

Key Points
SRF is essential for neutrophil migration in part by regulation of integrin homeostasis.
Several genes located on chromosome 5q are part of the SRF signaling pathway implicating dysfunction of SRF in myelodysplasia.
Abstract
Serum response factor (SRF) is a ubiquitously expressed transcription factor and master regulator of the actin cytoskeleton. We have previously shown that SRF is essential for megakaryocyte maturation and platelet formation and function. Here we elucidate the role of SRF in neutrophils, the primary defense against infections. To study the effect of SRF loss in neutrophils, we crossed Srffl/fl mice with select Cre-expressing mice and studied neutrophil function in vitro and in vivo. Despite normal neutrophil numbers, neutrophil function is severely impaired in Srf knockout (KO) neutrophils. Srf KO neutrophils fail to polymerize globular actin to filamentous actin in response to N-formyl-methionine-leucine-phenylalanine, resulting in significantly disrupted cytoskeletal remodeling. Srf KO neutrophils fail to migrate to sites of inflammation in vivo and along chemokine gradients in vitro. Polarization in response to cytokine stimuli is absent and Srf KO neutrophils show markedly reduced adhesion. Integrins play an essential role in cellular adhesion, and although integrin expression levels are maintained with loss of SRF, integrin activation and trafficking are disrupted. Migration and cellular adhesion are essential for normal cell function, but also for malignant processes such as metastasis, underscoring an essential function for SRF and its pathway in health and disease.
Introduction
Neutrophil function is impaired in inherited disorders such as in leukocyte adhesion deficiency and in acquired disorders such as myelodysplasia, resulting in severe morbidity and mortality resulting from infectious complications. In myelodysplasia, genetic abnormalities are frequent and can include loss of part or all of chromosome 5. Chromosome 5q contains several genes that are part of the “serum response factor (SRF) pathway.” The diaphanous-related formin mDIA and catenin α-1 are upstream regulators of SRF-mediated transcriptional regulation, whereas early growth response gene 1, which has been implicated in both neutrophil function and tumor suppression, lies downstream of SRF.1-4 The commonly deleted region on 5q also includes microRNAs 143 and 145,5 which are both directly regulated by SRF.6,7 MicroRNAs 143 and 145 have been implicated as tumor suppressors in epithelial and hematopoietic cells by modulating the MDM2-p53 pathway8 and may regulate neutrophil differentiation.9
SRF is ubiquitously expressed and, as master regulator of the actin cytoskeleton, performs critical functions in cellular migration, contractility, cell growth, and differentiation.10,11 Specificity of SRF-mediated transcriptional regulation is achieved by recruitment of different transcriptional cofactors belonging to 2 families of proteins: the myocardin-related family of transcription factors and ternary complex factors. The myocardin family includes myocardin and megakaryoblastic leukemias 1 and 2. The ternary complex factors include the ETS domain–containing proteins ELK1, ELK3, and ELK4.12 Cell- and context-dependent recruitment of SRF itself to different genomic regions, such as gene promoters or enhancers, by lineage-specific transcription factors, provides an additional layer of transcriptional regulatory specificity.13
SRF is widely expressed in the hematopoietic system and we and others have characterized its essential role in megakaryopoiesis and platelet formation and function.14,15 Deletion of SRF specifically in the megakaryocytic lineage leads to decreased megakaryocyte ploidy and defective proplatelet formation and platelet function. Loss of SRF impairs the interaction between hematopoietic stem cells and their niche,16 lymphocyte development,17 and macrophage function.13 Here we show that SRF is also critical for neutrophil function. SRF-deleted neutrophils fail to migrate to sites of inflammation in vivo. They fail to chemotax in vitro and exhibit markedly abnormal polarization and cellular adhesion. Importantly, although reduced expression of integrins has been postulated to be the underlying mechanism for defective adhesion in stem cells, we identify a novel role here for SRF in integrin homeostasis. In the absence of SRF, 1 of the 2 major neutrophil integrin complexes, Mac1, which consists of a CD11b/CD18 heterodimer, upon neutrophil activation, exhibits markedly reduced ligand binding and trafficking that are essential to directional migration. We thus identify a novel role for SRF in regulation of cell–cell and cell–extracellular matrix interactions.
Methods
Mice
All procedures were performed in compliance with relevant laws and institutional guidelines and approved by the Yale University Institutional Animal Care and Use Committee. Mice were obtained from Jackson Laboratory (Bar Harbor, ME). Srffl/fl mice18 were crossed with mice expressing Cre-recombinase under the Vav promoter19 for deletion in the hematopoietic stem cell or under the tetracycline-inducible Rosa26 promoter20 B6.Cg-Tg(tetO-cre)1Jaw/J x B6.Cg-Gt(ROSA)26Sortm1(rtTA*M2)Jae/J. A third transgene, Cre-inducible YFP expression, was crossed as reported previously to allow tracking of Srf-deleted cells.14,21 Mice were kept in microisolator cages, given autoclaved food and water, and handled only with gloves in a biological safety cabinet. Mice were maintained on Sulfatrim in drinking water for 14 days after transplantation.
Hematologic parameters
Complete blood counts were performed on a Hemavet (Drew Scientific, Waterbury, CT) according to the manufacturer’s instructions.
Neutrophil isolation
Whole bone marrow (BM) was subjected to discontinuous Percoll (GE Healthcare, Waukesha, WI) gradient centrifugation. Neutrophils were collected from the band between the 81% and 62% Percoll layers.
Western blot
Whole cell lysates from isolated neutrophils were used with standard western blotting techniques. Neutrophils were stimulated with N-formyl-methionine-leucine-phenylalanine (fMLP; Sigma-Aldrich, St. Louis, MO) with or without fibrinogen (Sigma-Aldrich) or vehicle for indicated times. Antibodies against SRF (G-20x), integrin α-M (M-19), glyceraldehyde-3-phosphate dehydrogenase (FL-335) were from Santa Cruz Biotechnology (Dallas, TX), and ERK (137F5) and P-ERK (E-10) from Cell Signaling.
Neutrophil migration in transwell assay
Neutrophil migration in a transwell system in response to fMLP and chemokine (CXC motif) ligand 1 (CXCL1) (PeproTech, Rocky Hill, NJ) was assessed as described previously.22
LPS nebulization and BAL
Srf wild-type (WT) and knockout (KO) mice (transplant recipients of tetOCre−/RTTA/SRFfl/fl or tetOCre+/RTTA/Srffl/fl BM treated for 8 days with doxycycline) were nebulized with 12.5 mg lipopolysaccharide (LPS; Sigma-Aldrich) in 5 mL phosphate-buffered saline for 15 minutes and analyzed at indicated time points as previously described.23 Bronchoalveolar lavage (BAL) was performed with phosphate-buffered saline supplemented with protease inhibitor cocktail (Roche, Basel, Switzerland). BAL specimens were analyzed morphologically by Wright Giemsa stain on cytospins and by flow cytometry. Donor cells were identified by CD45.2 vs CD45.1 and neutrophil- (Gr-1, 7/4) and macrophage- (F4/80) specific surface antigen staining.
In vivo neutrophil peritonitis assay
Srf WT and KO neutrophils were labeled with either far-red or violet cell tracking dyes (1.25 µM; Life Technologies, Grand Island, NY). WT and KO neutrophils were mixed 1:1 and injected retroorbitally into WT congenic recipient mice injected intraperitoneally 1.5 hours earlier with 1 mg monosodium urate crystals (Santa Cruz Biotechnology). Two hours after the intravenous neutrophil injection, mice were euthanized, and peritoneal lavage fluid (lavage), peripheral blood (PB), and BM were collected for analysis.
G- and F-actin formation assays
Neutrophils were incubated at 37°C with addition of 10 µM fMLP or vehicle for indicated times and stained with Alexa Fluor 647–conjugated Phalloidin and Alexa Fluor 488–conjugated deoxyribonuclease I (DNase I; Life Technologies) in the presence of 20 mg/mL L-lysolecithin (Sigma-Aldrich) and analyzed by flow cytometry.
Flow cytometry, cell sorting, and ICAM-1 binding assay
Neutrophils were isolated by flow-sorting as described previously.22 Isolated neutrophils were stained with antibodies against CD11a (BD Biosciences), CD11b (eBioscience, San Diego, CA), CD18 (BioLegend, San Diego, CA), CXCR2 (BioLegend), and Gr-1 (BD), 7/4 (Cedarlane, Burlington, ON, Canada). For intercellular adhesion molecule-1 (ICAM-1) binding, primary neutrophils were isolated, treated with vehicle or fMLP while incubated with antigen-presenting cell–conjugated ICAM-1, fixed after 2 or 5 minutes, and assessed by flow cytometry with concurrent staining for CD11b and CD11a.
DNA and RNA analysis and RNA sequencing
DNA and RNA were extracted and complementary DNA prepared according to standard methods. Quantitative reverse-transcription polymerase chain reaction (qRT-PCR) was performed on a CFX96 C1000 thermal cycler (Bio-Rad, Berkeley, CA) with Taqman Gene Expression Assays (Life Technologies) or specific primers (CXCR2 F-5′-AGCAAACAC CTCTACTACCCTCTA-3′ and R-5′-GGGCTGCATCAATTCAAATACCA-3′, fMLP receptor1 (FPR1) F-5′-GGTTCTCTCCTTTGTTGTGGCT-3′ and R-5′GATTGTGGATATGAGGGCCACT-3′ with 18S as internal control F-5′-ATGGCCGTTCTTAGTTGGTG-3′ R-5′CAATCT CGGGTGGCTGAA-3′), or gene-specific Taqman Probes (Life Technologies). Library preparation and sequencing were performed by the Yale Stem Cell Genomics Core Facility using the Illumina TruSeq RNA Sample Preparation Kit (Illumina, San Diego, CA). Samples were sequenced on an Illumina HiSequation 2000 using 50-cycle single-end sequencing. Fastq format sequencing reads were aligned to the mm10 genome using TopHat (version 2.0.0) software (http://tophat.cbcb.umd.edu). The cufflinks and cuffdiff (version 2.0.0) programs were used to identify differentially expressed transcripts (http://cufflinks.cbcb.umd.edu/). Data are publicly available through Gene Expression Omnibus (GSE55090).
Immunofluorescence and confocal imaging
Isolated neutrophils adhered to glass coverslips were stimulated with fMLP or vehicle for indicated times. Neutrophils were fixed, permeabilized with saponin 0.5%, and stained with respective antibodies and mounted with ProLong Gold Antifade Reagent with 4′6 diamidino-2-phenylindole (DAPI; Cell Signaling), and imaged with a Ti-E Eclipse confocal microscope (Nikon, Waltham, MA) equipped with Velocity software (Perkin Elmer, Waltham, MA). Neutrophils were stained with Phalloidin-Alexa Fluor 647 and antibodies against CD11b (BD Biosciences), clathrin (C-20, Santa Cruz Biotechnology), and kindlin (Abcam, Cambridge, MA). For internalization, neutrophils were stained with rat anti-CD11b fluorescein isothiocyanate (BioLegend), stimulated with 10 µM fMLP or vehicle, and incubated at 37°C for indicated times. Cells were fixed with addition of 4% paraformaldehyde and directly stained without permeabilization with wheat germ agglutinin conjugate 647 (Invitrogen) to label the membrane.
Statistics
Where indicated, statistical analysis was performed by calculating means and standard error of the mean; differences between groups were evaluated with the multiple Student t tests using GraphPad Prism 5; a P value <.05 was considered to be statistically significant.
Results
Srf−/− mice have normal neutrophil numbers and morphology
We have previously shown that loss of Srf restricted to the megakaryocytic lineage leads to thrombocytopenia and platelet dysfunction. Deletion of Srf in the hematopoietic stem cell by Vav-Cre–mediated excision recapitulates the severe platelet defect with thrombocytopenia and bleeding diathesis, but in addition leads to early death in the newborn period resulting from profound anemia (data not shown). Because SRF has been shown to regulate cell migration, we decided to look at the role of SRF in neutrophil function and specifically in neutrophil migration.10,13,24 Analysis of the white blood cell count (WBC) and morphology did not show significant differences in neutrophils between Vav-Cre-/Srffl/fl (Vav-Cre/Srf WT) and Vav-Cre+/Srffl/fl (Vav-Cre/Srf KO) mice (Figure 1A; supplemental Figure 1A on the Blood Web site); although this is consistent with a previous report,17 lymphocyte numbers were decreased in Vav-Cre/Srf KO mice (Figure 1A).
Figure 1.

White blood cell counts and differential in Srf WT and KO mice and efficiency of Srf deletion. (A) WBC and white cell differential in primary Vav-Cre/Srf WT and KO mice. PB from Vav-Cre/Srf WT and KO mice aged 3-12 days was obtained and WBC and WBC differential assessed on Hemavet. (B) qRT-PCR on neutrophils flow sorted based on Gr-1high 7/4high granularityhigh criteria from Vav-Cre/Srf WT and KO mice. Srf expression is successfully abrogated in Vav-Cre/Srf KO neutrophils (100-fold, P < .0005; n = 4). *P < .05, **P < .005, ***P < .0005.
Because early postnatal death in Vav-Cre Srf KO mice posed difficulty in studying the role of SRF in the myeloid lineage in vivo, we devised a strategy to circumvent this limitation. We crossed Srffl/fl mice with mice with doxycycline-inducible Cre expression, performed BM transplantations, and administered doxycycline for 8-12 days to induce Srf deletion restricted to the hematopoietic system in the adult recipient mice, engrafted for at least 6 weeks before treatment with doxycycline.
In both models, we verified highly efficient deletion of Srf in neutrophils by YFP reporter gene expression as shown previously14,25 and qRT-PCR and western blot analysis for Srf expression (Figure 1B; supplemental Figure 1). Slight differences in WBC differential in the Dox-inducible Srf KO mice compared with Vav-Cre/Srf KO mice are likely because of persistent recipient lymphocytes, the adult vs newborn age, and that Srf deletion is of shorter duration than in Vav-Cre/Srf KO mice in which Srf is deleted during embryogenesis. All future studies were thus performed in Dox-inducible Srf WT and KO littermate–derived BM recipients (referred to hereafter as Srf KO and WT mice).
Srf KO mice have decreased cellular response to lung inflammation
To assess whether Srf KO neutrophils could be recruited to inflammatory sites in vivo, we challenged Srf WT and KO mice with LPS via nebulization, initiating an innate immune response analogous to that induced by gram-negative infection of the lung. To assess the cellular response to LPS, we collected BAL specimens from animals that underwent control or LPS nebulization and assessed total cell numbers and cellular composition by morphology and flow cytometry. Without LPS nebulization, neutrophils were absent from Srf WT and KO BAL samples (Figure 2A-B; supplemental Figure 2). Four and 24 hours after LPS nebulization, Srf WT mice showed a significant increase in total BAL cell numbers with marked predominance of neutrophils (Figure 2A-C). In contrast, LPS did not induce significant recruitment of neutrophils to the lungs of Srf KO mice (total cells in BAL: 0.568 ± 0.093 × 106 vs 0.128 ± 0.024 × 106 at 4 hours and 1.337 ± 0.369 × 106 vs 0.347 ± 0.045 × 106 at 24 hours in WT vs KO mice, respectively; P = .0026 at 4 hours, P = .04 at 24 hours). Although WT mice showed a significant shift in the macrophage/neutrophil ratio in favor of neutrophils after exposure to LPS (neutrophil %: 79.2 ± 4.85 and 86.75 ± 4.73 at 4 and 24 hours, respectively), KO mice maintained an abnormally low total number and percentage of neutrophils throughout (neutrophil %: 5.8 ± 5.8 and 48.25 ± 10.84). Neutrophils exit the blood stream by an intricate mechanism consisting of engagement with the endothelium, followed by rolling and transmigration from the vascular lumen into the tissue to reach the site of inflammation. The decreased neutrophils in BAL from LPS-treated Srf KO mice could be due to decreased neutrophil recruitment to infectious sites secondary to a monocyte/macrophage defect, a decrease in cytokine production to LPS, a primary neutrophil defect, or a combination of defects. We therefore chose to directly test neutrophil migration in vitro and in vivo.
Figure 2.

Assessment of neutrophil recruitment in vivo secondary to LPS-induced inflammation in the lung in Srf WT and KO mice. BAL was performed in Srf WT and KO mice before and 4 and 24 hours after LPS nebulization. Total (A), neutrophil (B), and macrophage (C) cell numbers were determined in BAL. (Combined data from 2 independent experiments: 0 hours, n = 4; 4 hours, n = 5; 24 hours, n = 4; *P < .05; **P < .005; ***P < .0005). Macrophage and neutrophil percentages were determined on Wright Giemsa–stained cytospins and by flow cytometry. (D-F) Migration of Srf WT and KO neutrophils in vitro in a transwell assay and chemokine receptor expression. Srf WT and KO neutrophils were allowed to migrate across 3-µm pore membrane toward an fMLP (D) and CXCL1 (E) gradient at indicated concentrations. Representative experiments performed in triplicate of at least 3 independent experiments (**P < .005; ***P < .0005). mRNA expression of chemokine receptors was assessed in Srf WT and KO neutrophils (F).
Srf KO neutrophils have a migratory defect in vitro
We analyzed chemotaxis of Srf WT and KO neutrophils in vitro in response to 2 chemotactic agents, fMLP, which binds to FPR, and CXCL1, which binds to CXCR2, using a standard transwell assay. Significantly fewer mature primary Srf KO neutrophils migrated across the membrane at all concentrations of fMLP and CXCL1 compared with WT neutrophils (Figure 2D-E). To ensure that the migratory defect was not due to lack of either FPR or CXCR2, we verified receptor expression levels in Srf WT and KO neutrophils by qRT-PCR. We detected equal messenger RNA (mRNA) expression of FPR1 and a small but significant decrease in CXCR2 (Figure 2F), prompting analysis of CXCR2 protein expression on the cell surface, which was equal in WT and KO neutrophils (supplemental Figure 3A). To better understand the nature of the migratory defect, we performed real-time imaging using a Dunn chamber, confirming the migratory defect in Srf KO neutrophils (supplemental Figure 3B-C; supplemental Videos).
Lack of Srf KO neutrophils at inflammatory sites in vivo is due to a migratory defect
Our in vitro data suggest that the defect observed in Srf KO mice is due to a migratory defect, but we cannot exclude failure of neutrophils to egress from the BM or defective cytokine production at the site of inflammation. We thus tested neutrophil migration in a peritonitis model, in which isolated, differentially labeled mature WT and KO neutrophils are simultaneously injected into the blood stream of WT animals after peritonitis is induced. This model has the advantage of specifically testing neutrophil transmigration in an otherwise normal host. Neutrophils isolated from Srf WT and KO mice were labeled with either far-red or violet membrane dyes and injected retroorbitally into WT recipients with monosodium urate crystal–induced inflammatory peritonitis. Injected neutrophils were allowed to reach the site of inflammation for 2 hours after which lavage, PB, and BM were harvested for analysis by flow cytometry (Figure 3A). As shown in Figure 3B, fewer Srf KO neutrophils were recovered in blood and BM compared with WT neutrophils. Although WT neutrophils successfully migrated to the peritoneal cavity, Srf KO neutrophils were almost completely lacking from the site of inflammation in the peritoneal cavity (lavage). This decrease in KO cells in the peritoneal fluid is out of proportion to the reduced recovery of neutrophils in blood and BM, consistent with a defect in transmigration.
Figure 3.
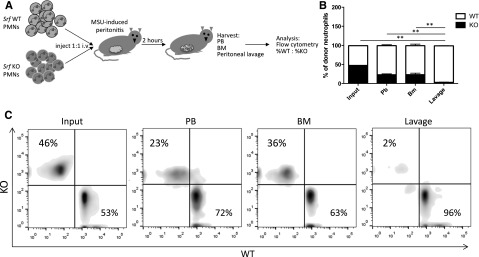
In vivo peritonitis model. (A) Peritonitis was induced in WT recipients and Srf WT, and KO neutrophils labeled with membrane dyes of 2 different colors were simultaneously injected intravenously and allowed to localize to tissues. PB, BM, and lavage fluid were harvested and percentages of donor WT vs KO neutrophils determined by flow cytometry detecting differential membrane staining (B-C). The experiment shown in panel B is representative of 4 independent experiments with n = 4 recipient mice of WT and KO neutrophils from 2 donors per experiment. Membrane dyes were alternated for WT and KO cells; **P < .005. (C) Primary flow data from 1 recipient showing donor KO and WT neutrophil distribution.
Reorganization of actin in neutrophils in response to cytokine stimulation
Rapid polymerization and depolymerization of actin (actin treadmilling), essential to cell migration (reviewed in Pollard and Cooper26 and Pick et al27), is dependent on several genes regulated by SRF.10,28 To elucidate the mechanism underlying the decreased migration of Srf KO neutrophils, we assessed actin polymerization in Srf WT and KO neutrophils in response to fMLP. In Srf WT neutrophils, actin rapidly polymerized at the leading edge after stimulation with fMLP as shown by phalloidin staining of F-actin. Such organized accumulation of F-actin was absent from Srf KO neutrophils (Figure 4A-B). Quantitation of G- (Figure 4C) and F-actin (Figure 4D) by DNAse1 and phalloidin staining, respectively, revealed no significant difference in G-actin in unstimulated and fMLP-stimulated Srf KO neutrophils when compared with WT. However, total F-actin increased significantly in Srf WT neutrophils within 30 seconds of fMLP stimulation. In Srf KO neutrophils, F-actin was markedly reduced not only before fMLP stimulation, but also failed to increase upon fMLP stimulation, corroborating the confocal imaging data.
Figure 4.

Srf KO neutrophils lack polarization with decreased f-actin polymerization when activated with fMLP. Srf WT and KO neutrophils were allowed to attach to poly-l-lysine–coated coverslips and stimulated with fMLP for 2 minutes. (A) F-actin was stained with phalloidin (middle) in Cre-expressing neutrophils marked by YFP (bottom); merge with DAPI is shown in the top panels. Quantification of polarized cells from 3 independent experiments; Srf WT, n = 71; Srf KO, n = 91 (B). G-actin (C) and F-actin (D) were assessed at 0, 30, 60, and 90 seconds after stimulation with fMLP by staining with DNAse I (G-actin) and phalloidin (F-actin) and mean fluorescence intensity (MFI) determined by flow cytometry. Representative of 3 independent experiments performed in triplicate.
Srf controls integrin function in neutrophils via inside-out signaling
We have previously identified novel targets of SRF essential for megakaryocyte differentiation and function and thus established an essential role for SRF-mediated transcriptional regulation in myelopoiesis.14 To identify potential downstream targets of SRF in neutrophils that may be responsible for the observed phenotype, we performed RNA deep sequencing on Srf WT and KO neutrophils isolated via fluorescence-activated cell sorting as described previously.22 More than 800 genes were significantly (P < .005) downregulated >2-fold in Srf KO compared with WT neutrophils. As expected, a significant number of actin cytoskeletal genes, known to be direct targets of SRF, were downregulated (Table 1 ; supplemental Table 1). In addition, the Janus kinase-signal transducer and activator of transcription, mitogen-activated protein kinase, focal adhesion kinase, and Toll-like receptor signaling pathways were enriched for significantly downregulated genes (Table 1; supplemental Table 2), suggesting alteration of lineage-specific gene expression as previously suggested by Sullivan et al.13 A previous study had shown downregulation of integrin expression in Srf KO hematopoietic stem cells resulting in loss of Srf KO stem cells from the BM niche.16
Table 1.
Deletion of Srf affects key signaling pathways
| Down-regulated genes | Up-Regulated genes | ||
|---|---|---|---|
| Pathways | Genes | Pathways | Genes |
| Regulation of actin cytoskeleton | ACTB, ITGAL, SSH1, SSH2, PIK3CD, WASF2, RAF1, ACTN1, PIP5K1A, MYH9, IQGAP1, ACTG1, CHRM3, ITGA8, SOS2, PPP1R12A, FGD3, DIAP1, PIK3R2, MYH10 | Natural killer cell mediated cytotoxicity | PRF1, KLRA18, KLRK1, KLRA33, GZMB, NCR1, LAT, SH2D1A, KLRA9, KLRA8, IFNG, KLRA4, KLRA7, FASL, KLRD1, KLRB1C, KLRC1 |
| Jak-STAT signaling pathway | IL9R, SOCS3, IL4RA, CREBBP, PIK3CD, STAT5B, CBL, IL6RA, AKT1, PIAS4, SOS2, JAK1, CSF2RB, CSF3R, IL13RA1, PIK3R2 | Cytokine-cytokine receptor interaction | IL2RB, IL1R1, CXCL9, CCL8, PF4, CCL5, KDR, TNFSF8, CCR9, VEGFC, TNFRSF9, CXCL16, IFNG, PDGFC, EPOR, FASL, XCL1, MPL |
| MAPK signaling pathway | IL1R2, TGFBR2, MAP4K2, RAF1, MKNK1, HSPA1A, SRF, FLNA, AKT1, FOS, MAP4K4, RPS6KA3, CASP3, DUSP1, MAP3K3, ARRB1, RASGRP4, RPS6KA2, JUN, SOS2, IL1B, MAP3K14 | Graft-versus-host disease | PRF1, KLRA9, KLRA8, IFNG, KLRA7, GZMB, FASL, KLRD1, KLRC1, CD28 |
| Focal adhesion | ACTB, TLN1, PIK3CD, RAF1, ACTN1, HGF, FLNA, ACTG1, AKT1, ITGA8, JUN, SOS2, PPP1R12A, ZYX, THBS1, COL11A2, DIAP1, PIK3R2 | Hematopoietic cell lineage | IL1R1, GP5, CD3G, CD8A, CD3E, CD59A, CD4, ANPEP, EPOR, CD5 |
| Toll-like receptor signaling pathway | AKT1, FOS, CD80, JUN, PIK3CD, TLR2, IL1B, TLR4, TLR5, TLR6, TLR8, PIK3R2 | T cell receptor signaling pathway | PRKCQ, LAT, CD3G, CD8A, CD3E, ICOS, IFNG, CTLA4, CD4, PDCD1, CD28 |
| Renal cell carcinoma | AKT1, HIF1A, JUN, SOS2, PIK3CD, CREBBP, SLC2A1, RAF1, HGF, PIK3R2 | Cell adhesion molecules (CAMs) | CADM1, CD8A, ICOS, CTLA4, CD4, ESAM, CD226, PDCD1, SDC3, CD28 |
| Acute myeloid leukemia | AKT1, PPARD, SOS2, STAT5B, PIK3CD, RAF1, RARA, ZBTB16, PIK3R2 | Complement and coagulation cascades | VWF, C3AR1, HC, C4B, CD59A, C2, F2R |
| Cytokine-cytokine receptor interaction | IL1R2, IL18RAP, IL9R, CCR1, IL4RA, TGFBR2, BMPR2, CXCR1, CXCR2, TNFRSF14, HGF, IL17RA, IL6RA, CCL6, INHBA, IL1B, CSF3R, CSF2RB, IL13RA1, LTB | Vascular smooth muscle contraction | PRKCQ, ADORA2A, MYLK3, CALD1, ADCY6, MRVI1, GUCY1A3, GUCY1B3 |
| Pathways in cancer | DVL2, PPARD, PTGS2, TGFBR2, CREBBP, PIK3CD, STAT5B, CBL, RAF1, HGF, ZBTB16, AKT1, FOS, CASP3, HIF1A, PIAS4, JUN, SLC2A1, SOS2, JAK1, CSF3R, RARA, TRAF5, PIK3R2 | Chemokine signaling pathway | CCR9, CXCL16, ADCY6, CXCL9, CCL8, PF4, GNG11, XCL1, CCL5 |
CD11a/CD18 (LFA1) and CD11b/CD18 (Mac1) represent the major integrin complexes responsible for neutrophil adhesion and trafficking.27,29,30 Although in our neutrophil data set, expression of Itgal (CD11a), Itgam (CD11b), and Itgb2 (CD18), the predominantly expressed integrins in neutrophils, were modestly downregulated (0.6- to 2-fold) (supplemental Table 3) qRT-PCR on additional sets of sorted neutrophils showed maintained integrin expression (supplemental Figure 4A). We assessed cell surface expression of these key integrins by flow cytometry in Srf WT and KO neutrophils. Surprisingly, CD11b expression was significantly increased on Srf KO neutrophils (Figure 5A).
Figure 5.

Integrin homeostasis in Srf WT and KO neutrophils. (A) Expression of integrin CD11b, CD18, and CD11a on the cell surface of Srf WT and KO neutrophils by flow cytometry. Srf WT and KO neutrophils were incubated with fluorescently labeled ICAM-1 (B) and stimulated with fMLP for 0, 2, and 5 minutes; (C) CD11b surface expression was determined at the same time. (Representative experiment of 3 independent experiments performed in triplicate; **P < .005, ***P < .0005.) (D) Srf WT and KO neutrophils were stimulated with vehicle or fMLP for 15 minutes at 37°C and lysates probed for Itgam, Erk, and P-Erk, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as loading control. Lysates are from 1 experiment; vertical lines have been inserted to indicate a repositioned gel lane. Neg ctrl, negative control.
Integrins assume a high-affinity conformation secondary to inside-out signaling upon fMLP stimulation.31,32 We therefore tested whether the increased Itgam cell surface expression would translate into increased ICAM-1 binding. However, ICAM-1 binding was significantly reduced in Srf KO neutrophils upon fMLP stimulation (Figure 5B) despite increased Itgam cell surface expression (Figure 5C-D; supplemental Figure 4). This suggests that the Itgam/Itgb2 heterodimer fails to assume an activated configuration upon fMLP stimulation. We verified intact signaling downstream of fMLP by determination of Erk phosphorylation (Figure 5D). We assessed expression of several genes essential to integrin activation, such as Kindlin (Fermt3),33,34 Talin,35,36 L-plastin (Lcp1),37,38 and cytohesin39 without differences between WT and KO neutrophils. Interestingly, confocal imaging reveals focal accumulation of Kindlin (leading edge, Figure 6C; supplemental Figure 5) and Talin (leading and trailing edges, data not shown) in WT and KO neutrophils, suggesting that their polarization is independent of Srf regulation.
Figure 6.

Integrin localization in Srf WT and KO neutrophils. Srf WT (left) and KO (right) neutrophils were (A) allowed to adhere to glass coverslips, stained with CD11b fluorescein isothiocyanate, and then stimulated with fMLP for 0 (none) and 15 (fMLP) minutes and stained with wheat germ agglutinin antibody as a membrane stain (without permeabilization) and DAPI (A). (B-C) Srf WT and KO neutrophils were allowed to adhere to glass coverslips, stimulated with fMLP for 0 (none) and 15 (fMLP) minutes and stained with antibodies against CD11b and Clathrin (B) and CD11b and Kindlin (C). (A-B) The top 3 panels show single sections of the Z-stack; the bottom panel shows a merge of the Z-stack. (C) Three sequential Z-stack sections for each image. DAPI stains nuclei. Scale bar = 6 µm; >, leading edge; *, trailing edge.
Srf affects integrin trafficking
To ensure spatiotemporally regulated localization of activated integrin complexes at both the leading and trailing edges with cell migration, integrins are internalized and either recycled or degraded via clathrin-dependent or clathrin-independent mechanisms (reviewed in Caswell et al40). We therefore assessed CD11b integrin localization in neutrophils in response to fMLP activation. As shown in Figure 6A, in response to fMLP activation, CD11b clusters at the leading and trailing edges in WT neutrophils, whereas in KO neutrophils CD11b remains localized uniformly in the cell membrane. Clustering of clathrin at the leading and trailing edges and colocalization with CD11b integrin at the leading edge in WT cells (Figure 6B) suggests, as shown previously (reviewed in Caswell et al40), that active endosomal recycling may be necessary to allow spatiotemporal localization of integrins. Such clustering is absent in Srf KO neutrophils pointing toward deficient integrin trafficking.
Discussion
SRF is considered a master regulator of the actin cytoskeleton. SRF functions downstream of several pathways, including the ρ-A and mitogen-activated protein kinase pathways, fulfilling essential roles in cell-cycle regulation and cell division, cell differentiation, and tissue-specific cell function (reviewed in Miano et al10).
We show here that SRF is essential for mature neutrophil migration without significant defects in neutrophil differentiation. Srf KO neutrophils show normal morphology, and neutrophil numbers are preserved early upon SRF deletion. Srf KO neutrophils fail to migrate in vitro and in vivo toward chemotactic stimuli. Induction of lung inflammation via LPS nebulization results in significantly reduced neutrophil recruitment to inflamed lungs 4 and 24 hours after LPS stimulation in Srf KO mice. Because all immune cells are defective in Srf KO mice, we first assessed neutrophil migration in vitro. WT neutrophils efficiently migrate toward a chemokine gradient, whereas significantly fewer Srf KO neutrophils migrate through the 3-µm pore membrane into the bottom well. Direct visualization in the Dunn chamber confirms this in vitro migration defect. In an in vivo peritonitis model, where except for the injected neutrophils, all other immune cells are host-derived and thus normally express Srf, WT neutrophils migrate rapidly into the inflamed peritoneal space, whereas significantly fewer Srf KO neutrophils migrate into the inflamed peritoneal cavity. There is also a decrease in Srf KO neutrophils recovered from the PB and BM relative to injected cells compared with WT cells. Because mature neutrophils are injected into the retroorbital sinuses of WT recipients, this is independent of BM production or endothelial “transmigration.” To ensure that differential cell labeling had no effect on detection of WT or KO neutrophils, each experiment was performed in duplicate with labeling of WT and KO cells with far-red and violet cell dyes. This suggests that KO neutrophils harbor an additional defect that potentially compromises their behavior in vivo, such as a defect in adhesion (supplemental Figure 4C).
To assess the mechanism by which lack of Srf leads to the neutrophil migration defect, we assessed the actin cytoskeleton.10 Expression analysis revealed reduced mRNA expression of G-actin, a known direct transcriptional target of SRF, whereas flow-cytometric quantitation of G-actin protein by DNAse 1 staining revealed abundant G-actin in both WT and KO cells. However, F-actin polymerization, essential to cellular movement, was reduced in KO neutrophils, confirming a defect in actin polymerization. Confocal imaging confirmed the quantitative defect in F-actin polymerization in addition to markedly reduced polarization in response to fMLP activation in Srf KO neutrophils.
By RNA sequencing, we confirmed that numerous known targets of SRF essential to actin polymerization and depolymerization are significantly downregulated in Srf KO neutrophils10,11,41 and we identified additional potential targets within the actin cytoskeletal and inflammatory pathways.
LFA1 (CD11a/CD18) and Mac1 (CD11b/CD18) represent the major integrin complexes in neutrophils and are essential to neutrophil adhesion, rolling, arrest, and directional migration. The defects in adhesion and migration in Srf KO neutrophils are likely to be in part the result of lack of focal adhesion formation and cell polarization from defects in the actin cytoskeleton. Although downregulation of integrin expression has been suggested to lead to defective hematopoietic stem cell adhesion with loss of SRF,16 we observe here an increase in CD11b/CD18 on the cell surface of Srf KO neutrophils despite normal mRNA expression. This led us to hypothesize that integrin homeostasis must be disrupted with loss of SRF. We show that despite upregulation of CD11b on the plasma membrane, ICAM-1 binding is reduced. This suggests that CD11b fails to assume the activated conformation secondary to inside-out signaling upon fMLP stimulation, despite focal accumulation of kindlin and talin. It has been previously suggested that the cytoskeleton actively regulates the binding conformation of CD11b/CD18 and its mobility within the membrane, and inactivation of RhoA inhibits chemokine-induced neutrophil adhesion.42,43 Because Srf transcriptional activity is regulated downstream of RhoA, loss of SRF is likely to affect inside-out signaling-mediated activation of integrins.
Integrin complexes in both the inactive and activated state undergo endocytotic recycling via clathrin-dependent and clathrin-independent mechanisms to direct integrins to the leading edge of the cell or to lysosomes for degradation, both essential to allowing forward movement of the cell (reviewed in Caswell et al40 and Margadant et al44). Our data suggest that such directional trafficking of CD11b does not occur in Srf KO neutrophils. Before stimulation with fMLP, CD11b in WT neutrophils is localized circumferentially on the plasma membrane. After fMLP stimulation, CD11b clusters predominantly at the leading edge. This clustering of CD11b is accompanied by polarized localization of clathrin and endosomes (data not shown), suggesting active trafficking of integrins to and from the leading and trailing edges. In KO neutrophils, clustering of integrins and clathrin-coated vesicles at the leading or trailing edges is near absent. Clathrin-mediated endocytosis is highly dependent on tightly regulated recruitment, activity, and disassembly of many proteins. Actin and other cytoskeletal proteins such as actin-binding protein 1, coronin, cofilin, and profilin are needed to provide force to pull the formed vesicle out from the planar plasma membrane, allowing a final scission step to occur (reviewed in Weinberg and Drubin45). Thus deregulated expression of these key actin regulatory proteins is likely to compromise clathrin-mediated endocytosis.
In summary, we have shown a severe neutrophil function defect from loss of Srf in mice. SRF directs a gene network including several genes on chromosome 5q, one of the most common cytogenetic abnormalities in myelodysplasia. We have extended the known function of SRF in hematopoiesis to 1 of the key cells of the innate immune response. We have identified a mechanism, namely integrin activation and trafficking, in addition to the known function in actin polymerization by which loss of SRF contributes to lack of cell migration and by which it may affect cell–cell and cell–microenvironment interactions.
Supplementary Material
Acknowledgments
The authors thank Gouzel Toukmolina and Geoff Lyons in the Yale Cell Sorter Facility for flow cytometric cell sorting; Dr Mei Zhong, Director of the Genomics Core of Yale Stem Cell Center for making the mRNA library and performing the sequence service; and Dr David Calderwood for his valuable advice.
This work was supported by a career development award from the Yale Claude D. Pepper Older Americans Independence Center (P30AG021342) (S.H.), the Yale Comprehensive Cancer Center (S.H.), and the National Institutes of Health (National Institute of Diabetes, Digestive and Kidney Disease, grant K08 DK073366) (S.H.), (National Heart, Lung and Blood Institute, grant RO1 HL093004 [E.M.B.], and grant R15 HL104593 [P.G.]).
Footnotes
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE55090).
The online version of this article contains a data supplement.
There is an Inside Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: A.T. designed and performed research, collected and analyzed data, and wrote the manuscript; W.T. and P.X.Z. performed research; E.B. designed and performed research and analyzed the data; A.L. analyzed data; P.G. and D.W. provided important reagents and provided valuable advice; and S.H. initiated, designed, and performed research, collected and analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Stephanie Halene, Section of Hematology, Department of Internal Medicine and Yale Comprehensive Cancer Center, Yale University School of Medicine, 300 George St 786E, New Haven, CT 06511; e-mail: stephanie.halene@yale.edu.
References
- 1.Cullen EM, Brazil JC, O’Connor CM. Mature human neutrophils constitutively express the transcription factor EGR-1. Mol Immunol. 2010;47(9):1701–1709. doi: 10.1016/j.molimm.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 2.Eisenmann KM, Dykema KJ, Matheson SF, et al. 5q- myelodysplastic syndromes: chromosome 5q genes direct a tumor-suppression network sensing actin dynamics. Oncogene. 2009;28(39):3429–3441. doi: 10.1038/onc.2009.207. [DOI] [PubMed] [Google Scholar]
- 3.Joslin JM, Fernald AA, Tennant TR, et al. Haploinsufficiency of EGR1, a candidate gene in the del(5q), leads to the development of myeloid disorders. Blood. 2007;110(2):719–726. doi: 10.1182/blood-2007-01-068809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gineitis D, Treisman R. Differential usage of signal transduction pathways defines two types of serum response factor target gene. J Biol Chem. 2001;276(27):24531–24539. doi: 10.1074/jbc.M102678200. [DOI] [PubMed] [Google Scholar]
- 5.Starczynowski DT, Kuchenbauer F, Argiropoulos B, et al. Identification of miR-145 and miR-146a as mediators of the 5q- syndrome phenotype. Nat Med. 2010;16(1):49–58. doi: 10.1038/nm.2054. [DOI] [PubMed] [Google Scholar]
- 6.Xin M, Small EM, Sutherland LB, et al. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 2009;23(18):2166–2178. doi: 10.1101/gad.1842409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cordes KR, Sheehy NT, White MP, et al. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460(7256):705–710. doi: 10.1038/nature08195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J, Sun Q, Zhang Z, Ge S, Han ZG, Chen WT. Loss of microRNA-143/145 disturbs cellular growth and apoptosis of human epithelial cancers by impairing the MDM2-p53 feedback loop. Oncogene. 2013;32(1):61–69. doi: 10.1038/onc.2012.28. [DOI] [PubMed] [Google Scholar]
- 9.Batliner J, Buehrer E, Fey MF, Tschan MP. Inhibition of the miR-143/145 cluster attenuated neutrophil differentiation of APL cells. Leuk Res. 2012;36(2):237–240. doi: 10.1016/j.leukres.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 10.Miano JM, Long X, Fujiwara K. Serum response factor: master regulator of the actin cytoskeleton and contractile apparatus. Am J Physiol Cell Physiol. 2007;292(1):C70–C81. doi: 10.1152/ajpcell.00386.2006. [DOI] [PubMed] [Google Scholar]
- 11.Miano JM. Serum response factor: toggling between disparate programs of gene expression. J Mol Cell Cardiol. 2003;35(6):577–593. doi: 10.1016/s0022-2828(03)00110-x. [DOI] [PubMed] [Google Scholar]
- 12.Wang Z, Wang DZ, Hockemeyer D, McAnally J, Nordheim A, Olson EN. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature. 2004;428(6979):185–189. doi: 10.1038/nature02382. [DOI] [PubMed] [Google Scholar]
- 13.Sullivan AL, Benner C, Heinz S, et al. Serum response factor utilizes distinct promoter- and enhancer-based mechanisms to regulate cytoskeletal gene expression in macrophages. Mol Cell Biol. 2011;31(4):861–875. doi: 10.1128/MCB.00836-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Halene S, Gao Y, Hahn K, et al. Serum response factor is an essential transcription factor in megakaryocytic maturation. Blood. 2010;116(11):1942–1950. doi: 10.1182/blood-2010-01-261743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ragu C, Boukour S, Elain G, et al. The serum response factor (SRF)/megakaryocytic acute leukemia (MAL) network participates in megakaryocyte development. Leukemia. 2010;24(6):1227–1230. doi: 10.1038/leu.2010.80. [DOI] [PubMed] [Google Scholar]
- 16.Ragu C, Elain G, Mylonas E, et al. The transcription factor Srf regulates hematopoietic stem cell adhesion. Blood. 2010;116(22):4464–4473. doi: 10.1182/blood-2009-11-251587. [DOI] [PubMed] [Google Scholar]
- 17.Fleige A, Alberti S, Gröbe L, et al. Serum response factor contributes selectively to lymphocyte development. J Biol Chem. 2007;282(33):24320–24328. doi: 10.1074/jbc.M703119200. [DOI] [PubMed] [Google Scholar]
- 18.Miano JM, Ramanan N, Georger MA, et al. Restricted inactivation of serum response factor to the cardiovascular system. Proc Natl Acad Sci USA. 2004;101(49):17132–17137. doi: 10.1073/pnas.0406041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Georgiades P, Ogilvy S, Duval H, et al. VavCre transgenic mice: a tool for mutagenesis in hematopoietic and endothelial lineages. Genesis. 2002;34(4):251–256. doi: 10.1002/gene.10161. [DOI] [PubMed] [Google Scholar]
- 20.Belteki G, Haigh J, Kabacs N, et al. Conditional and inducible transgene expression in mice through the combinatorial use of Cre-mediated recombination and tetracycline induction. Nucleic Acids Res. 2005;33(5):e51. doi: 10.1093/nar/gni051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stadtfeld M, Graf T. Assessing the role of hematopoietic plasticity for endothelial and hepatocyte development by non-invasive lineage tracing. Development. 2005;132(1):203–213. doi: 10.1242/dev.01558. [DOI] [PubMed] [Google Scholar]
- 22.Halene S, Gaines P, Sun H, et al. C/EBPepsilon directs granulocytic-vs-monocytic lineage determination and confers chemotactic function via Hlx. Exp Hematol. 2010;38(2):90–103. doi: 10.1016/j.exphem.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bruscia EM, Zhang PX, Ferreira E, et al. Macrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator-/- mice. Am J Respir Cell Mol Biol. 2009;40(3):295–304. doi: 10.1165/rcmb.2008-0170OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leitner L, Shaposhnikov D, Mengel A, et al. MAL/MRTF-A controls migration of non-invasive cells by upregulation of cytoskeleton-associated proteins. J Cell Sci. 2011;124(Pt 24):4318–4331. doi: 10.1242/jcs.092791. [DOI] [PubMed] [Google Scholar]
- 25.Srinivas S, Watanabe T, Lin CS, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pollard TD, Cooper JA. Actin, a central player in cell shape and movement. Science. 2009;326(5957):1208–1212. doi: 10.1126/science.1175862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pick R, Brechtefeld D, Walzog B. Intraluminal crawling versus interstitial neutrophil migration during inflammation. Mol Immunol. 2013;55(1):70–75. doi: 10.1016/j.molimm.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 28.Sotiropoulos A, Gineitis D, Copeland J, Treisman R. Signal-regulated activation of serum response factor is mediated by changes in actin dynamics. Cell. 1999;98(2):159–169. doi: 10.1016/s0092-8674(00)81011-9. [DOI] [PubMed] [Google Scholar]
- 29.Hepper I, Schymeinsky J, Weckbach LT, et al. The mammalian actin-binding protein 1 is critical for spreading and intraluminal crawling of neutrophils under flow conditions. J Immunol. 2012;188(9):4590–4601. doi: 10.4049/jimmunol.1100878. [DOI] [PubMed] [Google Scholar]
- 30.Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwartz MA. Integrin signaling revisited. Trends Cell Biol. 2001;11(12):466–470. doi: 10.1016/s0962-8924(01)02152-3. [DOI] [PubMed] [Google Scholar]
- 32.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 33.Moser M, Bauer M, Schmid S, et al. Kindlin-3 is required for beta2 integrin-mediated leukocyte adhesion to endothelial cells. Nat Med. 2009;15(3):300–305. doi: 10.1038/nm.1921. [DOI] [PubMed] [Google Scholar]
- 34.Moser M, Legate KR, Zent R, Fässler R. The tail of integrins, talin, and kindlins. Science. 2009;324(5929):895–899. doi: 10.1126/science.1163865. [DOI] [PubMed] [Google Scholar]
- 35.Tadokoro S, Shattil SJ, Eto K, et al. Talin binding to integrin beta tails: a final common step in integrin activation. Science. 2003;302(5642):103–106. doi: 10.1126/science.1086652. [DOI] [PubMed] [Google Scholar]
- 36.Simonson WT, Franco SJ, Huttenlocher A. Talin1 regulates TCR-mediated LFA-1 function. J Immunol. 2006;177(11):7707–7714. doi: 10.4049/jimmunol.177.11.7707. [DOI] [PubMed] [Google Scholar]
- 37.Chen H, Mocsai A, Zhang H, et al. Role for plastin in host defense distinguishes integrin signaling from cell adhesion and spreading. Immunity. 2003;19(1):95–104. doi: 10.1016/s1074-7613(03)00172-9. [DOI] [PubMed] [Google Scholar]
- 38.Jones SL, Wang J, Turck CW, Brown EJ. A role for the actin-bundling protein L-plastin in the regulation of leukocyte integrin function. Proc Natl Acad Sci USA. 1998;95(16):9331–9336. doi: 10.1073/pnas.95.16.9331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kinashi T. Integrin regulation of lymphocyte trafficking: lessons from structural and signaling studies. Adv Immunol. 2007;93:185–227. doi: 10.1016/S0065-2776(06)93005-3. [DOI] [PubMed] [Google Scholar]
- 40.Caswell PT, Vadrevu S, Norman JC. Integrins: masters and slaves of endocytic transport. Nat Rev Mol Cell Biol. 2009;10(12):843–853. doi: 10.1038/nrm2799. [DOI] [PubMed] [Google Scholar]
- 41.Sun Q, Chen G, Streb JW, et al. Defining the mammalian CArGome. Genome Res. 2006;16(2):197–207. doi: 10.1101/gr.4108706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anderson SI, Hotchin NA, Nash GB. Role of the cytoskeleton in rapid activation of CD11b/CD18 function and its subsequent downregulation in neutrophils. J Cell Sci. 2000;113(Pt 15):2737–2745. doi: 10.1242/jcs.113.15.2737. [DOI] [PubMed] [Google Scholar]
- 43.Laudanna C, Campbell JJ, Butcher EC. Role of Rho in chemoattractant-activated leukocyte adhesion through integrins. Science. 1996;271(5251):981–983. doi: 10.1126/science.271.5251.981. [DOI] [PubMed] [Google Scholar]
- 44.Margadant C, Monsuur HN, Norman JC, Sonnenberg A. Mechanisms of integrin activation and trafficking. Curr Opin Cell Biol. 2011;23(5):607–614. doi: 10.1016/j.ceb.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 45.Weinberg J, Drubin DG. Clathrin-mediated endocytosis in budding yeast. Trends Cell Biol. 2012;22(1):1–13. doi: 10.1016/j.tcb.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.