancer patients. BMC Cancer. 2012;12:426. doi: 10.1186/1471-2407-12-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Srivastava P. Interaction of heat shock proteins with peptides and antigen presenting cells: chaperoning of the innate and adaptive immune responses. Annu Rev Immunol. 2002;20:395–425. doi: 10.1146/annurev.immunol.20.100301.064801. [DOI] [PubMed] [Google Scholar]
- 41.Facciponte JG, Wang XY, MacDonald IJ, Park JE, Arnouk H, Grimm MJ, Li Y, Kim H, Manjili MH, Easton DP, et al. Heat shock proteins HSP70 and GP96: structural insights. Cancer Immunol Immunother. 2006;55:339–346. doi: 10.1007/s00262-005-0020-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Demine R, Walden P. Testing the role of gp96 as peptide chaperone in antigen processing. J Biol Chem. 2005;280:17573–17578. doi: 10.1074/jbc.M501233200. [DOI] [PubMed] [Google Scholar]
- 43.Randow F, Seed B. Endoplasmic reticulum chaperone gp96 is required for innate immunity but not cell viability. Nat Cell Biol. 2001;3:891–896. doi: 10.1038/ncb1001-891. [DOI] [PubMed] [Google Scholar]
- 44.Schild H, Rammensee HG. gp96-the immune system’s Swiss army knife. Nat Immunol. 2000;1:100–101. doi: 10.1038/77770. [DOI] [PubMed] [Google Scholar]
- 45.Yang Y, Li Z. Roles of heat shock protein gp96 in the ER quality control: redundant or unique function? Mol Cells. 2005;20:173–182. [PubMed] [Google Scholar]
- 46.Lin CY, Lin TY, Wang HM, Huang SF, Fan KH, Liao CT, Chen IH, Lee LY, Li YL, Chen YJ, et al. GP96 is over-expressed in oral cavity cancer and is a poor prognostic indicator for patients receiving radiotherapy. Radiat Oncol. 2011;6:136. doi: 10.1186/1748-717X-6-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parshad R, Sanford KK. Radiation-induced chromatid breaks and deficient DNA repair in cancer predisposition. Crit Rev Oncol Hematol. 2001;37:87–96. doi: 10.1016/s1040-8428(00)00111-6. [DOI] [PubMed] [Google Scholar]
- 48.Barcellos-Hoff MH, Park C, Wright EG. Radiation and the microenvironment-tumorigenesis and therapy. Nat Rev Cancer. 2005;5:867–875. doi: 10.1038/nrc1735. [DOI] [PubMed] [Google Scholar]
- 49.Engels H, Wambersie A. Relative biological effectiveness of neutrons for cancer induction and other late effects: a review of radiobiological data. Recent Results Cancer Res. 1998;150:54–87. doi: 10.1007/978-3-642-78774-4_3. [DOI] [PubMed] [Google Scholar]
- 50.Kaiser HE, Nasir A, Groger AM, Link CJ. The etiology of second primary neoplasms. In Vivo. 1998;12:89–93. [PubMed] [Google Scholar]
- 51.Perel Y, Leverger G, Carrere A, Caudry M, Garabedian EN, Ansoborlo S, Vergnes P. Second thyroid neoplasms after prophylactic cranial irradiation for acute lymphoblastic leukemia. Am J Hematol. 1998;59:91–94. doi: 10.1002/(sici)1096-8652(199809)59:1<91::aid-ajh18>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 52.Roszkowski K, Gackowski D, Rozalski R, Dziaman T, Siomek A, Guz J, Szpila A, Foksinski M, Olinski R. Small field radiotherapy of head and neck cancer patients is responsible for oxidatively damaged DNA/oxidative stress on the level of a whole organism. Int J Cancer. 2008;123:1964–1967. doi: 10.1002/ijc.23700. [DOI] [PubMed] [Google Scholar]
- 53.Aziz NM, Rowland JH. Trends and advances in cancer survivorship research: challenge and opportunity. Semin Radiat Oncol. 2003;13:248–266. doi: 10.1016/S1053-4296(03)00024-9. [DOI] [PubMed] [Google Scholar]
- 54.Allan JM, Travis LB. Mechanisms of therapy-related carcinogenesis. Nat Rev Cancer. 2005;5:943–955. doi: 10.1038/nrc1749. [DOI] [PubMed] [Google Scholar]
- 55.Vissink A, Jansma J, Spijkervet FK, Burlage FR, Coppes RP. Oral sequelae of head and neck radiotherapy. Crit Rev Oral Biol Med. 2003;14:199–212. doi: 10.1177/154411130301400305. [DOI] [PubMed] [Google Scholar]
- 56.Roch-Lefèvre S, Pouzoulet F, Giraudet AL, Voisin P, Vaurijoux A, Gruel G, Grégoire E, Buard V, Delbos M, Voisin P, et al. Cytogenetic assessment of heterogeneous radiation doses in cancer patients treated with fractionated radiotherapy. Br J Radiol. 2010;83:759–766. doi: 10.1259/bjr/21022597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rusin P, Olszewski J, Morawiec-Bajda A, Przybylowska K, Kaczmarczyk D, Golinska A, Majsterek I. Comparative study of DNA damage and repair in head and neck cancer after radiation treatment. Cell Biol Int. 2009;33:357–363. doi: 10.1016/j.cellbi.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 58.Chistiakov DA, Voronova NV, Chistiakov PA. Genetic variations in DNA repair genes, radiosensitivity to cancer and susceptibility to acute tissue reactions in radiotherapy-treated cancer patients. Acta Oncol. 2008;47:809–824. doi: 10.1080/02841860801885969. [DOI] [PubMed] [Google Scholar]
- 59.Thacker J, Zdzienicka MZ. The XRCC genes: expanding roles in DNA double-strand break repair. DNA Repair (Amst) 2004;3:1081–1090. doi: 10.1016/j.dnarep.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 60.Werbrouck J, De Ruyck K, Duprez F, Veldeman L, Claes K, Van Eijkeren M, Boterberg T, Willems P, Vral A, De Neve W, et al. Acute normal tissue reactions in head-and-neck cancer patients treated with IMRT: influence of dose and association with genetic polymorphisms in DNA DSB repair genes. Int J Radiat Oncol Biol Phys. 2009;73:1187–1195. doi: 10.1016/j.ijrobp.2008.08.073. [DOI] [PubMed] [Google Scholar]
- 61.Pratesi N, Mangoni M, Mancini I, Paiar F, Simi L, Livi L, Cassani S, Buglione M, Grisanti S, Almici C, et al. Association between single nucleotide polymorphisms in the XRCC1 and RAD51 genes and clinical radiosensitivity in head and neck cancer. Radiother Oncol. 2011;99:356–361. doi: 10.1016/j.radonc.2011.05.062. [DOI] [PubMed] [Google Scholar]
- 62.Faust F, Kassie F, Knasmüller S, Boedecker RH, Mann M, Mersch-Sundermann V. The use of the alkaline comet assay with lymphocytes in human biomonitoring studies. Mutat Res. 2004;566:209–229. doi: 10.1016/j.mrrev.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 63.Kanvah S, Joseph J, Schuster GB, Barnett RN, Cleveland CL, Landman U. Oxidation of DNA: damage to nucleobases. Acc Chem Res. 2010;43:280–287. doi: 10.1021/ar900175a. [DOI] [PubMed] [Google Scholar]
- 64.Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G----T and A----C substitutions. J Biol Chem. 1992;267:166–172. [PubMed] [Google Scholar]
- 65.Cooke MS, Evans MD, Dove R, Rozalski R, Gackowski D, Siomek A, Lunec J, Olinski R. DNA repair is responsible for the presence of oxidatively damaged DNA lesions in urine. Mutat Res. 2005;574:58–66. doi: 10.1016/j.mrfmmm.2005.01.022. [DOI] [PubMed] [Google Scholar]
- 66.Olinski R, Rozalski R, Gackowski D, Foksinski M, Siomek A, Cooke MS. Urinary measurement of 8-OxodG, 8-OxoGua, and 5HMUra: a noninvasive assessment of oxidative damage to DNA. Antioxid Redox Signal. 2006;8:1011–1019. doi: 10.1089/ars.2006.8.1011. [DOI] [PubMed] [Google Scholar]
- 67.Paz-Elizur T, Ben-Yosef R, Elinger D, Vexler A, Krupsky M, Berrebi A, Shani A, Schechtman E, Freedman L, Livneh Z. Reduced repair of the oxidative 8-oxoguanine DNA damage and risk of head and neck cancer. Cancer Res. 2006;66:11683–11689. doi: 10.1158/0008-5472.CAN-06-2294. [DOI] [PubMed] [Google Scholar]
- 68.Kadam SB, Shyama SK, Almeida VG. Evaluation of the in vivo genotoxic effects of gamma radiation on the peripheral blood leukocytes of head and neck cancer patients undergoing radiotherapy. Mutat Res. 2013;752:42–46. doi: 10.1016/j.mrgentox.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 69.Bonassi S, Znaor A, Ceppi M, Lando C, Chang WP, Holland N, Kirsch-Volders M, Zeiger E, Ban S, Barale R, et al. An increased micronucleus frequency in peripheral blood lymphocytes predicts the risk of cancer in humans. Carcinogenesis. 2007;28:625–631. doi: 10.1093/carcin/bgl177. [DOI] [PubMed] [Google Scholar]
- 70.Murgia E, Ballardin M, Bonassi S, Rossi AM, Barale R. Validation of micronuclei frequency in peripheral blood lymphocytes as early cancer risk biomarker in a nested case-control study. Mutat Res. 2008;639:27–34. doi: 10.1016/j.mrfmmm.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 71.Fenech M, Morley A. Solutions to the kinetic problem in the micronucleus assay. Cytobios. 1985;43:233–246. [PubMed] [Google Scholar]
- 72.Majer BJ, Laky B, Knasmüller S, Kassie F. Use of the micronucleus assay with exfoliated epithelial cells as a biomarker for monitoring individuals at elevated risk of genetic damage and in chemoprevention trials. Mutat Res. 2001;489:147–172. doi: 10.1016/s1383-5742(01)00068-0. [DOI] [PubMed] [Google Scholar]
- 73.Albertini RJ, Anderson D, Douglas GR, Hagmar L, Hemminki K, Merlo F, Natarajan AT, Norppa H, Shuker DE, Tice R, et al. IPCS guidelines for the monitoring of genotoxic effects of carcinogens in humans. International Programme on Chemical Safety. Mutat Res. 2000;463:111–172. doi: 10.1016/s1383-5742(00)00049-1. [DOI] [PubMed] [Google Scholar]
- 74.León-Mejía G, Espitia-Pérez L, Hoyos-Giraldo LS, Da Silva J, Hartmann A, Henriques JA, Quintana M. Assessment of DNA damage in coal open-cast mining workers using the cytokinesis-blocked micronucleus test and the comet assay. Sci Total Environ. 2011;409:686–691. doi: 10.1016/j.scitotenv.2010.10.049. [DOI] [PubMed] [Google Scholar]
- 75.Minicucci EM, Kowalski LP, Maia MA, Pereira A, Ribeiro LR, de Camargo JL, Salvadori DM. Cytogenetic damage in circulating lymphocytes and buccal mucosa cells of head-and-neck cancer patients undergoing radiotherapy. J Radiat Res. 2005;46:135–142. doi: 10.1269/jrr.46.135. [DOI] [PubMed] [Google Scholar]
- 76.Bentzen SM, Parliament M, Deasy JO, Dicker A, Curran WJ, Williams JP, Rosenstein BS. Biomarkers and surrogate endpoints for normal-tissue effects of radiation therapy: the importance of dose-volume effects. Int J Radiat Oncol Biol Phys. 2010;76:S145–S150. doi: 10.1016/j.ijrobp.2009.08.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Suzuki K, Yamashita S. Low-dose radiation exposure and carcinogenesis. Jpn J Clin Oncol. 2012;42:563–568. doi: 10.1093/jjco/hys078. [DOI] [PubMed] [Google Scholar]
- 78.Ghazali N, Shaw RJ, Rogers SN, Risk JM. Genomic determinants of normal tissue toxicity after radiotherapy for head and neck malignancy: a systematic review. Oral Oncol. 2012;48:1090–1100. doi: 10.1016/j.oraloncology.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 79.Safwat A, Bentzen SM, Turesson I, Hendry JH. Deterministic rather than stochastic factors explain most of the variation in the expression of skin telangiectasia after radiotherapy. Int J Radiat Oncol Biol Phys. 2002;52:198–204. doi: 10.1016/s0360-3016(01)02690-6. [DOI] [PubMed] [Google Scholar]
- 80.Sonis ST, Fey EG. Oral complications of cancer therapy. Oncology (Williston Park) 2002;16:680–686; discussion 686, 691-692; 695. [PubMed] [Google Scholar]
- 81.Sonis ST, Elting LS, Keefe D, Peterson DE, Schubert M, Hauer-Jensen M, Bekele BN, Raber-Durlacher J, Donnelly JP, Rubenstein EB. Perspectives on cancer therapy-induced mucosal injury: pathogenesis, measurement, epidemiology, and consequences for patients. Cancer. 2004;100:1995–2025. doi: 10.1002/cncr.20162. [DOI] [PubMed] [Google Scholar]
- 82.Huang FS, Kemp CJ, Williams JL, Erwin CR, Warner BW. Role of epidermal growth factor and its receptor in chemotherapy-induced intestinal injury. Am J Physiol Gastrointest Liver Physiol. 2002;282:G432–G442. doi: 10.1152/ajpgi.00166.2001. [DOI] [PubMed] [Google Scholar]
- 83.Andreassen CN, Alsner J. Genetic variants and normal tissue toxicity after radiotherapy: a systematic review. Radiother Oncol. 2009;92:299–309. doi: 10.1016/j.radonc.2009.06.015. [DOI] [PubMed] [Google Scholar]
- 84.Barnett GC, West CM, Dunning AM, Elliott RM, Coles CE, Pharoah PD, Burnet NG. Normal tissue reactions to radiotherapy: towards tailoring treatment dose by genotype. Nat Rev Cancer. 2009;9:134–142. doi: 10.1038/nrc2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Parliament MB, Murray D. Single nucleotide polymorphisms of DNA repair genes as predictors of radioresponse. Semin Radiat Oncol. 2010;20:232–240. doi: 10.1016/j.semradonc.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 86.Alsbeih G, Al-Harbi N, Al-Hadyan K, El-Sebaie M, Al-Rajhi N. Association between normal tissue complications after radiotherapy and polymorphic variations in TGFB1 and XRCC1 genes. Radiat Res. 2010;173:505–511. doi: 10.1667/RR1769.1. [DOI] [PubMed] [Google Scholar]
- 87.Shiiba M, Shinozuka K, Saito K, Fushimi K, Kasamatsu A, Ogawara K, Uzawa K, Ito H, Takiguchi Y, Tanzawa H. MicroRNA-125b regulates proliferation and radioresistance of oral squamous cell carcinoma. Br J Cancer. 2013;108:1817–1821. doi: 10.1038/bjc.2013.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lefebvre JL. Current clinical outcomes demand new treatment options for SCCHN. Ann Oncol. 2005;16 Suppl 6:vi7–vi12. doi: 10.1093/annonc/mdi452. [DOI] [PubMed] [Google Scholar]
- 89.Pui CH, Relling MV. Can the genotoxicity of chemotherapy be predicted? Lancet. 2004;364:917–918. doi: 10.1016/S0140-6736(04)17038-4. [DOI] [PubMed] [Google Scholar]
- 90.Adamo V, Ferraro G, Pergolizzi S, Sergi C, Laudani A, Settineri N, Alafaci E, Scimone A, Spano F, Spitaleri G. Paclitaxel and cisplatin in patients with recurrent and metastatic head and neck squamous cell carcinoma. Oral Oncol. 2004;40:525–531. doi: 10.1016/j.oraloncology.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 91.Zhang J, Tuckett RP. Comparison of paclitaxel and cisplatin effects on the slowly adapting type I mechanoreceptor. Brain Res. 2008;1214:50–57. doi: 10.1016/j.brainres.2008.01.069. [DOI] [PubMed] [Google Scholar]
- 92.Hoffmann TK. Systemic therapy strategies for head-neck carcinomas: Current status. GMS Curr Top Otorhinolaryngol Head Neck Surg. 2012;11:Doc03. doi: 10.3205/cto000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Califano J, van der Riet P, Westra W, Nawroz H, Clayman G, Piantadosi S, Corio R, Lee D, Greenberg B, Koch W, et al. Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Res. 1996;56:2488–2492. [PubMed] [Google Scholar]
- 94.Desai B, Gadhia P. Chromosomal rearrangements in lymphocytes of head and neck squamous cell carcinoma treated with chemotherapeutic agents. Neoplasma. 2012;59:463–468. doi: 10.4149/neo_2012_059. [DOI] [PubMed] [Google Scholar]
- 95.Minicucci EM, Ribeiro DA, de Camargo B, Costa MC, Ribeiro LR, Favero Salvadori DM. DNA damage in lymphocytes and buccal mucosa cells of children with malignant tumours undergoing chemotherapy. Clin Exp Med. 2008;8:79–85. doi: 10.1007/s10238-008-0161-3. [DOI] [PubMed] [Google Scholar]
- 96.Ha PK, Califano JA. The molecular biology of mucosal field cancerization of the head and neck. Crit Rev Oral Biol Med. 2003;14:363–369. doi: 10.1177/154411130301400506. [DOI] [PubMed] [Google Scholar]
- 97.Ronchetti D, Arisi E, Neri A, Pruneri G, Digiuni B, Sambataro G, Gallo O, Pignataro L. Microsatellite analyses of recurrence or second primary tumor in head and neck cancer. Anticancer Res. 2005;25:2771–2775. [PubMed] [Google Scholar]
- 98.García Sar D, Montes-Bayón M, Aguado Ortiz L, Blanco-González E, Sierra LM, Sanz-Medel A. In vivo detection of DNA adducts induced by cisplatin using capillary HPLC-ICP-MS and their correlation with genotoxic damage in Drosophila melanogaster. Anal Bioanal Chem. 2008;390:37–44. doi: 10.1007/s00216-007-1634-z. [DOI] [PubMed] [Google Scholar]
- 99.Bhana S, Hewer A, Phillips DH, Lloyd DR. p53-dependent global nucleotide excision repair of cisplatin-induced intrastrand cross links in human cells. Mutagenesis. 2008;23:131–136. doi: 10.1093/mutage/gen001. [DOI] [PubMed] [Google Scholar]
- 100.Knox RJ, Friedlos F, Lydall DA, Roberts JJ. Mechanism of cytotoxicity of anticancer platinum drugs: evidence that cis-diamminedichloroplatinum(II) and cis-diammine-(1,1-cyclobutanedicarboxylato)platinum(II) differ only in the kinetics of their interaction with DNA. Cancer Res. 1986;46:1972–1979. [PubMed] [Google Scholar]
- 101.Danesi CC, Dihl RR, Bellagamba BC, de Andrade HH, Cunha KS, Guimarães NN, Lehmann M. Genotoxicity testing of combined treatment with cisplatin, bleomycin, and 5-fluorouracil in somatic cells of Drosophila melanogaster. Mutat Res. 2012;747:228–233. doi: 10.1016/j.mrgentox.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 102.Heidelberger C, Chaudhuri NK, Danneberg P, Mooren D, Griesbach L, Duschinsky R, Schnitzer RJ, Pleven E, Scheiner J. Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature. 1957;179:663–666. doi: 10.1038/179663a0. [DOI] [PubMed] [Google Scholar]
- 103.Peters GJ, van Triest B, Backus HH, Kuiper CM, van der Wilt CL, Pinedo HM. Molecular downstream events and induction of thymidylate synthase in mutant and wild-type p53 colon cancer cell lines after treatment with 5-fluorouracil and the thymidylate synthase inhibitor raltitrexed. Eur J Cancer. 2000;36:916–924. doi: 10.1016/s0959-8049(00)00026-5. [DOI] [PubMed] [Google Scholar]
- 104.Danesi CC, Bellagamba BC, Dihl RR, de Andrade HH, Cunha KS, Spanó MA, Reguly ML, Lehmann M. Mutagenic evaluation of combined paclitaxel and cisplatin treatment in somatic cells of Drosophila melanogaster. Mutat Res. 2010;696:139–143. doi: 10.1016/j.mrgentox.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 105.Hong WK, Lippman SM, Itri LM, Karp DD, Lee JS, Byers RM, Schantz SP, Kramer AM, Lotan R, Peters LJ. Prevention of second primary tumors with isotretinoin in squamous-cell carcinoma of the head and neck. N Engl J Med. 1990;323:795–801. doi: 10.1056/NEJM199009203231205. [DOI] [PubMed] [Google Scholar]
- 106.Minard CG, Spitz MR, Wu X, Hong WK, Etzel CJ. Evaluation of glutathione S-transferase polymorphisms and mutagen sensitivity as risk factors for the development of second primary tumors in patients previously diagnosed with early-stage head and neck cancer. Cancer. 2006;106:2636–2644. doi: 10.1002/cncr.21928. [DOI] [PubMed] [Google Scholar]
- 107.Lothaire P, de Azambuja E, Dequanter D, Lalami Y, Sotiriou C, Andry G, Castro G, Awada A. Molecular markers of head and neck squamous cell carcinoma: promising signs in need of prospective evaluation. Head Neck. 2006;28:256–269. doi: 10.1002/hed.20326. [DOI] [PubMed] [Google Scholar]
- 108.Khan AJ, King BL, Smith BD, Smith GL, DiGiovanna MP, Carter D, Haffty BG. Characterization of the HER-2/neu oncogene by immunohistochemical and fluorescence in situ hybridization analysis in oral and oropharyngeal squamous cell carcinoma. Clin Cancer Res. 2002;8:540–548. [PubMed] [Google Scholar]
- 109.Rusnak DW, Lackey K, Affleck K, Wood ER, Alligood KJ, Rhodes N, Keith BR, Murray DM, Knight WB, Mullin RJ, et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Ther. 2001;1:85–94. [PubMed] [Google Scholar]
- 110.Aissat N, Le Tourneau C, Ghoul A, Serova M, Bieche I, Lokiec F, Raymond E, Faivre S. Antiproliferative effects of rapamycin as a single agent and in combination with carboplatin and paclitaxel in head and neck cancer cell lines. Cancer Chemother Pharmacol. 2008;62:305–313. doi: 10.1007/s00280-007-0609-2. [DOI] [PubMed] [Google Scholar]
- 111.Yadav A, Kumar B, Teknos TN, Kumar P. Sorafenib enhances the antitumor effects of chemoradiation treatment by downregulating ERCC-1 and XRCC-1 DNA repair proteins. Mol Cancer Ther. 2011;10:1241–1251. doi: 10.1158/1535-7163.MCT-11-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sonis S, Haddad R, Posner M, Watkins B, Fey E, Morgan TV, Mookanamparambil L, Ramoni M. Gene expression changes in peripheral blood cells provide insight into the biological mechanisms associated with regimen-related toxicities in patients being treated for head and neck cancers. Oral Oncol. 2007;43:289–300. doi: 10.1016/j.oraloncology.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 113.Kuhnert F, Davis CR, Wang HT, Chu P, Lee M, Yuan J, Nusse R, Kuo CJ. Essential requirement for Wnt signaling in proliferation of adult small intestine and colon revealed by adenoviral expression of Dickkopf-1. Proc Natl Acad Sci USA. 2004;101:266–271. doi: 10.1073/pnas.2536800100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hall EJ, Wuu CS. Radiation-induced second cancers: the impact of 3D-CRT and IMRT. Int J Radiat Oncol Biol Phys. 2003;56:83–88. doi: 10.1016/s0360-3016(03)00073-7. [DOI] [PubMed] [Google Scholar]
- 115.Berrington de Gonzalez A, Gilbert E, Curtis R, Inskip P, Kleinerman R, Morton L, Rajaraman P, Little MP. Second solid cancers after radiation therapy: a systematic review of the epidemiologic studies of the radiation dose-response relationship. Int J Radiat Oncol Biol Phys. 2013;86:224–233. doi: 10.1016/j.ijrobp.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]