

Abstract
Aims/Introduction: Postprandial serum C‐peptide levels are readily determined in clinical practice and have a good correlation with serum C‐peptide levels after glucagon load; the measurement is often used as an index of endogenous insulin secretion. However, the factors affecting postprandial serum C‐peptide levels remain to be evaluated.
Materials and Methods: To investigate the clinical factors affecting postprandial serum C‐peptide, 2‐h postprandial C‐peptide levels after breakfast (PPCPR) were analyzed retrospectively for comparison with glucagon‐stimulated C‐peptide (CPR‐6min) levels measured during hospital admission in 273 Japanese patients with type 2 diabetes.
Results: Multiple regression analysis showed that years from diagnosis, body mass index (BMI) and HbA1c were the major independent variables predicting PPCPR (R2 = 0.315). HbA1c was a major factor predicting PPCPR, but did not predict CPR‐6min. In addition, HbA1c was negatively correlated with PPCPR (r = −0.410, P < 0.0001) and PPCPR/CPR‐6min (r = −0.313, P < 0.0001).
Conclusions: PPCPR was correlated with common factors predicting CPR, including years from diagnosis and BMI, but also was negatively correlated with HbA1c, a unique factor. These results show that chronic elevation of the glucose level might impair endogenous insulin secretion after meal load, but might have little effect on endogenous insulin secretion after glucagon load. (J Diabetes Invest, doi: 10.1111/j.2040‐1124.2011.00126.x, 2011)
Keywords: C‐peptide, Meal load, HbA1c
Introduction
Type 2 diabetes is a heterogeneous disease characterized by insulin resistance and defective insulin secretion1, and is progressive in that the mode of therapy must be altered over the decades of diabetes; diet and exercise therapy alone might be adequate initially, but secondary oral hypoglycemic agent (OHA) treatment and insulin treatment are eventually required2,3. This is, at least in part, as a result of progressive loss of pancreatic β‐cell function. The results of the United Kingdom Progressive Diabetes Study (UKPDS) show that pancreatic β‐cell function (%β), assessed by Homeostasis Model Assessment (HOMA) in patients allocated to diet or OHA decreased approximately 25% in 5 years4. In addition, a decline in endogenous insulin secretion over more than several decades of diabetes in patients including insulin‐treated patients was observed in a cross‐sectional study5.
Determination of fasting serum C‐peptide level and stimulated serum C‐peptide level by intravenous glucagon is used widely to assess endogenous insulin secretory reserves6–9, and the utility of the indices using C‐peptide level in choosing insulin therapy has been shown10. The postprandial serum C‐peptide level can easily be measured in clinical practice and has a good correlation with the serum C‐peptide level after glucagon load11; it is often used as an index of endogenous insulin secretion, and can be used for both non‐insulin‐treated and insulin‐treated patients11–13. Duration of diabetes and body mass index (BMI) are the major factors in serum fasting and glucagon‐stimulated C‐peptide levels5,14, but the factors affecting postprandial serum C‐peptide levels remain to be evaluated.
In the present study of Japanese patients with type 2 diabetes, to evaluate the clinical factors affecting postprandial serum C‐peptide by cross‐sectional study, 2‐h postprandial C‐peptide levels after breakfast were analyzed and compared with glucagon‐stimulated C‐peptide levels.
Subjects and methods
Subjects
A total of 388 Japanese patients with type 2 diabetes who were admitted to Kyoto University Hospital between 1997 and 2002 for poor glycemic control were enrolled in the study. Patients with pancreatic or liver disease, taking diabetogenic medications, pregnant or with serum creatinine ≥1.3 mg/dL were excluded from the study. Type 2 diabetes mellitus was diagnosed based on the criteria of the American Diabetes Association (ADA)15. Patients with serum creatinine ≥1.3 mg/dL were excluded, as serum C‐peptide immunoreactivity (CPR) is elevated by decreased renal function16. Of these patients, 115 were excluded as a result of incomplete clinical examinations and the remaining 273 patients, including patients without diabetic medication, oral hypoglycemic agent‐treated patients and insulin‐treated patients, were analyzed. The clinical profiles of the patients are shown in Table 1.
Table 1. Clinical profiles of patients.
| No. patients | 273 |
|---|---|
| Male/female | 158/115 |
| Age (years) | 61.2 ± 12.2 |
| Years from diagnosis | 9.6 ± 9.6 |
| Systolic blood pressure (mmHg) | 121.8 ± 12.9 |
| Diastolic blood pressure (mmHg) | 73.6 ± 9.6 |
| BMI (kg/m2) | 23.9 ± 3.7 |
| HbA1c at admission (%) | 9.7 ± 2.0 |
| sCre (mg/dL) | 0.69 ± 0.18 |
| Glucagon load: FPG/PG‐6min (mg/dL) | 164.1 ± 47.9/180.6 ± 49.1 |
| Glucagon load: FCPR/CPR‐6min (ng/mL) | 1.80 ± 0.97/3.83 ± 1.76 |
| Meal load: FPG/PPPG (mg/dL) | 167.0 ± 54.8/271.5 ± 83.5 |
| Meal load: FCPR/PPCPR (ng/mL) | 1.76 ± 0.94/4.87 ± 2.41 |
BMI, body mass index; CPR‐6min, C‐peptide immunoreactivity 6 min after intravenous injection of 1 mg glucagon; FCPR, fasting CPR; FPG, fasting plasma glucose; OHA, oral hypoglycemic agents; PG‐6min, plasma glucose 6 min after glucagon load; PPCPR, postprandial CPR; PPPG, postprandial plasma glucose; sCre, serum creatinine.
Methods
On the first day in hospital, medical history, physical examination and laboratory evaluation including glycosylated hemoglobin were carried out. HbA1c was measured using high performance liquid chromatography (HA‐8180; Arcray, Kyoto, Japan). The HbA1c (%) value was estimated as a National Glycohemoglobin Standardization Program (NGSP) equivalent (%) calculated by the formula HbA1c (%) = HbA1c (Japan Diabetes Society [JDS]) (%) + 0.4%, considering the relational expression of HbA1c (JDS) (%) measured by the previous Japanese standard substance and measurement methods and HbA1c (NGSP)17. β‐Cell function was evaluated within 1 week after an overnight fast by measuring fasting CPR (FCPR), CPR 6min after intravenous injection of 1 mg glucagon (CPR‐6min)6 and postprandial CPR. Serum CPR was measured by radioimmunoassay (Daiichi III; Daiichi Radioisotope Laboratories, Osaka, Japan). Postprandial CPR 2 h after breakfast (PPCPR) was determined. The meal at breakfast was prescribed as nutritional therapy according to the treatment guide for diabetes of the JDS18, which included 516.6 ± 67.7 kcal (mean ± SD) energy consisting of 49% carbohydrate, 16% protein and 35% fat. In patients taking OHA, medication was stopped for measurement of CPR, but was maintained until 1 day before to prevent hyperglycemia during the test5. Plasma glucose was measured by the glucose oxidase method.
The study protocol was approved by the ethics committee of Kyoto University.
Statistical Analysis
Statistical analysis was carried out with the Stat View 5.0 system (SAS institute Inc., Cary, NC, USA). Data are presented as mean ± SD, unless otherwise noted. The relationship between the parametric clinical data and CPR values was investigated by Pearson’s analysis. The relationship between the non‐parametric clinical data and CPR values was investigated by Spearman’s analysis. Clinical parameters among three groups were compared by analysis of variance (anova). For comparison of two groups, Scheffé’s test was carried out. P‐values < 0.05 were considered statistically significant.
Results
Simple correlation coefficients between FCPR, CPR‐6min and PPCPR, and measures of variables (age, years from diagnosis, sex, BMI, systolic and diastolic blood pressure, HbA1c, serum creatinine and plasma glucose [PG]) were calculated and are shown in Table 2. Years from diagnosis and BMI were significantly correlated with all three measures of CPR. PG and HbA1c were significantly correlated with PPCPR (P < 0.0001, r = −0.410), but not with CPR‐6min (Figure 1).
Table 2. P‐values and r‐values of correlation between C‐peptide immunoreactivity and measures of variables.
| FCPR (ng/mL) | CPR‐6min (ng/mL) | PPCPR (ng/mL) | |
|---|---|---|---|
| Age (years) | 0.4257 (ND) | 0.0456 (−0.121) | 0.3896 (ND) |
| Years from diagnosis | 0.0024 (−0.182) | <0.0001 (−0.246) | 0.0007 (−0.205) |
| Sex | 0.0709 (ND) | 0.1879 (ND) | 0.8321 (ND) |
| BMI (kg/m2) | <0.0001 (0.435) | <0.0001 (0.367) | <0.0001 (0.311) |
| Systolic blood pressure (mmHg) | 0.5551 (ND) | 0.9388 (ND) | 0.0865 (ND) |
| Diastolic blood pressure (mmHg) | 0.5739 (ND) | 0.0327 (0.130) | 0.0705 (ND) |
| HbA1c (%) | 0.0443 (−0.122) | 0.1507 (ND) | <0.0001 (−0.410) |
| sCre (mg/dL) | 0.0104 (0.155) | 0.1641 (ND) | 0.0140 (0.148) |
| FPG (mg/dL) | 0.3764 (ND) | ND | ND |
| PG‐6min (mg/dL) | ND | 0.7333 (ND) | ND |
| PPPG (mg/dL) | ND | ND | <0.0001 (−0.285) |
All correlations except correlations between sex and C‐peptide immunoreactivity (CPR) were analyzed by Pearson’s analysis. Correlations between sex and CPR were analyzed by Spearman’s analysis. P‐values are shown. In parenthesis, r‐values are shown.
BMI, body mass index; CPR‐6min, C‐peptide immunoreactivity 6 min after intravenous injection of 1 mg glucagon; FCPR, fasting CPR; FPG, fasting plasma glucose; ND, not determined; PG‐6min, plasma glucose 6 min after glucagon load; PPCPR: postprandial CPR; PPPG: postprandial plasma glucose; sCre: serum creatinine.
Figure 1.
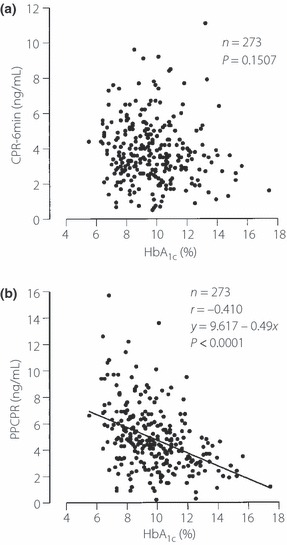
The relationship between HbA1c and (a) C‐peptide immunoreactivity 6 min after intravenous injection of 1 mg glucagon (CPR‐6min) and (b) 2‐h postprandial C‐peptide levels after breakfast (PPCPR).
Stepwise multiple regression analysis was carried out using the independent variables in Table 2 to predict CPR as a dependent variable (Table 3). FCPR was independently predicted by years from diagnosis, BMI and serum creatinine, accounting for 22.4% of the variability of FCPR. CPR‐6min was independently predicted by years from diagnosis and BMI, accounting for 17.9% of the variability of the dependent variables. PPCPR was independently predicted by years from diagnosis, BMI and HbA1c, accounting for 31.5% of the variability of the dependent variables. Thus, HbA1c is an important independent variable predicting PPCPR, but not FCPR or CPR‐6min.
Table 3. Stepwise multiple regression analysis for predictors of C‐peptide immunoreactivity.
| F‐value | Partial regression coefficient | Standard partial regression coefficient | R 2 (R) | |
|---|---|---|---|---|
| FCPR (ng/mL) | ||||
| Years from diagnosis | 9.4 | −0.017 | −0.170 | 0.224 (0.473) |
| BMI (kg/m2) | 55.2 | 0.108 | 0.406 | |
| sCre (mg/dL) | 7.3 | 0.823 | 0.149 | |
| CPR‐6min (ng/mL) | ||||
| Years from diagnosis | 14.6 | −0.039 | −0.214 | 0.179 (0.423) |
| BMI (kg/m2) | 38.9 | 0.170 | 0.349 | |
| PPCPR (ng/mL) | ||||
| Years from diagnosis | 23.4 | −0.063 | −0.252 | 0.315 (0.561) |
| BMI (kg/m2) | 27.5 | 0.178 | 0.270 | |
| HbA1c (%) | 68.7 | −0.516 | −0.431 | |
BMI, body mass index; CPR‐6min, C‐peptide immunoreactivity 6 min after intravenous injection of 1 mg glucagon; FCPR, fasting CPR; PPCPR, postprandial CPR; sCre, serum creatinine.
Because HbA1c might be involved in decreased PPCPR, the clinical data among three groups of increased HbA1c (≤8.5%, 8.6–10.3%, ≥10.4%) were compared, as shown in Table 4. Although there was no significant difference among these groups in FCPR and CPR‐6min, PPCRP was significantly reduced with increasing levels of HbA1c. CPR‐6min was significantly correlated with PPCPR (P < 0.0001, r = 0.564, PPCPR = 0.774 × CPR‐6min + 1.913; Figure 2a). PPCPR was correlated with CPR‐6min in each tertile group of HbA1c (HbA1c ≤ 8.5: P < 0.0001, r = 0.595, y = 2.159 + 0.970x, n = 90; 8.6% ≤ HbA1c ≤ 10.3%: P < 0.0001, r = 0.674, y = 1.587 + 0.829x, n = 92; 10.4% ≤ HbA1c: P < 0.0001, r = 0.494, y = 2.091 + 0.482x, n = 91). Because the higher HbA1c group was distributed mainly below the regression line of total patients and the lower HbA1c group above the line in the scattergram, and the increase in PPCPR per CPR‐6min in the regression line of each tertile group was lower in the higher HbA1c group, we examined the correlation between the ratio of PPCPR to CPR‐6min (PPCPR/CPR‐6min) and HbA1c. PPCPR/CPR‐6min was negatively correlated with HbA1c (P < 0.0001, r = −0.313; Figure 2b).
Table 4. Comparison of clinical characteristics and clinical profile among groups according to HbA1c at admission.
| Groups (HbA1c at admission) | I (≤8.5%) | II (8.6–10.3%) | III (≥10.4%) | P |
|---|---|---|---|---|
| No. patients | 90 | 92 | 91 | |
| HbA1c (%) | 7.6 ± 0.1 | 9.5 ± 0.1* | 12.0 ± 0.1*† | <0.0001 |
| Sex (male/female) | 53/37 | 55/37 | 50/41 | |
| Age (years) | 64.2 ± 1.2 | 61.6 ± 1.3 | 57.6 ± 1.3* | 0.0011 |
| BMI (kg/m2) | 24.2 ± 0.3 | 24.1 ± 0.4 | 23.5 ± 0.4 | 0.3579 |
| Years from diagnosis | 11.7 ± 1.2 | 9.7 ± 0.8 | 7.4 ± 0.8* | 0.0088 |
| bSBP (mmHg) | 122.9 ± 1.4 | 120.6 ± 1.3 | 121.3 ± 1.4 | 0.4746 |
| DBP (mmHg) | 72.9 ± 1.2 | 72.7 ± 1.0 | 75.3 ± 0.9 | 0.1302 |
| sCre (mg/dL) | 0.74 ± 0.02 | 0.70 ± 0.02 | 0.63 ± 0.02*† | <0.0001 |
| FPG (mg/dL) | 134.2 ± 3.7 | 163.4 ± 3.9* | 195.0 ± 5.4*† | <0.0001 |
| PG‐6min (mg/dL) | 152.4 ± 3.9 | 178.7 ± 4.0* | 211.1 ± 5.6*† | <0.0001 |
| PPPG (mg/dL) | 223.3 ± 6.4 | 268.2 ± 7.7* | 323.7 ± 8.9*† | <0.0001 |
| FCPR (ng/mL) | 1.92 ± 0.10 | 1.84 ± 0.10 | 1.66 ± 0.11 | 0.2004 |
| CPR‐6min (ng/mL) | 3.85 ± 0.18 | 3.97 ± 0.19 | 3.66 ± 0.18 | 0.5467 |
| PPCPR (ng/mL) | 5.90 ± 0.29 | 4.88 ± 0.24* | 3.86 ± 0.18*† | <0.0001 |
Data are presented as mean ± SE.
*P < 0.01 vs group I, †P < 0.01 vs group II.
BMI, body mass index; CPR‐6min, C‐peptide immunoreactivity 6 min after intravenous injection of 1 mg glucagon; DBP, diastolic blood pressure; FCPR, fasting CPR; FPG, fasting plasma glucose; PG‐6min, plasma glucose 6 min after glucagon load; PPCPR, postprandial CPR; PPPG, postprandial plasma glucose; SBP, systolic blood pressure; sCre, serum creatinine. FPG and FCPR are values when meal load was carried out.
Figure 2.
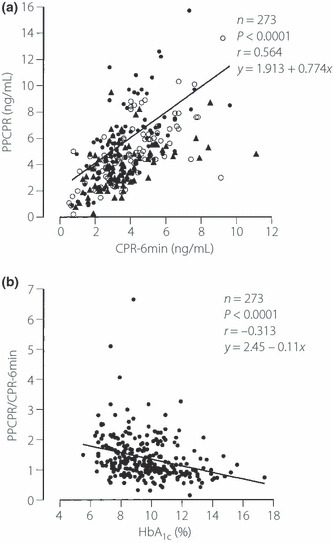
Relationship between (a) C‐peptide immunoreactivity 6 min after intravenous injection of 1 mg glucagon (CPR‐6min) and 2‐h postprandial C‐peptide levels after breakfast (PPCPR) and (b) PPCPR/CPR‐6min and HbA1c. Black circles, HbA1c ≤ 8.5%; white circles, 8.6% ≤ HbA1c ≤ 10.3%, black triangles, 10.4% ≤ HbA1c
Discussion
In the present study, HbA1c was negatively correlated with PPCPR, but not with FCPR or CPR‐6min, which suggests that chronic elevation of the glucose level might impair endogenous insulin secretion after a meal load.
Although meal load is not equivalent to glucose load, as it contains nutrients other than carbohydrates that modulate glucose‐induced insulin secretion, elevated glucose in plasma might play an important role in meal‐stimulated insulin secretion. Indeed, the plasma glucose level after a meal load was increased considerably to more than 100 mg/dL in average. In contrast, the increment of glucose after glucagon load was only approximately 15 mg/dL, indicating a small contribution of glucose elevation to increased insulin secretion by glucagon loading.
Because HbA1c was positively correlated with PPPG in the present study (P < 0.0001, r = 0.570), HbA1c reflects postprandial glucose level. In simple correlation, both HbA1c and PPPG were significantly correlated with PPCPR; whereas in stepwise regression analysis, HbA1c was important to predict PPCPR, but PPPG was not. In addition, in simple correlation to PPCPR, the r‐value for HbA1c (0.410) was larger compared with that for PPPG (0.285; Table 2). These results show that PPCPR is more strongly affected by chronic elevation of glucose levels than by transient elevation of glucose levels.
Multiple regression analysis showed that years from diagnosis, BMI and HbA1c were the major independent variables predicting PPCPR. This shows that years from diagnosis and BMI are common major factors predicting CPR. In contrast, HbA1c was the major factor predicting PPCPR, but not FCPR or CPR‐6min, and was negatively correlated with PPCPR. We hypothesized that CPR‐6min reflects reserve capacity of endogenous insulin secretion independent of glycemic control and that PPCPR is predicted by a fundamental factor independent of glycemic control and by a variable factor dependent of glycemic control. CPR‐6min predicted 31.8% of the variability of PPCPR as shown in Figure 2a. When a regression model using CPR‐6min and HbA1c as independent variables to predict PPCPR as a dependent variable was used, CPR‐6min and HbA1c predicted 44.9% of the variability of PPCPR (P < 0.0001, R = 0.670, PPCPR = 6.286 + 0.730 × CPR‐6min − 0.434 × HbA1c). The addition of HbA1c as an independent variable increased the prediction of the variability of PPCPR by 13.1%. In the present study, PPCPR/CPR‐6min was used as a putative index of variability dependent of glycemic control and was found to be correlated with HbA1c in the present study (Figure 2b). Furthermore, improvement of glycemic control by treatment ameliorates the CPR response after oral glucose load19–21. In addition, the CPR response after glucagon load is affected little by treatment to improve hyperglycemia and it is not correlated with the CPR response after oral glucose load before treatment, whereas it is well‐correlated with improved CPR response after oral glucose load after treatment21. Reversible impairment of endogenous insulin response after glucose load is explained by glucose toxicity, in which chronic hyperglycemia deteriorates meal‐induced and glucose‐induced insulin secretion and insulin‐sensitive glucose disposal22. Therefore, the chronic high glucose level shown by high HbA1c might impair endogenous insulin secretion after meal load, but has little effect on endogenous insulin secretion after glucagon load. The lack of influence of HbA1c on CPR‐6min might be helpful to evaluate reserve capacity of endogenous insulin secretion, even when glycemic control is poor enough to deteriorate postprandial insulin secretion. In contrast, PPCPR is affected by HbA1c and might reflect the state of deteriorated insulin secretion by glucose toxicity that may be recovered by improved glycemic control.
In stepwise regression analysis, HbA1c was not important to predict FCPR, but was important to predict PPCPR. In simple correlation, HbA1c was significantly negatively correlated not only with PPCPR, but also with FCPR, whereas the P‐value and r‐value for FCPR were larger and smaller, respectively, compared with those for PPCPR (Table 2). Taken together, these findings suggest that glucose toxicity might deteriorate not only postprandial insulin secretion, but also fasting insulin secretion, whereas postprandial insulin secretion might be more vulnerable to glucose toxicity than to fasting insulin secretion.
The suppressive effect of glucose toxicity on insulin secretion in vivo might be attributable to impairment of β‐cell responsiveness to glucose22 and to impairment of incretin effect23,24. However, it is important to understand why glucagon‐stimulated CPR is preserved despite severe impairment of glucose‐stimulated CPR before treatment to improve hyperglycemia21. This remains largely unknown, but our hypothesis based on an in vitro study is that deteriorated intracellular glucose metabolism plays an important role in impaired glucose‐induced insulin secretion25 and that increased intracellular cyclic adenosine 3′,5′‐monophosphate concentration derived from glucagon stimulation ameliorates impaired intracellular glucose metabolism to improve suppressed insulin secretion26.
A recent study showed that indices using CPR correlate well with β‐cell mass by analysis of β‐cell areas of samples obtained during pancreatectomy and serum levels of CPR before operation27. Thus, PPCPR might reflect not only β‐cell mass, but also reversible impairment of endogenous secretion as a result of chronic glucose elevation.
Acknowledgement
The authors declare no conflict of interest.
References
- 1.DeFronzo RA. Lilly lecture 1987. The triumvirate: β‐cell, muscle, liver. A collusion responsible for NIDDM. Diabetes 1988; 37: 667–687 [DOI] [PubMed] [Google Scholar]
- 2.Yki‐Järvinen H, Kauppila M, Kujansuu E, et al. Comparison of insulin regimens in patients with non‐insulin‐dependent diabetes mellitus. N Engl J Med 1992; 327: 1426–1433 [DOI] [PubMed] [Google Scholar]
- 3.Turner RC, Cull CA, Frighi V, et al. Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: progressive requirement for multiple therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS) Group. JAMA 1999; 281: 2005–2012 [DOI] [PubMed] [Google Scholar]
- 4.U.K. Prospective Diabetes Study Group . U.K. prospective diabetes study 16. Overview of 6 years’ therapy of type II diabetes: a progressive disease. Diabetes 1995; 44: 1249–1258 [PubMed] [Google Scholar]
- 5.Funakoshi S, Fujimoto S, Hamasaki A, et al. Analysis of factors influencing pancreatic β‐cell function in Japanese patients with type 2 diabetes: association with body mass index and duration of diabetic exposure. Diabetes Res Clin Pract 2008; 82: 353–358 [DOI] [PubMed] [Google Scholar]
- 6.Faber OK, Binder C. C‐peptide response to glucagon. A test for the residual β‐cell function in diabetes mellitus. Diabetes 1977; 26: 605–610 [DOI] [PubMed] [Google Scholar]
- 7.Hendriksen C, Faber OK, Drejer J, et al. Prevalence of residual B‐cell function in insulin‐treated diabetics evaluated by the plasma C‐peptide response to intravenous glucagon. Diabetologia 1977; 13: 615–619 [DOI] [PubMed] [Google Scholar]
- 8.Jayyab AK, Heding LG, Czyzyk A, et al. Serum C‐peptide and IRI levels after administration of glucagon and glucose in non‐insulin‐dependent diabetics. Horm Metab Res 1982; 14: 112–116 [DOI] [PubMed] [Google Scholar]
- 9.Gjessing HJ, Damsgaard EM, Matzen LE, et al. Reproducibility of β‐cell function estimates in non‐insulin‐dependent diabetes mellitus. Diabetes Care 1987; 10: 558–562 [DOI] [PubMed] [Google Scholar]
- 10.Funakoshi S, Fujimoto S, Hamasaki A, et al. Utility of indices using C‐peptide levels for indication of insulin therapy to achieve good glycemic control in Japanese patients with type 2 diabetes. J Diabetes Invest 2011. doi:10.1111/j.2040‐1124.2010.00096.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koskinen PJ, Viikari JS, Irjala KM. Glucagon‐stimulated and postprandial plasma C‐peptide values as measures of insulin secretory capacity. Diabetes Care 1988; 11: 318–322 [DOI] [PubMed] [Google Scholar]
- 12.Aoki Y. Variation of endogenous insulin secretion in association with treatment status: assessment by serum C‐peptide and modified urinary C‐peptide. Diabetes Res Clin Pract 1991; 14: 165–173 [DOI] [PubMed] [Google Scholar]
- 13.Aoki Y, Yanagisawa Y, Yazaki K, et al. Clinical significance of the two‐hour postprandial serum C‐peptide level in patients with diabetes mellitus. J Jpn Diabetes Soc 1992; 35: 811–818 (Japanese). [Google Scholar]
- 14.Chung JO, Cho DH, Chung DJ, et al. Plasma C‐peptide level is inversely associated with family history of type 2 diabetes in Korean type 2 diabetic patients. Endocr J 2010; 57: 931–938 [DOI] [PubMed] [Google Scholar]
- 15.American Diabetes Association . Diagnosis and classification of diabetes mellitus. Diabetes Care 2010; 33: S62–S69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kajinuma H, Tanabashi S, Ishiwata K, et al. Urinary excretion of C‐peptide in relation to renal function In: Baba S (ed.). Proinsulin, Insulin, C‐peptide. Excerpta Medica, Amsterdam, 1979; 183–189 [Google Scholar]
- 17.The Committee of Japan Diabetes Society on the diagnostic criteria of diabetes mellitus . Report of the Committee on the classification and diagnostic criteria of diabetes mellitus. J Diabetes Invest 2010; 1: 212–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Japan Diabetes Society . (ed.). Treatment Guide for Diabetes 2007. Japan Diabetes Society, Bunkodo, Tokyo, Japan, 2007. [Google Scholar]
- 19.Seino Y, Ikeda M, Kurahachi H, et al. Failure of suppress plasma glucagon concentrations by orally administered glucose in diabetic patients after treatment. Diabetes 1978; 27: 1145–1150 [DOI] [PubMed] [Google Scholar]
- 20.Kosaka K, Kuzuya T, Akanuma Y, et al. Increase in insulin response after treatment of overt maturity‐onset diabetes is independent of the mode of treatment. Diabetologia 1980; 18: 23–28 [DOI] [PubMed] [Google Scholar]
- 21.Iwasaki Y, Kondo K, Hasegawa H, et al. C‐peptide response to glucagon in type 2 diabetes mellitus: a comparison with oral glucose tolerance test. Diabetes Res 1994; 25: 129–137 [PubMed] [Google Scholar]
- 22.Rossetti L, Giaccari A, DeFronzo RA. Glucose toxicity. Diabetes Care 1990; 13: 610–630 [DOI] [PubMed] [Google Scholar]
- 23.Knop FK, Vilsbøll T, Højberg PV, et al. Reduced incretin effect in type 2 diabetes: cause or consequence of the diabetic state? Diabetes 2007; 56: 1951–1959 [DOI] [PubMed] [Google Scholar]
- 24.Højberg PV, Vilsbøll T, Zander M, et al. Four weeks of near‐normalization of blood glucose has no effect on postprandial GLP‐1 and GIP secretion, but augments pancreatic B‐cell responsiveness to a meal in patients with Type 2 diabetes. Diabet Med 2008; 25: 1268–1275 [DOI] [PubMed] [Google Scholar]
- 25.Fujimoto S, Nabe K, Takehiro M, et al. Impaired metabolism‐secretion coupling in pancreatic β‐cells: role of determinants of mitochondrial ATP production. Diabetes Res Clin Pract 2007; 77(Suppl. 1): S2–S10 [DOI] [PubMed] [Google Scholar]
- 26.Mukai E, Fujimoto S, Sato H, et al. Exendin‐4 suppresses Src activation and reactive oxygen species production in diabetic GK rat islets in an Epac‐dependent manner. Diabetes 2011; 60: 218–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meier JJ, Menge BA, Breuer TG, et al. Functional assessment of pancreatic β‐cell area in humans. Diabetes 2009; 58: 1595–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]