

Abstract
Pancreatic β‐cell failure resulting from decreased β‐cell mass or dysfunction is the ultimate step towards most types of diabetes. Even if insulin resistance exists, diabetes does not develop unless pancreatic β‐cell function or its adaptation is compromised. Classically, two types of cell death (apoptosis and necrosis) have been studied in the diabetes field. Recently, a third type of cell death (autophagy, sometimes called type 2 programmed cell death in comparison with apoptosis, type 1 programmed cell death) and its pathophysiological role have been recognized and are being investigated. In the present review, we will discuss the role of various types of cell death in the development of type 1 and type 2 diabetes. Specifically, we will briefly cover recent progress regarding the role of autophagy in diabetes, which is becoming a hot topic in diabetes and metabolism. (J Diabetes Invest, doi: 10.1111/j.2040‐1124.2010.0054.x, 2010)
Keywords: Diabetes, β‐cell death, Autophagy
Introduction
Pancreatic β‐cell failure emanating from β‐cell death or dysfunction is the critical precipitating event in the development of diabetes. In type 1 diabetes, β‐cell death is the final and critical step in the development of the disease, whereas both β‐cell death and dysfunction contribute to β‐cell failure in type 2 diabetes. Among diverse types of cell death, β‐cell apoptosis (type 1 programmed cell death) plays a major role in the reduction of β‐cell mass; however, macroautophagy (here referred to as autophagy), which is also called type 2 programmed cell death, might also play a role in the regulation or execution of β‐cell death1. Here, we describe the role of apoptosis and necrosis in the β‐cell loss of diabetes. Additionally, we review the role of autophagy in pancreatic β‐cell physiology and in the development of diabetes shown in recent publications.
β‐cell apoptosis in type 1 diabetes
Diverse types of cell death modes contribute to β‐cell injury in diabetes. Among them, apoptosis is the most prominent type of cell death in diabetes. In type 1 diabetes, it is generally agreed that apoptosis of pancreatic β‐cells is the most important and final step in the progression of the disease2,3. However, it has not been elucidated which molecule(s) are the real culprit(s) in pancreatic β‐cell apoptosis. Perforin, Fas ligand (FasL), tumor necrosis factor (TNF)‐α, interleukin (IL)‐1, interferon (IFN)‐γ and nitric oxide (NO) have been claimed as the effector molecules; however, they, as a single agent, might explain only part of β‐cell death in type 1 diabetes. Whereas FasL was initially considered as a strong candidate for the most important death effector4, subsequent experiments by others, including the present authors, showed discordant results2,5. Combinations or synergism between IFN‐γ and TNF‐α or IL‐1 are being revisited as possible death effectors and the molecular mechanism explaining such a synergism has been addressed in several recent papers6,7. However, it is still controversial as to which combination between IFN‐γ/TNF‐αvs IFN‐γ/IL‐1 is dominant in the development of type 1 diabetes in vivo. Signal transducer and activator of transcription‐1 (STAT1) is phosphorylated by IFN‐γ7 and modulates signal transduction downstream of both IFN‐γ/TNF‐α and IFN‐γ/IL‐1 combinations. The present authors and others have reported that mice with targeted disruption of STAT1 are resistant to the development of natural type 1 diabetes or that after multiple streptozotocin (STZ) treatment8,9 (Figure 1). However, because STAT1 is downstream of both IFN‐γ/TNF‐α and IFN‐γ/IL‐1, the abrogation of type 1 diabetes in STAT1‐knockout mice cannot tell which combination is dominant. Although nuclear factor‐κB (NF‐κB) can be activated by both TNF‐α and IL‐1, NF‐κB activation might have a different role in β‐cell death according to the death signals. NF‐κB activation by TNF‐α has been reported to play an anti‐apoptotic role10. However, NF‐κB activation could be detrimental to the cells after its activation by other (death) effectors, such as IL‐111 (Figure 1). Because of such differences, gene targeting of the NF‐κB‐related pathway might be able to tell which cytokine combination is more important in the development of type 1 diabetes. In fact, our data showed that transgenic mice expressing a NF‐κB inhibitor, IκBα‐superrepressor (IκBα‐SR), specifically in pancreatic β‐cells driven by rat insulin promoter, developed accelerated diabetes, suggesting that the net effect of NF‐κB in β‐cells in vivo is anti‐apoptotic12. These genetic approaches might be able to provide important information regarding the development of preventive or therapeutic agents in type 1 diabetes. For instance, inhibitors of STAT1 or its upstream Janus kinase 2 (JAK2) signaling could be used to inhibit the development of type 1 diabetes or recurrence of type 1 diabetes after islet transplantation. Downstream molecules of NF‐κB activation, such as X‐linked inhibitor of apoptosis protein, that have a strong antiapoptotic activity can be used to inhibit β‐cell death in vivo and to prevent the development or recurrence of type 1 diabetes13.
Figure 1.
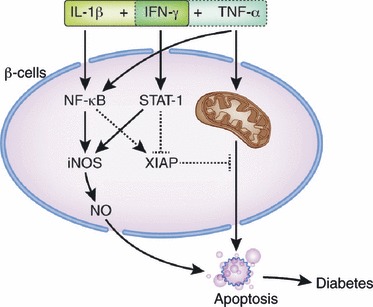
Effector cytokines and signal transduction in β‐cell death of type 1 diabetes. Both interleukin‐1β (IL‐1β) + interferon‐γ (IFN‐γ) and tumor necrosis factor‐α (TNF‐α) + IFN‐γ synergisms have been implicated in β‐cell death of type 1 diabetes. STAT1 is a common signal transducer in both cytokine synergism models. Nuclear factor (NF)‐κB is activated by both IL‐1β and TNF‐α. However, NF‐κB activation by IL‐1β is likely to play a pro‐apoptotic role by enhancing inducible nitric oxide synthase (iNOS) expression and producing nitric oxide (NO), whereas TNF‐α‐induced NF‐κB activation might have an anti‐apoptotic function by inducing expression of anti‐apoptotic molecules, such as X‐linked inhibitor of apoptosis protein (XIAP).
β‐cell apoptosis in type 2 diabetes
The role of pancreatic β‐cell death in type 2 diabetes is less clear. In the preclinical period of type 2 diabetes characterized by insulin resistance, hyperinsulinemia and β‐cell hyperplasia develop to compensate for the relative lack of insulin action, which is clearly shown in animal models of type 2 diabetes. For the development of overt type 2 diabetes, relative insulin deficiency is critical in addition to insulin resistance. Insulin deficiency of type 2 diabetes could be a result of decreased insulin release from β‐cells and/or decreased β‐cell mass. Decreased β‐cell mass in full‐blown or clinically overt type 2 diabetes has been reported in type 2 diabetes patients14,15. The mechanism of decreased β‐cell mass in type 2 diabetes is likely to be a result of pancreatic β‐cell apoptosis or death. However, only a few papers reported apoptosis of pancreatic β‐cells in type 2 diabetes14,16, which is due to very low probability of detecting ongoing β‐cell death in given pancreatic sections of slowly progressive type 2 diabetes.
Even more obscure is the effector molecule(s) in pancreatic β‐cell apoptosis of type 2 diabetes. Besides insulin, the concentration of several molecules such as TNF‐α or advanced glycation end‐products (AGE) is elevated, which could be potentially harmful to β‐cell function or β‐cell viability in the long term. Amylin secreted together with insulin17,18, endoplasmic reticulum (ER) stress resulting from prolonged high insulin production or lipid molecules, such as free fatty acids (FFA), might also contribute to the β‐cell apoptosis in type 2 diabetes.
FFA also has been reported as a possible effector of pancreatic β‐cell dysfunction or death (lipotoxicity). FFA released from the visceral fat of obese subjects is one of the strong culprits in the pathogenesis of insulin resistance that is a prerequisite for the development of type 2 diabetes19. The role of FFA in insulin resistance has been well established. Recently, papers suggesting the potential role of FFA in relative insulin deficiency, as well as in insulin resistance, were published. According to such papers, FFA plays a role as an effector of pancreatic β‐cell dysfunction or apoptosis (lipoapoptosis)20,21. However, the detailed molecular and cellular mechanism of lipoapoptosis is not elucidated. Previous papers have reported that ceramide produced from FFA plays an important role in lipoapotosis of pancreatic β‐cells in type 2 diabetes20,22. In such events, c‐Jun N‐terminal kinases (JNK) activation by lipid intermediates produced from FFA might contribute to the lipoapoptosis of pancreatic β‐cells in obesity‐induced diabetes23. JNK might also be involved in FFA‐induced pancreatic β‐cell dysfunction or decreased insulin production associated with obesity24. Thus, in addition to the well‐established role of JNK activation in insulin resistance, JNK might contribute to the β‐cell death and β‐cell dysfunction in lipid injury or those associated with obesity. Although ceramide has been implicated as a death effector in lipoapoptosis of pancreatic β‐cells, the role of lipid intermediates produced from FFA other than ceramide cannot be eliminated. For instance, diacylglyerol (DAG), triglyceride (TG) or other metabolites resulting from incomplete β‐oxidation of fatty acids have been implicated as the final effector molecules in FFA‐induced insulin resistance or lipotoxicity25–27. We have reported that lysophosphatidylcholine (LPC) generated from FFA might act as one of the final effector molecules in FFA‐induced lipoapoptosis of hepatocytes28. However, the role for LPC in lipoapoptosis of pancreatic β‐cells remains to be tested.
β‐cell necrosis in diabetes
Necrosis of pancreatic β‐cells might also contribute to the development of diabetes. In the case of diabetes induced by exogenous agents such as STZ or virus, necrosis might play a major role in the development of disease29. In the case of natural type 1 diabetes, β‐cells undergoing secondary necrosis after physiological apoptosis can contribute to the induction of insulitis. During the neonatal period in mice, the β‐cell population increases rapidly, and this postnatal expansion of β‐cell mass is followed by a transient wave of physiological β‐cell apoptosis30. Normally, these apoptotic β‐cells are engulfed by phagocytes, such as macrophages, thereby preventing the release of potentially cytotoxic or inflammatory cellular contents31. However, in non‐obese diabetic (NOD) mice with classic type 1 diabetes, the remaining apoptotic β‐cells that are not properly cleared due to defective phagocytosis of apoptotic cells might undergo secondary necrosis. Endogenous ligands from the cells undergoing secondary necrosis might be able to stimulate Toll‐like receptors 2 (TLR2) on antigen‐presenting cells (APC) in the pancreatic lymph nodes (PLN), which might ultimately lead to macrophage activation, dendritic cell (DC) maturation, and priming of naïve diabetogenic T cells. This model is substantiated by the significantly decreased incidence of diabetes and attenuated severity of insulitis in TLR2‐knockout NOD mice compared with wild‐type NOD mice32. In contrast, the development of type 1 diabetes was not inhibited in TLR4‐knockout NOD mice. These findings suggest that β‐cell necrosis and its sensing through TLR2 might be one of the initial events for the stimulation of APC and the development of type 1 diabetes.
Overview of autophagy
Autophagy is an intracellular bulk degradation/recycling process that promotes cell survival under nutrient deprivation, but can also execute the non‐apoptotic form of programmed cell death under certain conditions. Autophagy is derived from Greek roots: auto, meaning ‘self’, and phagy, ‘to eat’. Although the term literally translates to ‘self‐eating’, this non‐specific degradation process that involves recycling of cellular components plays a crucial role in cellular homeostasis. The ubiquitin‐proteasome system (UPS) and the autophagy‐lysosome pathway are the two main routes for eukaryotic intracellular protein clearance. Proteasomes predominantly degrade short‐lived, ubiquitinated, soluble proteins. In contrast, autophagy is critical in the sequestration of cytoplasm by forming double‐membrane vesicles called autophagosomes. The outer membrane of the autophagic vacuoles fuses with lysosomes to form autophagolysosomes, where their contents are degraded by lysosomal enzymes. Because intracellular protein degradation affects many cellular processes, dysregulated or impaired autophagy is likely to be involved in a wide variety of disease states, such as neurodegeneration or cancer.
By degrading intracellular proteins, the autophagy pathway functions in adaptation to cellular starvation. After nutrient stress, autophagy is activated to produce amino acids that can serve as an alternative energy source. Therefore, autophagy can act as a pro‐survival mechanism during nutrient starvation. In addition to the aforementioned stimuli, inhibition of the proteasome function has been shown to activate autophagy, suggesting that the two systems can act in a compensatory fashion to one another33, although inconsistent results have been reported. Even in the presence of sufficient nutrients or under basal conditions, autophagy is necessary for the sequestration of denatured long‐lived proteins and damaged cell organelles as a measure of organelle recycling constitutively or in response to the environmental insults.
Because autophagy is important for the supply of energy sources in cellular starvation, inhibition of autophagy might lead to a shortage of bioenergetic sources, such as NADPH2 and ATP, which might trigger apoptosis34. Thus, the constitutive turnover of proteins and organelles is important for the supply of basic nutrients and the maintenance of organelle function or cell viability. Diverse forms of autophagosome formation have been reported depending on the substrates or organelle being degraded, suggesting that autophagy is a heterogeneous process.
Autophagy of pancreatic β‐cells
The main function of pancreatic β‐cells is the regulation of glucose homeostasis. In response to elevated glucose, exocytosis of insulin‐containing secretory granules occurs in β‐cells. To replenish the lost insulin, insulin translation should be stimulated35. Even under non‐stimulatory glucose concentrations, insulin synthesis continues, showing that constant protein synthesis and degradation occur in pancreatic β‐cells. To maintain secretory granules of β‐cells in an optimal state, aged β‐cell granules should undergo degradation through autophagy or a related phenomenon, crinophagy35. Hence, it is possible that defects in the regulation of autophagy might play a role in the pathogenesis of diabetes.
In addition to rapid turnover of insulin granules, pancreatic β‐cells have to deal with an overwhelming accumulation of misfolded or aggregated proteins provoked by chronic ER or oxidative stress, such as hyperglycemia, cytokines or FFA. For example, chronic hyperglycemia itself induces the formation of ubiquitinated‐protein aggregates in a pancreatic β‐cell line (INS‐1 832/13)36, although the nature of those proteins needs to be clarified. These ubiquitinated proteins are cleared by a ubiquitin‐proteasome system or autophagy, showing that autophagy can act as a defense mechanism against diabetes‐induced cellular damage36. Furthermore, autophagy is important in the turnover of dysfunctional mitochondria in β‐cells in a process called ‘mitophagy’, and dysregulation of this process might predispose to diabetes37.
In addition to its role in the turnover of insulin granules, protein aggregates or dysfunctional organelles, several studies using cell lines and rodent models have shown the importance of autophagy in regulating β‐cell survival and function. In animal models, low‐level constitutive autophagy in β‐cells has been reported in C57BL/6 mice fed a standard diet; however, autophagy was upregulated in mice fed a high‐fat diet (HFD), suggesting compensatory autophagy activation to relieve metabolic stress of β‐cells38. Similarly, β‐cells of diabetic db/db mice contained large numbers of autophagosomes, compared with non‐diabetic control mice. To prove the role of autophagy in β‐cells, two groups reported mice with β‐cell specific deletion of the Atg7 (autophagy‐related 7)38,39. β‐cell specific Atg7‐knockout (Atg7Δβ‐cell) mice showed hypoinsulinemia and hyperglycemia. Atg7Δβ‐cell mice also showed increased apoptosis and decreased proliferation of β‐cells leading to decreased β‐cell mass (Figure 2a–d). Consistent with the decreased β‐cell mass, insulin content in the pancreas was significantly lower in Atg7Δβ‐cell mice compared with control mice (Figure 2e). Basal and high glucose‐stimulated insulin secretion from viable primary islets of Atg7Δβ‐cell mice was reduced compared with control mice (Figure 2f). Furthermore, glucose‐induced cytosolic Ca2+ transients were defective in primary islet cells from Atg7Δβ‐cell mice (Figure 2g), suggesting that β‐cell function is impaired, even in apparently healthy β‐cells of Atg7Δβ‐cell mice, and that autophagy is necessary for the physiological function of β‐cells. Electron microscopy (EM) showed more ultrastructural abnormalities, such as swelling of mitochondria and cisternal distension of rough ER and Golgi complex, even in apparently normal‐looking β‐cells of Atg7Δβ‐cell mice38,39. An accumulation of ubiquitinated proteins was also noted. These results suggest that autophagy is necessary to maintain structure, mass and function of pancreatic β‐cells. If the adaptive autophagic machinery is impaired, it might predispose to the development of type 2 diabetes. Autophagy might also function as a protective mechanism against insulin resistant states, such as obesity, and alteration of the autophagic process could be a predisposing factor to type 2 diabetes.
Figure 2.
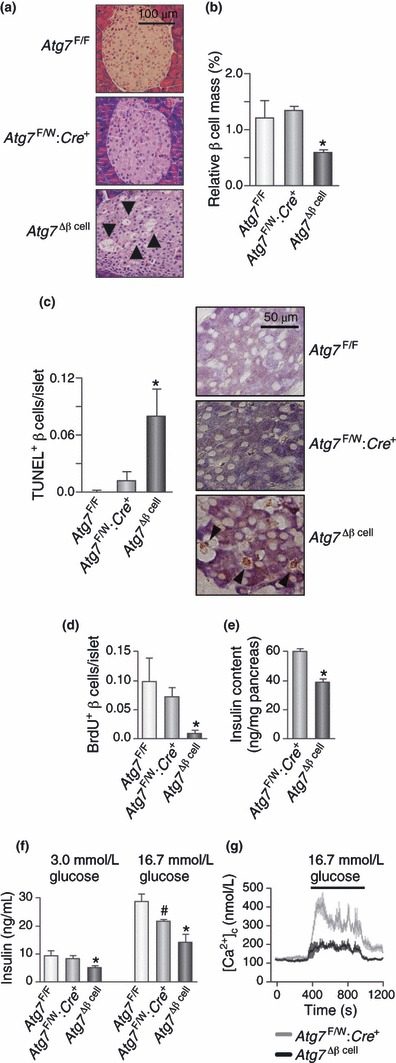
Morphological and functional changes in β‐cells of Atg7Δβ‐cell mice. (a) Hematoxylin–eosin staining showing variable‐sized vacuolated cells in pancreatic islets of Atg7Δβ‐cell mice (arrows). Some vacuolated cells stained positive for insulin. (b) Paraffin‐embedded pancreatic sections were subjected to insulin immunohistochemistry, and relative β‐cell area was determined by point counting (*P < 0.05 vs Atg7F/F and Atg7F/W; Cre+ mice; n = 9 each). (c) Combined insulin immunohistochemistry and TUNEL staining was carried out to count apoptotic β‐cell number/islet (*P < 0.05 vs Atg7F/F and Atg7F/W; Cre+ mice; n = 9 each). Representative apoptotic β‐cells are shown on the right (arrow heads). (d) Double immunohistochemistry for insulin and BrdU after intraperitoneal injection of BrdU was carried out to evaluate β‐cell proliferation (P < 0.05 by anova; *P < 0.05 vs Atg7F/F mice by post‐hoc analysis; n = 9 each). (e) Pancreatic insulin was extracted by acid‐ethanol, and insulin content was measured by radioimmunoassay (*P < 0.001 vs Atg7F/W; Cre+ mice; n = 8 each). (f) Insulin secretion ex vivo. Pancreatic islets from 20‐week‐old mice (n = 3 each) were incubated in KRBB containing 3.0 mmol/L glucose for 1 h and then in KRBB containing 16.7 mmol/L glucose for 1 h. Buffer was collected for insulin radioimmunoassay. Results are representative of two independent experiments carried out in triplicate (*P < 0.05 vs Atg7F/F and Atg7F/W; Cre+ mice; #P < 0.05 vs Atg7F/F mice). (g) Glucose‐induced Ca2+ transients in isolated islets (n = 5 each). [Ca2+]c was determined in Tyrode solution containing 3.0 or 16.7 mmol/L glucose (36°C). In panels (b)–(f), error bars represent SEM. Reproduced from the original paper in Cell Metabolism 2008; 8: 318–325.
In addition to the mouse model, β‐cells in human type 2 diabetes patients also show an accumulation of autophagic vacuoles and autophagosomes, which might represent the adaptive process rather than cause of β‐cell loss1. However, further studies are warranted to substantiate this point, because a blockade in the autophagic process after the completion of autophagosome formation can lead to the increased number of autophagosomes and increased autophagosome number does not necessarily mean increased autophagic activity. The protective role of autophagy against β‐cell lipoapoptosis in vitro that otherwise could be potentially involved in the decrease of β‐cell mass and β‐cell failure of type 2 diabetes has also been suggested40. However, this interpretation should be based on the cellular or environmental context. For example, a recent report suggested that inhibition of autophagy prolonged pancreatic β‐cell survival and delayed cell death in Pdx1‐deficient mice41.
Autophagy and cell death
Autophagy can promote cell survival, but under certain circumstances it might contribute to the execution of cell death. However, the molecular basis underlying its dual role remains unclear. It is also not certain whether or not autophagy is a real effector mechanism of cell death. The visualization of a large‐scale accumulation of autophagosomes in areas undergoing cell death has led to the belief that autophagy is a non‐apoptotic form of programmed cell death. In many cases, however, it is agreed that this autophagic activity in dying cells does not cause cell death, but it simply occurs as a death process. In fact, activation of autophagy machinery might actually be a compensatory survival mechanism of the cells to provide energy substrates. Furthermore, autophagy enhances the quality control of mitochondria, preventing reactive oxygen species (ROS) production and ROS‐mediated DNA damage. By preventing ROS accumulation, autophagy might have a role in the inhibition of the progression of cell death mode to necrosis. Most evidence shows that, at least in cells with intact apoptotic machinery, autophagy is primarily a pro‐survival rather than a pro‐death mechanism.
However, it can also mediate cellular demise, depending on cell context and specific circumstances. Massive autophagy might result in direct self‐destruction of cells, initially suggested as type 2 cell death. Alternatively, autophagy might somehow trigger cell death by switching pro‐survival signals to pro‐apoptotic signals. Further research will be necessary to fully define the complex interplay between autophagy and apoptosis pathways or even necrosis, because there are increasing research interests to develop autophagy‐enhancing drugs in various medical conditions, such as neurodegenerative diseases, diabetes and cancer.
Autophagy and age‐associated diseases
Because autophagy degrades aggregated proteins and recycles damaged organelles, the decline of autophagic activity might be one of the mechanisms leading to aging and age‐associated diseases42. In fact, autophagic activity has been reported to diminish with age, and research focusing on inducing autophagy to ameliorate age‐induced disease is now emerging. One of the mechanisms of anti‐aging effects of autophagy could be the elimination of damaged mitochondria. Increased mitochondrial production of ROS subsequent to the loss of mitochondrial function has been implicated in the aging process, which constitutes the ‘mitochondrial theory of aging’. Because impaired glucose tolerance and diabetes are frequently associated with aging, dysregulated autophagy might play a role in the development of diabetes in aged subjects or other age‐associated diseases, such as neurodegeneration.
Autophagy and insulin resistance
In addition to its role in pancreatic β‐cells, autophagy might have a function in insulin sensitive tissues. A recent paper reported that autophagy regulates lipid content in the liver. Singh et al.43 reported that starvation induced the recruitment of several autophagy‐related proteins to lipid droplets to form autolipophagosomes and induced triglyceride (TG) breakdown. However, the efficiency of ‘lipophagy’ was impaired when intracellular lipid content was chronically increased, such as HFD. In such a case, autophagic clearance was impaired, as shown by reduced association of autophagic vacuoles with lipid droplets in response to starvation in HFD‐fed mice. Subsequently, autophagy‐mediated breakdown of lipid stores was further diminished in the liver, inducing a vicious circle in which increased fat ingestion was associated with decreased fat removal and excessive lipid deposition in the liver43. However, seemingly inconsistent findings have also been published44. Although autophagy is likely to be involved in lipid metabolism and lipid injury, further studies will be necessary to understand the role of autophagy in lipid metabolism in more detail.
The role of autophagy in adipose tissue, another insulin target tissue, was also reported45,46. The mutant mice with a targeted deletion of Atg7 in adipose tissue were slim and contained less white adipose tissue mass. Autophagy‐deficient white adipocytes were smaller and contained more mitochondria, showing increased rates of β‐oxidation and reduced rates of hormone‐induced lipolysis. Consistently, the mutant mice had lower fatty acids and their levels decreased at faster rates on insulin stimuli. Those mutant mice were resistant to HFD‐induced obesity, suggesting that autophagy is important in adipogenesis.
Another paper reported that autophagy participates in the downregulation of insulin receptors and ER stress‐mediated insulin resistance as an adaptive process to ER stress47. Conversely, insulin resistance has been reported to suppress autophagy in the liver48. A recent paper showed the role of hepatic autophagy in insulin resistance in vivo49. In both genetic and dietary models of obesity, suppression of Atg7 was observed in the liver, resulting in defective insulin signaling and elevated ER stress. These metabolic abnormalities were restored by Atg7 expression, suggesting that loss of autophagy is a critical component of defective insulin action seen in obesity. How‐ever, it is not clear whether or not decreased expression of autophagy‐related genes in obesity is a general phenomenon or if autophagic activity could be restored by overexpression of such genes.
Conclusion
β‐cell death of various types play diverse and important roles in the development of type 1 and type 2 diabetes. Because β‐cell death is the ultimate final step in the development of diabetes, current investigation of anti‐apoptotic therapies targeting β‐cells is an emerging area in diabetes research. These results also show that autophagy is important in the maintenance of β‐cell mass, structure and function. Dysregulation of autophagy in insulin producing β‐cells or insulin target tissues might predispose to metabolic disorders, such as type 2 diabetes. Exploring measures to modulate autophagic activity to protect against β‐cell failure in type 2 diabetes or aging could be a promising area of research. Such approaches might also be applicable to insulin‐resistant states, such as obesity and metabolic syndrome.
Acknowledgments
This study was supported by a grant from the Korea Healthcare technology R&D Project, Ministry for Health, Welfare & Family Affairs, Korea (A080967) and the 21C Frontier Functional Proteomics Project of the Korean Ministry of Science & Technology (FPR08B1‐210). Myung‐Shik Lee is the recipient of the Bio R&D program (2008‐04090) and Global Research Laboratory Grant from the National Research Foundation of Korea. No potential conflict of interest was reported.
References
- 1.Masini M, Bugliani M, Lupi R, et al. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 2009; 52: 1083–1086 [DOI] [PubMed] [Google Scholar]
- 2.Kim YH, Kim S, Kim KA, et al. Apoptosis of pancreatic beta‐cells detected in accelerated diabetes of NOD mice: no role of Fas‐Fas ligand interaction in autoimmune diabetes. Eur J Immunol 1999; 29: 455–465 [DOI] [PubMed] [Google Scholar]
- 3.O’Brien BA, Huang Y, Geng X, et al. Phagocytosis of apoptotic cells by macrophages from NOD mice is reduced. Diabetes 2002; 51: 2481–2488 [DOI] [PubMed] [Google Scholar]
- 4.Chervonsky AV, Wang Y, Wong FS, et al. The role of Fas in autoimmune diabetes. Cell 1997; 89: 17–24 [DOI] [PubMed] [Google Scholar]
- 5.Kim S, Kim KA, Hwang DY, et al. Inhibition of autoimmune diabetes by Fas ligand: the paradox is solved. J Immunol 2000; 164: 2931–2936 [DOI] [PubMed] [Google Scholar]
- 6.Liu D, Pavlovic D, Chen MC, et al. Cytokines induce apoptosis in beta‐cells isolated from mice lacking the inducible isoform of nitric oxide synthase (iNOS−/−). Diabetes 2000; 49: 1116–1122 [DOI] [PubMed] [Google Scholar]
- 7.Suk K, Kim S, Kim YH, et al. IFN‐gamma/TNF‐alpha synergism as the final effector in autoimmune diabetes: a key role for STAT1/IFN regulatory factor‐1 pathway in pancreatic beta cell death. J Immunol 2001; 166: 4481–4489 [DOI] [PubMed] [Google Scholar]
- 8.Gysemans CA, Ladriere L, Callewaert H, et al. Disruption of the gamma‐interferon signaling pathway at the level of signal transducer and activator of transcription‐1 prevents immune destruction of beta‐cells. Diabetes 2005; 54: 2396–2403 [DOI] [PubMed] [Google Scholar]
- 9.Kim S, Kim HS, Chung KW, et al. Essential role for signal transducer and activator of transcription‐1 in pancreatic beta‐cell death and autoimmune type 1 diabetes of nonobese diabetic mice. Diabetes 2007; 56: 2561–2568 [DOI] [PubMed] [Google Scholar]
- 10.Beg AA, Baltimore D. An essential role for NF‐kappaB in preventing TNF‐alpha‐induced cell death. Science 1996; 274: 782–784 [DOI] [PubMed] [Google Scholar]
- 11.Giannoukakis N, Rudert WA, Trucco M, et al. Protection of human islets from the effects of interleukin‐1beta by adenoviral gene transfer of an Ikappa B repressor. J Biol Chem 2000; 275: 36509–36513 [DOI] [PubMed] [Google Scholar]
- 12.Kim S, Millet I, Kim HS, et al. NF‐kappa B prevents beta cell death and autoimmune diabetes in NOD mice. Proc Natl Acad Sci USA 2007; 104: 1913–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim HS, Kim S, Lee MS. IFN‐gamma sensitizes MIN6N8 insulinoma cells to TNF‐alpha‐induced apoptosis by inhibiting NF‐kappaB‐mediated XIAP upregulation. Biochem Biophys Res Commun 2005; 336: 847–853 [DOI] [PubMed] [Google Scholar]
- 14.Butler AE, Janson J, Bonner‐Weir S, et al. Beta‐cell deficit and increased beta‐cell apoptosis in humans with type 2 diabetes. Diabetes 2003; 52: 102–110 [DOI] [PubMed] [Google Scholar]
- 15.Yoon KH, Ko SH, Cho JH, et al. Selective beta‐cell loss and alpha‐cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab 2003; 88: 2300–2308 [DOI] [PubMed] [Google Scholar]
- 16.Kaneto H, Kajimoto Y, Miyagawa J, et al. Beneficial effects of antioxidants in diabetes: possible protection of pancreatic beta‐cells against glucose toxicity. Diabetes 1999; 48: 2398–2406 [DOI] [PubMed] [Google Scholar]
- 17.Janson J, Ashley RH, Harrison D, et al. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate‐sized toxic amyloid particles. Diabetes 1999; 48: 491–498 [DOI] [PubMed] [Google Scholar]
- 18.Lorenzo A, Razzaboni B, Weir GC, et al. Pancreatic islet cell toxicity of amylin associated with type‐2 diabetes mellitus. Nature 1994; 368: 756–760 [DOI] [PubMed] [Google Scholar]
- 19.Roden M, Price TB, Perseghin G, et al. Mechanism of free fatty acid‐induced insulin resistance in humans. J Clin Invest 1996; 97: 2859–2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shimabukuro M, Zhou YT, Levi M, et al. Fatty acid‐induced beta cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci USA 1998; 95: 2498–2502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou YP, Grill VE. Long‐term exposure of rat pancreatic islets to fatty acids inhibits glucose‐induced insulin secretion and biosynthesis through a glucose fatty acid cycle. J Clin Invest 1994; 93: 870–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paumen MB, Ishida Y, Muramatsu M, et al. Inhibition of carnitine palmitoyltransferase I augments sphingolipid synthesis and palmitate‐induced apoptosis. J Biol Chem 1997; 272: 3324–3329 [DOI] [PubMed] [Google Scholar]
- 23.Martinez SC, Tanabe K, Cras‐Meneur C, et al. Inhibition of Foxo1 protects pancreatic islet beta‐cells against fatty acid and endoplasmic reticulum stress‐induced apoptosis. Diabetes 2008; 57: 846–859 [DOI] [PubMed] [Google Scholar]
- 24.Solinas G, Naugler W, Galimi F, et al. Saturated fatty acids inhibit induction of insulin gene transcription by JNK‐mediated phosphorylation of insulin‐receptor substrates. Proc Natl Acad Sci USA 2006; 103: 16454–16459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koves TR, Ussher JR, Noland RC, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab 2008; 7: 45–56 [DOI] [PubMed] [Google Scholar]
- 26.Montell E, Turini M, Marotta M, et al. DAG accumulation from saturated fatty acids desensitizes insulin stimulation of glucose uptake in muscle cells. Am J Physiol Endocrinol Metab 2001; 280: E229–E237 [DOI] [PubMed] [Google Scholar]
- 27.Perseghin G, Scifo P, De Cobelli F, et al. Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: a 1H‐13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes 1999; 48: 1600–1606 [DOI] [PubMed] [Google Scholar]
- 28.Han MS, Park SY, Shinzawa K, et al. Lysophosphatidylcholine as a death effector in the lipoapoptosis of hepatocytes. J Lipid Res 2008; 49: 84–97 [DOI] [PubMed] [Google Scholar]
- 29.Takasu N, Komiya I, Asawa T, et al. Streptozocin‐ and alloxan‐induced H2O2 generation and DNA fragmentation in pancreatic islets. H2O2 as mediator for DNA fragmentation. Diabetes 1991; 40: 1141–1145 [DOI] [PubMed] [Google Scholar]
- 30.Bonner‐Weir S. Life and death of the pancreatic beta cells. Trends Endocrinol Metab 2000; 11: 375–378 [DOI] [PubMed] [Google Scholar]
- 31.Savill J, Dransfield I, Gregory C, et al. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol 2002; 2: 965–975 [DOI] [PubMed] [Google Scholar]
- 32.Kim HS, Han MS, Chung KW, et al. Toll‐like receptor 2 senses beta‐cell death and contributes to the initiation of autoimmune diabetes. Immunity 2007; 27: 321–333 [DOI] [PubMed] [Google Scholar]
- 33.Ding WX, Ni HM, Gao W, et al. Linking of autophagy to ubiquitin‐proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 2007; 171: 513–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maiuri MC, Zalckvar E, Kimchi A, et al. Self‐eating and self‐killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8: 741–752 [DOI] [PubMed] [Google Scholar]
- 35.Uchizono Y, Alarcon C, Wicksteed BL, et al. The balance between proinsulin biosynthesis and insulin secretion: where can imbalance lead? Diabetes Obes Metab 2007; 9(Suppl. 2): 56–66 [DOI] [PubMed] [Google Scholar]
- 36.Kaniuk NA, Kiraly M, Bates H, et al. Ubiquitinated‐protein aggregates form in pancreatic beta‐cells during diabetes‐induced oxidative stress and are regulated by autophagy. Diabetes 2007; 56: 930–939 [DOI] [PubMed] [Google Scholar]
- 37.Twig G, Elorza A, Molina AJ, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 2008; 27: 433–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ebato C, Uchida T, Arakawa M, et al. Autophagy is important in islet homeostasis and compensatory increase of Beta cell mass in response to high‐fat diet. Cell Metab 2008; 8: 325–332 [DOI] [PubMed] [Google Scholar]
- 39.Jung HS, Chung KW, Won KJ, et al. Loss of autophagy diminishes pancreatic Beta cell mass and function with resultant hyperglycemia. Cell Metab 2008; 8: 318–324 [DOI] [PubMed] [Google Scholar]
- 40.Choi SE, Lee SM, Lee YJ, et al. Protective role of autophagy in palmitate‐induced INS‐1 beta‐cell death. Endocrinology 2009; 150: 126–134 [DOI] [PubMed] [Google Scholar]
- 41.Fujimoto K, Hanson PT, Tran H, et al. Autophagy regulates pancreatic beta cell death in response to Pdx1 deficiency and nutrient deprivation. J Biol Chem 2009; 284: 27664–27673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cavallini G, Donati A, Taddei M, et al. Evidence for selective mitochondrial autophagy and failure in aging. Autophagy 2007; 3: 21–27 [DOI] [PubMed] [Google Scholar]
- 43.Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature 2009; 458: 1131–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shibata M, Yoshimura K, Furuya N, et al. The MAP1‐LC3 conjugation system is involved in lipid droplet formation. Biochem Biophys Res Commun 2009; 382: 419–423 [DOI] [PubMed] [Google Scholar]
- 45.Singh R, Xiang Y, Wang Y, et al. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest 2009; 119: 3329–3339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Goldman S, Baerga R, et al. Adipose‐specific deletion of autophagy‐related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci USA 2009; 106: 19860–19865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou L, Zhang J, Fang Q, et al. Autophagy‐mediated insulin receptor down‐regulation contributes to endoplasmic reticulum stress‐induced insulin resistance. Mol Pharmacol 2009; 76: 596–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu HY, Han J, Cao SY, et al. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1‐dependent expression of key autophagy genes by insulin. J Biol Chem 2009; 284: 31484–31492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang L, Li P, Fu S, et al. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab 2010; 11: 467–478 [DOI] [PMC free article] [PubMed] [Google Scholar]