

Abstract
Neonatal diabetes mellitus (NDM) is the term commonly used to describe diabetes with onset before 6 months‐of‐age. It occurs in approximately one out of every 100,000–300,000 live births. Although this term encompasses diabetes of any etiology, it is recognized that NDM diagnosed before 6 months‐of‐age is most often monogenic in nature. Clinically, NDM subgroups include transient (TNDM) and permanent NDM (PNDM), as well as syndromic cases of NDM. TNDM often develops within the first few weeks of life and remits by a few months of age. However, relapse occurs in 50% of cases, typically in adolescence or adulthood. TNDM is most frequently caused by abnormalities in the imprinted region of chromosome 6q24, leading to overexpression of paternally derived genes. Mutations in KCNJ11 and ABCC8, encoding the two subunits of the adenosine triphosphate‐sensitive potassium channel on the β‐cell membrane, can cause TNDM, but more often result in PNDM. NDM as a result of mutations in KCNJ11 and ABCC8 often responds to sulfonylureas, allowing transition from insulin therapy. Mutations in other genes important to β‐cell function and regulation, and in the insulin gene itself, also cause NDM. In 40% of NDM cases, the genetic cause remains unknown. Correctly identifying monogenic NDM has important implications for appropriate treatment, expected disease course and associated conditions, and genetic testing for at‐risk family members. Early recognition of monogenic NDM allows for the implementation of appropriate therapy, leading to improved outcomes and potential societal cost savings. (J Diabetes Invest, doi:10.1111/j.2040‐1124.2011.00106.x, 2011)
Keywords: Neonatal diabetes mellitus, Mutation, β‐Cell
Introduction
Diabetes mellitus (DM) is a heterogeneous group of disorders characterized by hyperglycemia and varying degrees of metabolic derangement as a result of β‐cell insufficiency or impaired insulin secretion and/or action. Most often, diabetes mellitus is polygenic in nature, with the most prevalent diabetes disorders being type 1 and type 2 diabetes. Type 1A diabetes is caused by autoimmune destruction of β‐cells, which is typically manifested by the presence of autoantibodies directed against the pancreatic β‐cells. β‐Cell mass and insulin secretion are markedly reduced at the time when classic symptoms of diabetes first manifest. The natural course is ongoing destruction of the β‐cells with absolute insulin deficiency and the need for exogenous insulin in physiological replacement doses. Human leukocyte antigen (HLA) haplotype has a major contributing role in susceptibility to type 1 diabetes, and although several other genetic factors also influence this risk, none of these are independently sufficient to cause type 1 diabetes1. Similarly, type 2 diabetes is caused by the combined effects of multiple environmental factors including obesity, ethnicity, age and metabolic influences, in addition to a poorly characterized genetic predisposition partly accounted for by polymorphisms in genes mostly important in β‐cell function2. Both impaired insulin secretion and insulin resistance are seen in type 2 diabetes, resulting in relative insulin deficiency.
In distinction to these polygenic forms of diabetes, monogenic diabetes disorders are caused by highly penetrant inherited or sporadic mutations in single genes that are critical for β‐cell function. Monogenic diabetes is a rare, but important, cause of diabetes, accounting for an estimated 1–2% of all diabetes cases3. However, the diagnosis is underrecognized, and most patients are incorrectly determined to have type 1 diabetes or type 2 diabetes, leading to unnecessary use of insulin and a failure to identify additional at‐risk family members. Monogenic diabetes phenotypes include maturity‐onset diabetes of the young (MODY), neonatal diabetes and syndromic diabetes that might have diabetes onset in the neonatal or infancy period4.
The term MODY refers to the general phenotype of dominantly inherited, non‐ketotic diabetes typically diagnosed before age 25 years. Several genes encoding proteins important to β‐cell function or regulation have been identified that lead to MODY forms of monogenic diabetes (Table 1). Neonatal diabetes mellitus (NDM) occurs in approximately 1:100,000–300,000 live births3. Traditionally, it has been defined as persistent hyperglycemia, with onset within the first months of life requiring insulin management. The diagnosis of NDM is most often applied to diabetes with onset before 6 months‐of‐age; however, the age limit for NDM varies in the literature and has, in some instances, been extended to 12 months‐of‐age5,6. This variation stems from a focus on capturing monogenic forms of NDM, which can present beyond 6 months‐of‐age. For the purposes of this review, we define NDM as having onset within the first 6 months‐of‐life, with the recognition that a small percentage of monogenic NDM will present between 6 and 12 months‐of‐age5. Despite a readily distinguishable phenotype of diabetes onset before 6 months‐of‐age, monogenic forms of NDM are still often misdiagnosed as type 1 diabetes as a result of lack of genetic testing. Correctly distinguishing monogenic NDM from type 1 diabetes presenting in infancy critically impacts treatment decisions, surveillance of complications and associated conditions, and has important genetic implications for siblings and offspring of affected individuals.
Table 1. Maturity‐onset diabetes of the young.
| Gene | Protein | Clinical features | |
|---|---|---|---|
| MODY1 | HNF4A | Hepatocyte nuclear factor 4‐alpha | Sensitive to sulfonylureas |
| MODY2 | GCK | Glucokinase | Stable, non‐progressive elevated fasting blood glucose; typically does not require treatment |
| MODY3 | HNF1A | Hepatocyte nuclear factor 1‐alpha | Sensitive to sulfonylureas |
| MODY4 | PDX1 (IPF1) | Pancreatic duodenal homeobox 1 (insulin promoter factor 1) | Rare, diabetes appears to be mild |
| MODY5 | HNF1B | Hepatocyte nuclear factor 1‐beta | Renal and genitourinary abnormalities; atrophy of the pancreas |
| MODY6 | NEUROD1 | Neurogenic differentiation 1 | |
| MODY7 | CEL | Carboxyl‐ester lipase | Atrophy of the pancreas |
| MODY8 | INS | Insulin |
MODY, maturity‐onset diabetes of the young.
Onset of DM before 6 months‐of‐age is unlikely to represent autoimmune disease. Studies in patients with NDM have shown that they generally do not have HLA class II haplotypes conferring increased susceptibility to type 1 diabetes, and protective HLA haplotypes have been shown in many cases of NDM7–10. Additionally, markers of immune intolerance in the form of anti‐islet or anti‐β‐cell autoantibodies are an extremely rare finding, being described in only a few cases of monogenic NDM11. The immaturity of the immune system before 6 months‐of‐age is, by itself, an argument against type 1A diabetes in this population. The one exception is immune dysregulation, polyendocrinopathy, enteropathy, X‐linked (IPEX) syndrome, caused by mutations in the FOXP3 gene, resulting in a loss of T regulatory cell function and leading to NDM associated with other autoimmune features including hypothyroidism, eczema and enteritis, often with an early demise12. Additionally, approximately 50% of TNDM have seemingly permanent remission, and during remission most individuals do not have evidence of β‐cell dysfunction or insulin resistance13. These findings argue strongly against type 1A diabetes when onset is in the immediate neonatal period, and type 1A diabetes will remain a rare cause up to 6 months‐of‐age. After 6 months‐of‐age, autoimmune diabetes will become an increasingly more common etiology of diabetes, but a small percentage of diabetes presenting after 6 months‐of‐age will be a result of the same single gene mutations that cause onset in early infancy. Thus, all diabetes with onset before 6 months‐of‐age should prompt investigation for a single gene mutation, and diabetes after 6 months‐of‐age might warrant evaluation for a monogenic cause, especially if antibody negative.
Various single gene and chromosomal abnormalities have been identified that cause different manifestations of NDM. Clinically, NDM can be divided into three subgroups: (i) transient NDM (TNDM); (ii) permanent NDM (PNDM); and (iii) NDM existing as part of a syndrome (syndromic NDM). This phenotypic classification is useful, as the most common causative genetic abnormalities differ by each subtype, though overlap exists. Approximately 50% of NDM is transient and 50% is permanent4. TNDM remits, on average, by 12 weeks‐of‐age; however, approximately 50% of individuals will relapse, typically in adolescence or young adulthood14. There are several syndromes that include NDM as a manifestation. The additional features of each syndrome typically aid in establishing the correct genetic diagnosis, but the clinical heterogeneity in some syndromes can lead to a mild phenotype that delays consideration and recognition of the syndrome.
Transient Neonatal Diabetes Mellitus
TNDM typically presents within the first several days to weeks of life. Intrauterine growth retardation (IUGR) is commonly seen in affected individuals. The insulin dose requirement is often lower than that needed in PNDM. TNDM resolves at a median age of 12 weeks; however, approximately 50% of cases will ultimately relapse, typically during adolescence or young adulthood.
A total 70% of TNDM is caused by defects causing overexpression of paternally expressed genes in the imprinted region of chromosome 6q2414. Three known mechanisms can cause 6q24‐related TNDM, including paternal uniparental isodisomy of chromosome 6, paternally inherited duplication of 6q24 and maternal methylation defects14. Cases are usually sporadic, but paternal transmission can occur15. Various abnormalities leading to overexpression of paternally expressed genes in the 6q24 region result in TNDM. The exact mechanism by which 6q24 region abnormalities result in TNDM remains unclear, but there are two implicated genes in this region –ZAC and HYMAI (see Table 2). ZAC (Z finger protein that regulates apoptosis and cell cycle arrest; also known as PLAG1– pleomorphic adenoma gene‐like 1) is a transcriptional regulator of the type 1 receptor for pituitary adenylate cyclase‐activating polypeptide, which is important in insulin secretion regulation16–18. The function of the HYMAI (hydatiform mole‐associated and imprinted transcript) is unknown17,19. The TNDM29 transgenic mouse overexpresses an artificial chromosome that contains the human ZAC and HYMAI genes, recapitulating a phenotype similar to 6q24 TNDM. Paternal, but not maternal, transmission of the chromosome results in hyperglycemia in 2–8 day‐old mice, which then remits with normal glucose tolerance seen from 1.5–2 months‐of‐age. At 6–10 months, the mice show normal fasting glycemia, but hyperglycemia after glucose challenge. In contrast, approximately half of patients with 6q24‐related TNDM experience relapse of diabetes during adolescence or young adulthood. It might be that the insulin resistance and increased insulin requirements seen during puberty and in pregnancy are a trigger for relapse of diabetes14. As aforementioned, during remission, most individuals do not show impairment in β‐cell function in the fasting state. Insulin secretory response to intravenous glucose loading might be subnormal or abnormal in those destined to have relapse of diabetes13. Studies in the mouse model suggest that altered β‐cell mass and insulin content might play a role; however, the molecular pathophysiology has not yet been fully delineated20. TNDM caused by 6q24 abnormalities is virtually always associated with IUGR. Additional distinguishing features might include umbilical hernia and macroglossia, which were present in 9 and 30% of subjects, respectively, with 6q24‐related TNDM in one series21.
Table 2. Neonatal diabetes mellitus.
| Gene or syndrome | Affected protein | Inheritance | Additional features | Treatment |
|---|---|---|---|---|
| Transient NDM | ||||
| 6q24 abnormalities ZAC (PLAG1), HYMA1 implicated | Implicated proteins: pleomorphic adenoma gene‐like 1, hydatiform mole‐associated and imprinted transcript | Spontaneous, AD | Low birthweight, macroglossia, umbilical hernia | Insulin, relapsed cases might respond to sulfonylureas and other oral medications |
| ABCC8 | SUR1 | Spontaneous, AD | Low birthweight | Responsive to sulfonylureas |
| KCNJ11 | Kir6.2 | Spontaneous, AD | Low birthweight, developmental delay; seizures | Responsive to sulfonylureas |
| INS | (Pro)insulin hormone | Recessive | Low birthweight | Insulin |
| Permanent NDM | ||||
| KCNJ11 | Kir6.2 | Spontaneous, AD | Low birthweight, developmental delay; seizures | Responsive to sulfonylureas |
| ABCC8 | SUR1 | Spontaneous, AD, AR | Low birthweight | Responsive to sulfonylureas |
| INS | (Pro)insulin hormone | Spontaneous, AD, AR | Low birthweight | Insulin |
| GCK | Glucokinase | AR | Low birthweight, parents have GCK MODY (MODY2) | Insulin ± oral therapies |
| Syndromic NDM | ||||
| FOXP3, IPEX syndrome | Forkhead box P3 | X‐linked | Autoimmune diabetes and thyroid disease, immune dysregulation, enteropathy, exfoliative dermatitis, often early demise | Insulin |
| EIF2AK3, Wolcott–Rallison Syndrome | Eukaryotic translation initiation factor 2‐alpha kinase 3 | AR | Epiphyseal dysplasia, exocrine pancreatic insufficiency | Insulin |
| PDX1 (IPF1) | Pancreatic duodenal homeobox 1 (insulin promoter factor 1) | AR | Agenesis of the pancreas, parents have IPF1 MODY (MODY4) | Insulin |
| PTF1A | Pancreas transcription factor 1A | AR | Pancreatic hypoplasia, cerebellar hypoplasia | Insulin |
| GLIS3 | Glioma‐associated oncogene similar 3 | AR | Congenital hypothyroidism, glaucoma, kidney cysts, hepatic fibrosis | Insulin |
| NEUROD1 | Neurogenic differentiation 1 | AR | Cerebellar hypoplasia, deafness | Insulin |
| HNF1B | Hepatocyte nuclear factor 1‐beta | Spontaneous, AD | Renal and genitourinary abnormalities, atrophy of the pancreas | Insulin |
AD, autosomal dominant; AR, autosomal recessive; NDM, neonatal diabetes mellitus.
Mutations in the adenosine triphosphate (ATP)‐sensitive potassium channel account for approximately 25% of TNDM, but are more commonly seen in cases of PNDM and will be discussed in further detail later. TNDM caused by either KCNJ11 or ABCC8 mutations cause a clinical picture that can be indistinguishable from 6q24‐related TNDM, although birthweight is typically lower in 6q24 TNDM and diabetes onset occurs at an earlier age, as does remission21. However, overlap occurs in all parameters between 6q24 and KCNJ11 and ABCC8‐related TNDM, making a genetic diagnosis necessary. Spontaneous and autosomal dominant HNF1B mutations, a well‐recognized cause of MODY, have rarely also been found to cause syndromic TNDM and are discussed later22–25. Recently, recessive mutations in the INS gene encoding insulin were found to be a cause of both PNDM and TNDM, with TNDM described in five patients. Diabetes occurs due to decreased insulin biosynthesis as a result of homozygous mutations. As expected, birthweight was markedly reduced, and median age at diagnosis was 1 week. Remission occurred at a median age of 26 weeks. Relapse occurred in one of the five patients at an age of 1 year26.
Permanent Neonatal Diabetes Mellitus
Permanent NDM is most commonly a result of mutations in the β‐cell ATP‐sensitive potassium channel (KATP) caused by heterozygous activating mutations in KCNJ11 and ABCC8, which account for 31 and 10% of all PNDM cases, respectively, in the Exeter series27,28. Mutations in the insulin gene itself cause 12% of PNDM. KCNJ11 encodes Kir6.2 (inwardly rectifying potassium channel) and ABCC8 encodes SUR1 (the type 1 subunit of the sulfonylurea receptor, a member of the ATP‐binding cassette transporter family), the two subunits of the KATP channel. The KATP channel is an octameric structure composed of four SUR1 subunits surrounding four Kir6.2 subunits that form a central pore. This channel links glucose metabolism to insulin secretion by closing in response to ATP. Glucose uptake into the β‐cell results in glycolysis leading to ATP generation. The increased ATP/adenosine diphosphate ratio causes closure of the KATP channel, preventing potassium efflux, which leads to depolarization of the β‐cell membrane. Voltage‐gated calcium channels then open, allowing influx of calcium, which prompts exocytosis of insulin‐containing granules from the β‐cell. In most cases, activating mutations in KCNJ11 or ABCC8 appear to render the KATP channel relatively less sensitive to ATP, leaving more channels in an open state after increased glucose. The net result is the failure of insulin response to hyperglycemia leading to NDM.
KCNJ11 Neonatal Diabetes Mellitus
Mutations in KCNJ11 are de novo in 80% of cases and inherited in an autosomal dominant pattern in the remaining cases. Diabetes presents from birth to 26 weeks‐of‐age with an average age at onset of 5 weeks. Low birthweight is common as a result of in utero insulin deficiency. KCNJ11 mutations result in decreased sensitivity of the Kir6.2 subunits to ATP, with failure of channel closure and consequently insufficient or absent insulin release from the β‐cell 27. In addition to the pancreas, Kir6.2 is also expressed in other tissues including neurons, brain and muscle. As a consequence, some patients with KCNJ11 mutations have neurological features with varying degrees of dysfunction and impairment, though the majority have isolated NDM (80% of KCNJ11 cases). The neurological features in their most severe form comprise a syndrome known as DEND (developmental delay, epilepsy and neonatal diabetes). Mild facial dysmorphisms have been seen in patients with DEND, including prominence of the metopic suture, bilateral ptosis and down‐turned mouth. Additionally, hypsarrhythmia has been a described electroencephalogram (EEG) finding in several patients with the DEND syndrome27,29. When milder neurodevelopmental perturbations occur without seizure disorder, this is termed intermediate DEND (iDEND). The V59M mutation is most frequently associated with iDEND, but several other mutations, including ones at the R201 residue, seem to be associated with learning disorders. Patients may also show hypotonia, muscle weakness and balance problems29. Several patients have hyperactivity and an attention‐deficit hyperactivity disorder (ADHD)‐like picture. Recent work with a mouse model expressing the V59M mutation selectively in brain or muscle tissue suggests that the muscle weakness seen in DEND cases is a result of malfunction of the KATP channel in the central nervous system, rather than in the peripheral nerves or myocytes30.
ABCC8 Neonatal Diabetes Mellitus
ABCC8‐related PNDM can be caused by dominant activating mutations, as well as recessive activating mutations and compound heterozygosity for activating and inactivating mutations of the SUR1 subunits of the KATP channel31. The majority of ABCC8 mutations are de novo; however, dominant and recessive inheritance occurs as well. Nucleotide binding and/or hydrolysis on SUR1 leads to opening of the KATP channel, whereas binding of adenine nucleotides to Kir6.2 closes the channel. Together, the SUR1 and Kir6.2 subunits of the KATP channel determine the low open channel probability; P0. ABCC8 mutations lead to channels with an increased P0. When the KATP channel remains open, glucose‐dependent insulin secretion fails to occur, resulting in diabetes28,31. ABCC8 mutations can lead to TNDM or PNDM, but more frequently cause TNDM. In contrast to KCNJ11‐related PNDM, the DEND/iDEND syndrome occurs more rarely with ABCC8 mutations. This might reflect differences in SUR1 vs SUR2 expression in the brain, but the precise involved region is unknown. Neurological complications, such as minor dystonia or visual and spatial dyspraxia, have been shown to occur in some patients with TNDM caused by KCNJ11 or ABCC8 mutations; however, this does not appear to be a consistent feature28.
INS Neonatal Diabetes Mellitus
Dominant mutations in INS, encoding the preproinsulin molecule, cause PNDM as well as MODY and autoantibody‐negative type 1 diabetes32–36. PNDM caused by dominant INS mutations seems to result from misfolding of proinsulin molecules, which then accumulate in the endoplasmic reticulum (ER) causing ER stress, and lead to β‐cell dysfunction and apoptosis as a result of the unfolded protein response32,36–40. Spontaneous mutations occur in 80% of cases. Patients present at a median age of 9 weeks‐old (generally somewhat older than patients with KATP channel mutations) and require treatment with insulin therapy. Birthweight is reduced, as expected, as a result of decreased In utero insulin secretion. Recessive mutations in INS can also cause PNDM as a result of decreased insulin biosynthesis. The phenotype is more severe than with dominant mutations, with earlier presentation and lower birthweight26. Two mouse models, the Akita and Munich strains, have non‐obese diabetes as a result of insulin gene mutations41–43.
GCK Neonatal Diabetes Mellitus
Homozygous or compound heterozygous mutations in GCK encoding glucokinase cause complete glucokinase deficiency leading to PNDM, whereas autosomal dominant glucokinase mutations lead to stable, non‐progressive mild hyperglycemia that rarely requires treatment (MODY2)44,45. This molecular etiology of NDM should be suspected in instances of known consanguinity or in families with a history of glucose intolerance or mild diabetes. Patients with glucokinase‐related PNDM are treated with insulin, but there has been a report of sulfonylureas added to insulin therapy resulting in increased basal and stimulated insulin secretion46. Additionally, glucokinase activators might eventually be the treatment of choice in glucokinase PNDM and would be another striking example of applied pharmacogenetics in NDM treatment47–49.
Syndromic Neonatal Diabetes Mellitus
The constellation of abnormalities seen in syndromes with PNDM as a feature can help to direct appropriate genetic analysis. Syndromes associated with PNDM include IPEX syndrome, Wolcott–Rallison syndrome, Wolfram syndrome and syndromes associated with mutations in the genes PDX/IPF1, PTF1A, GLIS3, NEUROD1 and HNF1B.23,24,50,51 IPEX is an X‐linked syndrome that presents in infancy with autoimmune endocrinopathy (diabetes and thyroid disease), enteropathy and exfoliative dermatitis. It is caused by mutations in the FOXP3 gene encoding forkhead box P3, which is important in regulatory T cell function. The enteropathy usually causes intractable diarrhea with villous atrophy and clinically presents as failure to thrive. Most children affected by this syndrome die in the first year of life from overwhelming sepsis as a result of the immune dysfunction. However, milder cases are being uncovered by current sequencing approaches to NDM. Thus, syndromic monogenic NDM should be considered when KATP channelopathies and other mutations have been excluded12. Wolcott–Rallison syndrome is an autosomal recessive disorder caused by mutations in the EIF2AK3 gene encoding eukaryotic translation initiation factor 2‐alpha kinase 3, leading to NDM and spondyloepiphyseal dysplasia. A mouse model of this disease suggests that EIF2AK3 mutations result in an accumulation of misfolded proteins in the endoplasmic reticulum, leading to increased apoptosis after the unfolded protein response52,53. Affected patients have been shown to have hypoplastic pancreatic islets, and exocrine pancreatic insufficiency can rarely occur in addition to DM. Patients might also have hepatomegaly, renal failure, severe cognitive delays and early death54. Wolfram syndrome is an autosomal recessive syndrome characterized by diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD), as well as neurodegenerative disease. Mutations in the WFS1 gene encoding wolframin have been identified in 90% of patients with a clinical diagnosis of Wolfram syndrome. Diabetes mellitus is typically the first feature to appear in this syndrome and occurs at a median age of 6 years, but can be seen as early as 3 weeks‐of‐age. Diabetes is due to insulinopenia as a result of β‐cell degeneration, and is characterized by negative autoantibodies and frequent occurrence of HLA‐DR2 haplotype55. PDX1 mutations encoding pancreatic duodenal homeobox 1 (also termed IPF1, encoding insulin promoter factor 1) in the heterozygous state are a cause of MODY4 and in the recessive state result in agenesis of the pancreas, leading to endocrine and exocrine pancreatic dysfunction and PNDM56,57. Milder phenotypes of PDX1‐related NDM have been recently described58. Homozygous mutations in the gene encoding pancreas transcription factor 1A (PTF1A) leads to a syndrome of NDM as a result of pancreatic hypoplasia and cerebellar hypoplasia59,60. A mouse model suggests that reduced gene expression levels impair pancreas specification and growth and impact β‐cell mass61. Homozygous mutations in GLIS3 (glioma‐associated oncogene similar family zinc finger protein 3) cause a syndrome of NDM, hypothyroidism, congenital glaucoma, kidney cysts and hepatic fibrosis62,63. Recently, mutations in NEUROD1 encoding neurogenic differentiation 1 have been shown to cause PNDM. Heterozygous mutations are a cause of MODY6. Two patients were described to have NEUROD1‐related PNDM associated with severe cerebellar hypoplasia, sensorineural deafness, visual impairment and learning difficulties51. Interestingly, three of the four heterozygous parents in their report had normal glucose tolerance. NEUROD1 is known to play an important role in the regulation of insulin gene transcription, and the clinical findings in NEUROD1 NDM highlight its important role in central nervous system development as well51,64. Heterozygous mutations in HNF1B are a rare cause of syndromic transient NDM with early relapse23–25. As in MODY5 cases, HNF1B‐related NDM is associated with renal and genitourinary abnormalities, as well as atrophy of the pancreas. Edghill et al.23 described NDM in one patient associated with pancreatic atrophy, mild exocrine insufficiency and low birthweight with remission of DM at 17 days and subsequent relapse at 8 years. There are other extremely rare syndromes described in single families with NDM as a feature where the underlying genetic cause is unknown65–67.
Pharmacogenetics: Sulfonylurea Treatment in Neonatal Diabetes Mellitus
As stated earlier, activating mutations in Kir6.2 and SUR1 cause the KATP channel to persist in an open state as a result of unresponsiveness of the channel to increased intracellular ATP. Sulfonylureas bind to the SUR1 subunits and close the KATP channel in an ATP‐independent manner. A majority of patients with KCNJ11 (approximately 90%) and ABCC8 mutations (85% in one series) are able to transition from insulin to oral sulfonylureas with better glycemic control68–71. Patients treated with sulfonylureas have been shown to secrete insulin in a meal‐dependent manner. The mechanisms underlying this are unclear, but might be related, in part, to improved response of the β‐cells to incretins. Sulfonylurea‐induced insulin secretion is increased more by oral glucose or mixed meal than by intravenous glucose68. Episodes of hypoglycemia appear to be less frequent with sulfonylurea therapy than insulin, despite improved glycemic control with transition to sulfonylureas, as evidenced by improved glycated hemoglobin68,72.
Some patients with intermediate DEND syndrome have shown improvement of their neurological abnormalities with sulfonylurea treatment73–76. The V59M mouse model exploring the role of KCNJ11 mutations in the iDEND syndrome suggests that this improvement might be a result of sulfonylurea action on Kir6.2 subunits within the central nervous system. Thus, those sulfonylureas with the highest blood–brain barrier permeability might result in the best outcomes for patients with iDEND and DEND30. Sulfonylureas can be moderately specific to SUR1 or bind both SUR1 and SUR2 subunits in vitro. SUR1 subunits are expressed in the pancreatic β‐ and α‐cells and in the brain and peripheral nerves, whereas SUR2 subunits are expressed in skeletal, cardiac and smooth muscle. Given that the DEND and iDEND syndromes appear to be entirely mediated by subunits located in the central nervous system, an additional implication of the findings from the V59 mouse model is that SUR1‐specific sulfonylureas might be favorable over SUR1/SUR2 sulfonylureas, although the degree of selectivity in vivo is uncertain and might be insignificant at relevant doses. In type 2 diabetes, it is thought that SUR2 receptor binding might underlie possible adverse cardiovascular outcomes seen with sulfonylurea use. Whether this has anything in common with non‐obese individuals without pre‐existing insulin resistance and atherosclerosis is unknown, but several individuals on prolonged, high‐dose sulfonylurea usage have done well without adverse cardiovascular events77. It is also important to note that of the limited side‐effects described with NDM‐sulfonylurea use69,78,79, cardiovascular adverse outcomes have not been reported in the context of sulfonylurea treatment of NDM, but are a theoretical concern underscoring the importance of following such patients with national registries80.
A small fraction of patients with KCNJ11 and ABCC8 mutations are unable to transition from insulin to sulfonylureas. The age at transition appears to play a role, as does the nature of the specific mutation. In a series of 49 patients with KCNJ11 mutations attempting transfer from insulin to oral sulfonylureas, five patients (10%) were unsuccessful. In two patients who were unsuccessful in transitioning from insulin to sulfonylureas at ages 27 and 43 years, their children, 3‐ and 11‐months‐old, respectively, were successfully transitioned, implicating duration of disease rather than the particular mutations (G53R and R201C). In the remaining three patients with Q52R, I296L and L164P mutations, lack of successful transfer was correlated with poor in vitro response of KATP channels containing corresponding Kir6.2 mutations expressed in xenopus oocytes to tolbutamide block. Such mutations affecting channel kinetics were less responsive to sulfonylureas than mutations affecting ATP sensitivity without affecting channel kinetics. The proportion of patients with neurological features was also higher in those unsuccessful at transitioning than in those who were able to transition from insulin to sulfonylureas (80% vs 14%), suggesting a correlation between severe phenotype and mutations expected to be less responsive to sulfonylureas68.
In addition to treatment of KATP channel PNDM, preliminary data suggest that relapsed TNDM as a result of KCNJ11 and ABCC8, as well as 6q24, might respond to sulfonylurea therapy (in combination with other oral medications in 6q24 relapsed diabetes)21,28,81. It should be emphasized that the pathophysiology of 6q24 mutations is not entirely clear; thus, the most appropriate therapy remains unclear.
Genetic Implications
The risk of recurrence differs between the various etiologies of transient and permanent forms of NDM, with dominant mutations conferring a 50% risk of transmission to children and recessive mutations conferring a 25% transmission risk. Thus, genetic counseling should be provided to families with TNDM and PNDM after a genetic diagnosis is established in the propositus and ideally after the parents have been tested for the mutation as well.
KATP Channel Neonatal Diabetes Mellitus
Most cases of NDM are sporadic resulting from de novo mutations. However, in some of these cases, there is a recurrence risk to a second child as a result of germline mosaicism82. In such cases, the mutation, though present in the gonads, might not be detectable in the blood of parents. Parents must be made aware of this possibility in apparently spontaneous cases. Autosomal recessive inheritance can occur in ABCC8 mutations, with a 25% risk of recurrence in each subsequent pregnancy. Additionally, compound heterozygosity for activating and inactivating ABCC8 mutations could result in a family with NDM as well as hyperinsulinemia of infancy31.
GCK Neonatal Diabetes Mellitus
Glucokinase NDM is autosomal recessive or due to compound heterozygosity; both carry a 25% recurrence risk for NDM. Offspring not affected with NDM could have a MODY phenotype of glucokinase DM (MODY2) and the parents would be expected to have glucokinase MODY.
INS Neonatal Diabetes Mellitus
Most dominant activating INS mutations are sporadic, but some will be inherited in an autosomal dominant pattern. Recessive INS mutations will have a 25% chance of recurrence.
HNF1B Neonatal Diabetes Mellitus
The two described cases of NDM as a result of HNF1B mutations were spontaneous and due to germline mosaicism23,24. Germline mosaicism would be expected to present a risk of recurrence in offspring. In the described case of NDM with HNF1B mutation (S148W) from germline mosaicism, a second sibling had the same mutation, but with a phenotype of neonatal polycystic, dysplastic kidneys without diabetes. As the phenotype of individuals carrying HNF1B mutations can vary greatly, a pedigree with non‐diabetes‐related renal disease, as well as neonatal and/or MODY diabetes, should prompt screening for HNF1B mutations22–24.
6q24 Imprinting Abnormalities
Paternal uniparental disomy accounts for 50% of sporadic cases, and methylation defects also cause sporadic TNDM14. Familial cases result from paternal duplication of 6q2415. In these cases, males have a 50% chance of passing on the duplication resulting in TNDM in their children. However, if a woman passes on this duplication (50% chance in each pregnancy), her children will not have TNDM, but her male children will have a 50% chance of transmitting TNDM to their children.
Syndromic Neonatal Diabetes Mellitus
With the exception of IPEX and HNF1B‐related diabetes, the syndromic causes of NDM are autosomal recessive, carrying a 25% recurrence risk in subsequent children. Mothers carrying a FOXP3 mutation will have a 50% chance of transmitting the mutation to each child. All affected males will have IPEX; affected daughters will be carriers. Fathers with IPEX syndrome will have no affected sons and all daughters will be carriers.
Approach to Genetic Testing for Neonatal Diabetes Mellitus
Given the enormous genetic and treatment implications that a diagnosis of monogenic NDM carries, all patients with diabetes onset before 6 months‐of‐age should undergo molecular genetic analysis for a monogenic etiology83. Recent assessment of the cost‐effectiveness of routine molecular genetic screening for KATP channel mutations in all children diagnosed before 6 months‐of‐age showed that such a strategy is cost saving84. This is due to the high rate of successful transition from insulin to sulfonylureas in NDM as a result of KATP channel mutations, with lower costs compared to insulin and insulin delivery equipment. Cost savings are also due to improved health outcomes and decreased complications expected from the improvement in glycated hemoglobin seen after transition. Although other NDM etiologies will continue to require insulin therapy, the prevalence of KATP channel mutations in all PNDM cases, as well as the promising response of relapsed TNDM caused by KATP channel mutations and 6q24 abnormalities to sulfonylurea therapy with additional oral medications as needed, suggest that establishing a molecular genetic diagnosis in all patients diagnosed with diabetes before 6 months‐of‐age might have societal cost benefits.
The presence of pancreatic autoantibodies should not dissuade genetic testing in diagnoses under 6 months‐of‐age, as pancreatic autoantibodies have been described in a few patients with molecular diagnoses of monogenic diabetes5,11,85. Although this scenario raises a question of so‐called ‘double diabetes’, data suggest that such patients have monogenic diabetes in isolation and the autoantibodies are likely a reflection of increased β‐cell apoptosis with resultant increased exposure of β‐cell antigens to antigen‐presenting cells, or might not be causally‐related at all. This is supported by patients with monogenic NDM and pancreatic autoantibodies who were successfully transitioned from insulin to sulfonylurea therapy with improved glycemic control, which would not be possible in type 1A diabetes5,11,86. In cases of proven monogenic NDM with pancreatic autoantibodies, additional investigation, such as HLA typing, might be prudent before attempting insulin‐to‐sulfonylurea transition to exclude type 1A diabetes, with anticipated progression to absolute insulinopenia. Transitions in these unclear situations should occur in the hospital setting under the guidance of physicians with experience in transitioning patients from insulin to sulfonylureas. All transitions should include the input of experienced physicians, but select unambiguous cases can occur in the outpatient setting80.
As there are no clinical features to reliably distinguish TNDM and PNDM at the time of presentation, in non‐syndromic patients, screening should begin with KCNJ11 and 6q24 abnormalities and, if negative, should proceed with testing for INS and ABCC8 mutations (see Figure 1)83. A similar approach should be taken with PNDM with or without neurological features, with the exclusion of 6q24 testing. In suspected consanguinity, autosomal recessive causes of NDM should be assessed for, most of which lead to syndromic NDM with additional features that help direct testing. Glucokinase NDM should be assessed in the setting of consanguinity and/or a family history in one or both parents of impaired fasting glucose, mild type diabetes or known glucokinase MODY diagnosis. Clearly, if a patient presents with features consistent with a syndrome, testing for the relevant mutation should be carried out first. However, if negative, testing for the most prevalent causes in descending order should be carried out. Given the possibility of a mild IPEX presentation12, it is also reasonable to screen for this disorder in all males diagnosed with NDM before 6 months‐of‐age, particularly if pancreatic autoantibodies are present. In known TNDM, 6q24 should be assessed first, as it causes 70% of all TNDM cases. If 6q24 screening is negative, testing for mutations in ABCC8 and KCNJ11 should be carried out. If a cause is not identified, INS and HNF1B mutations are a consideration.
Figure 1.
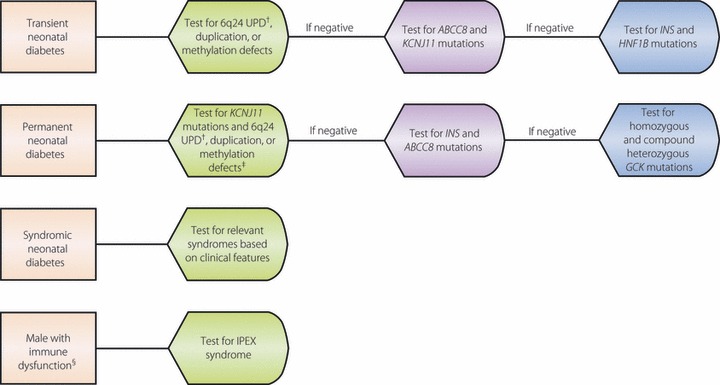
Approach to genetic testing for neonatal diabetes. †UPD, uniparental isodisomy. ‡If it is unclear if diabetes is permanent or transient, testing for both KCNJ11, the most common cause of permanent neonatal diabetes, and 6q24 chromosome abnormalities, the most common cause of transient neonatal diabetes, should be pursued. §Immune dysregulation, polyendocrinopathy, enteropathy, X‐linked (IPEX) should be considered as a potential cause of syndromic neonatal diabetes in males when associated with immune dysregulation, enteropathy and autoimmune endocrinopathies. It additionally can cause antibody‐positive neonatal diabetes.
Conclusions
Because diabetes presenting within the first 6 months of life is unlikely to represent type 1 diabetes, but rather, diabetes as a result of a single gene mutation, molecular genetic analysis of this group of patients should be seen as compulsory. Additionally, individuals with a diagnosis of diabetes after 6 months‐of‐age should also receive genetic analysis if their diabetes presentation is atypical for type 1 diabetes, has features consistent with a known monogenic cause, or if there are additional affected family members with a history of NDM or a pattern that suggests Mendelian inheritance. The application of pharmacogenetics to KATP channel‐related NDM allows for transition from insulin injections to sulfonylureas in most patients. The life‐changing impact of this transition for patients and their families cannot be overstated. It might be that in the near future glucokinase activators and agents ameliorating ER stress will offer the same life‐changing therapeutic option to patients with GCK and INS‐related diabetes, respectively. Of equal importance to these therapeutic considerations are the genetic implications of a diagnosis of monogenic NDM for parents, siblings, offspring and the more distant relatives of an identified subject. Families with a molecular genetic diagnosis of TNDM or PNDM should receive genetic counseling to ensure full understanding of the recurrence risk of NDM, which varies, based on molecular etiology. As monogenic etiologies of NDM are uncovered with increasing frequency, we will improve our methods to identify affected individuals and test at‐risk family members, find more applications for pharmacogenetics resulting in improved therapy and outcomes, and we will learn more about the important regulators of pancreatic development, β‐cell mass and regeneration, and insulin processing and secretion. This knowledge will not only forward the field of monogenic diabetes, but will undoubtedly have an impact on polygenic forms of diabetes as well.
Future Directions
A large number of patients with phenotypic monogenic diabetes remain without a genetic diagnosis after DNA sequencing of relevant genes. Using cutting‐edge technologies, such as whole exome and even whole genome, sequencing could help to identify gene mutations in a substantial portion of individuals who are likely to have a monogenic cause of their diabetes. It is likely that in the years to come such technologies will be inexpensive and sensitive enough to become first‐line diagnostic tools to establish the molecular genetic causes of monogenic diabetes. Clearly, improvement in quality control measures and establishment of systems to handle the volume of data generated will be important initial steps toward wider application of these technologies. The age that most appropriately defines ‘neonatal diabetes’ is questionable. As cases accumulate with diagnoses after 6 months‐of‐age, after infancy and even into early childhood, as notably illustrated by diagnoses of KCNJ11‐related diabetes at 22‐months‐old (Greeley SA, Worrell H, Naylor R, Paz V, Philipson LH, Bell GI, 2010, unpublished data) and at 4‐years‐old87, the age at which to consider genetic testing will be extended. HLA haplotyping and antibody testing will help to guide the selection of appropriate older cases for screening. By following these patients longitudinally through monogenic diabetes registries, new definitions of what is currently termed ‘neonatal diabetes’ will emerge.
Acknowledgement
The authors declare that there is no conflict of interest associated with this manuscript.
References
- 1.Concannon P, Rich SS, Nepom GT. Genetics of type 1A diabetes. N Engl J Med 2009; 360: 1646–1654 [DOI] [PubMed] [Google Scholar]
- 2.Voight BF, Scott LJ, Steinthorsdottir V, et al. Twelve type 2 diabetes susceptibility loci identified through large‐scale association analysis. Nat Genet 2010; 42: 579–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murphy R, Ellard S, Hattersley AT. Clinical implications of a molecular genetic classification of monogenic beta‐cell diabetes. Nat Clin Pract Endocrinol Metab 2008; 4: 200–213 [DOI] [PubMed] [Google Scholar]
- 4.Edghill EL, Hattersley AT. Genetic disorders of the pancreatic beta cell and diabetes (permanent neonatal diabetes and maturity‐onset diabetes of the young) In: Seino S, Bell GI (eds). Pancreatic Beta Cell in Health and Disease. Springer, Japan, 2008; 389–420 [Google Scholar]
- 5.Stoy J, Greeley SAW, Paz VP, et al. Diagnosis and treatment of neonatal diabetes: an United States experience. Pediatr Diabetes 2008; 9: 450–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greeley SA, Tucker SE, Naylor RN, et al. Neonatal diabetes mellitus: a model for personalized medicine. Trends Endocrinol Metab 2010; 21: 464–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shield JP, Gardner RJ, Wadsworth EJ, et al. Aetiology and genetic basis of neonatal diabetes. Arch Dis Child Fetal Neonatal Ed 1997; 76: F39–F42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marquis E, Le Monnier de Gouville I, Bouvattier C, et al. HLA‐DRB 1 and DQB1 genotypes in patients with insulin‐dependent neonatal diabetes mellitus. A study of 13 cases. Tissue Antigens 2000; 56: 217–222 [DOI] [PubMed] [Google Scholar]
- 9.Iafusco D, Stazi MA, Cotichini R, et al. Permanent diabetes mellitus in the first year of life. Diabetologia 2002; 45: 798–804 [DOI] [PubMed] [Google Scholar]
- 10.Edghill El, Dix RJ, Flanagan SE, et al. HLA genotyping supports a nonautoimmune etiology in patients diagnosed with diabetes under the age of 6 months. Diabetes 2006; 55: 1895–1898 [DOI] [PubMed] [Google Scholar]
- 11.Gach A, Wyka K, Pietrzak I, et al. Neonatal diabetes in a child positive for islet cell antibodies at onset and Kir6.2 activating mutation. Diabetes Res Clin Pract 2009; 86: e25–e27 [DOI] [PubMed] [Google Scholar]
- 12.Rubio‐Cabezas O, Minton JA, Caswell R, et al. Clinical heterogeneity in patients with FOXP3 mutations presenting with permanent neonatal diabetes. Diabetes Care 2009; 32: 111–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shield JP, Temple IK, Sabin M, et al. An assessment of pancreatic endocrine function and insulin sensitivity in patients with transient neonatal diabetes in remission. Arch Dis Child Fetal Neonatal Ed 2004; 89: F341–F343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Temple IK, Gardner RJ, Mackay DJ, et al. Transient neonatal diabetes mellitus: widening our understanding of the aetiopathogenesis of diabetes. Diabetes 2000; 49: 1359–1366 [DOI] [PubMed] [Google Scholar]
- 15.Temple IK, James RS, Crolla JA, et al. An imprinted gene(s) for diabetes? Nat Genet 1995; 9: 110–112 [DOI] [PubMed] [Google Scholar]
- 16.Kamiya M, Judson H, Okazaki Y, et al. The cell cycle control gene ZAC/PLAG1 is imprinted‐a strong candidate gene for transient neonatal diabetes. Hum Mol Genet 2000; 9: 453–460 [DOI] [PubMed] [Google Scholar]
- 17.Temple IK, Shield JP. Transient neonatal diabetes, a disorder of imprinting. J Med Genet 2002; 39: 872–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barz T, Hoffmann A, Panhuysen M, et al. Peroxisome proliferator‐activated receptor gamma is a Zac target gene mediating Zac antiproliferation. Cancer Res 2006; 66: 11975–11982 [DOI] [PubMed] [Google Scholar]
- 19.Arima T, Drewell RA, Arney KL, et al. A conserved imprinting control region at the HYMAI/ZAC domain is implicated in transient neonatal diabetes mellitus. Hum Mol Genet 2001; 10: 1475–1483 [DOI] [PubMed] [Google Scholar]
- 20.Ma D, Shield JP, Dean W, et al. Impaired glucose homeostasis in transgenic mice expressing the human transient neonatal diabetes mellitus locus, TNDM. J Clin Invest 2004; 114: 339–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flanagan SE, Patch AM, Mackay DJG, et al. Mutations in ATP‐sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes 2007; 56: 1930–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edghill EL, Bingham C, Ellard S, et al. Mutations in hepatocyte nuclear factor‐1beta and their related phenotypes. J Med Genet 2006; 43: 84–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edghill EL, Bingham C, Slingerland AS, et al. Hepatocyte nuclear factor‐1 beta mutations cause neonatal diabetes and intrauterine growth retardation: support for a critical role of HNF‐1beta in human pancreatic development. Diabet Med 2006; 23: 1301–1306 [DOI] [PubMed] [Google Scholar]
- 24.Yorifuji T, Kurokawa K, Mamada M, et al. Neonatal diabetes mellitus and neonatal polycystic, dysplastic kidneys: Phenotypically discordant recurrence of a mutation in the hepatocyte nuclear factor‐1beta gene due to germline mosaicism. J Clin Endocrinol Metab 2004; 89: 2905–2908 [DOI] [PubMed] [Google Scholar]
- 25.D’Amato E, Lorini R. Letter Re: neonatal diabetes mellitus and mutation in the HNF‐1beta gene. J Clin Endocrinol Metab 2005; 90: 5906–5907 [DOI] [PubMed] [Google Scholar]
- 26.Garin I, Edghill EL, Akerman I, et al. Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. PNAS 2010; 107: 3105–3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gloyn AL, Pearson ER, Antcliff JF, et al. Activating mutations in the gene encoding the ATP‐sensitive potassium‐channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med 2004; 350: 1838–1849 [DOI] [PubMed] [Google Scholar]
- 28.Babenko AP, Polak M, Cave H, et al. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med 2006; 355: 456–466 [DOI] [PubMed] [Google Scholar]
- 29.Gloyn AL, Diatloff‐Zito C, Edghill EL, et al. KCNJ11 activating mutations are associated with developmental delay, epilepsy, and neonatal diabetes syndrome and other neurological features. Eur J Hum Genet 2006; 14: 824–830 [DOI] [PubMed] [Google Scholar]
- 30.Clark RH, McTaggart JS, Webster R, et al. Muscle dysfunction caused by a KATP channel mutation in neonatal diabetes in neuronal in origin. Science 2010; 329: 458–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ellard S, Flanagan SE, Girard CA, et al. Permanent neonatal diabetes caused by dominant, recessive, or compound heterozygous SUR1 mutations with opposite functional effects. Am J Hum Genet 2007; 81: 375–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Støy J, Edghill EL, Flanagan SE, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. PNAS 2007; 104: 15040–15044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Polak M, Dechaume A, Cave H, et al. Heterozygous missense mutations in the insulin gene are linked to permanent diabetes appearing in the neonatal period or in early infancy: a report from the French ND (Neonatal Diabetes) study group. Diabetes 2008; 57: 1115–1119 [DOI] [PubMed] [Google Scholar]
- 34.Edghill EL, Flanagan SE, Patch AM, et al. Insulin mutation screening in 1,044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes 2008; 57: 1034–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Molven A, Ringdal M, Nordbo AM, et al. Mutations in the insulin gene can cause MODY and autoantibody‐negative type 1 diabetes. Diabetes 2008; 57: 1131–1135 [DOI] [PubMed] [Google Scholar]
- 36.Meur G, Simon A, Harun N, et al. Insulin gene mutations resulting in early‐onset diabetes: marked differences in clinical presentation, metabolic status, and pathogenic effect through endoplasmic reticulum retention. Diabetes 2010; 59: 653–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rajan S, Eames SC, Park SY, et al. In vitro processing and secretion of mutant insulin proteins that cause permanent neonatal diabetes. Am J Physiol Endocrinol Metab 2010; 298: E403–E410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park SY, Ye H, Steiner DF, et al. Mutant proinsulin proteins associated with neonatal diabetes are retained in the endoplasmic reticulum and not efficiently secreted. Biochem Biophys Res Commun 2010; 391: 1449–1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Colombo C, Porzio O, Liu M, et al. Seven mutations in the human insulin gene linked to permanent neonatal/infancy‐onset diabetes mellitus. J Clin Invest 2008; 118: 2148–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hodish I, Liu M, Rajpal G, et al. Misfolded proinsulin affects bystander proinsulin in neonatal diabetes. J Biol Chem 2010; 285: 685–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zuber C, Fan JY, Guhl B, et al. Misfolded proinsulin accumulates in expanded pre‐Golgi intermediates and endoplasmic reticulum subdomains in pancreatic beta cells of Akita mice. FASEB J 2004; 18: 917–919 [DOI] [PubMed] [Google Scholar]
- 42.Izumi T, Yokota‐Hashimoto H, Zhao S, et al. Dominant negative pathogenesis by mutant proinsulin in the Akita diabetic mouse. Diabetes 2003; 52: 409–416 [DOI] [PubMed] [Google Scholar]
- 43.Herbach N, Rathkolb B, Kemter E, et al. Dominant‐negative effects of a novel mutated Ins2 allele causes early‐onset diabetes and severe beta‐cell loss in Munich Ins2C95S mutant mice. Diabetes 2007; 56: 1268–1276 [DOI] [PubMed] [Google Scholar]
- 44.Velho G, Froguel P. Genetic, metabolic and clinical characteristics of maturity onset diabetes of the young. Eur J Endocrinol 1998; 138: 233–239 [DOI] [PubMed] [Google Scholar]
- 45.Njolstad PR, Oddmund S, Cuesta‐Munoz A, et al. Permanent neonatal diabetes mellitus due to glucokinase deficiency: an inborn error of the glucose/insulin signaling pathway. N Engl J Med 2001; 344: 1588–1592 11372010 [Google Scholar]
- 46.Turkkahraman D, Bircan I, Tribble ND, et al. Permanent neonatal diabetes mellitus caused by a novel homozygous (T168A) glucokinase (GCK) mutation; initial response to oral sulphonylurea therapy. J Pediatr 2008; 153: 122–126 [DOI] [PubMed] [Google Scholar]
- 47.Pal M. Medicinal chemistry approaches for glucokinase activation to treat type 2 diabetes. Curr Med Chem 2009; 16: 3858–3874 [DOI] [PubMed] [Google Scholar]
- 48.Haynes NE, Corbett WL, Bizzarro FT, et al. Discovery, structure‐activity relationships, pharmacokinetics, and efficacy of glucokinase activator (2R)‐3‐cyclopentyl‐2‐(4‐methanesulfonylphenyl)‐N‐thiazol‐2‐yl‐propionamide (RO0281675). J Med Chem 2010; 53: 3618–3625 [DOI] [PubMed] [Google Scholar]
- 49.Ohyama S, Takano H, Iino T, et al. A small‐molecule glucokinase activator lowers blood glucose in the sulfonylurea‐desensitized rat. Eur J Pharmacol 2010; 640: 250–256 [DOI] [PubMed] [Google Scholar]
- 50.Hattersley A, Bruining J, Shield J, et al. ISPAD Clinical Practice Consensus Guidelines 2006–2007. The diagnosis and management of monogenic diabetes in children. Pediatr Diabetes 2006; 7: 352–360 [DOI] [PubMed] [Google Scholar]
- 51.Rubio‐Cabezas O, Minton JA, Kantor I, et al. Homozygous mutations in NEUROD1 are responsible for a novel syndrome of permanent neonatal diabetes and neurological abnormalities. Diabetes 2010; 59: 2326–2331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harding HP, Zeng H, Zhang Y, et al. Diabetes mellitus and exocrine pancreatic dysfunction in perk −/− mice reveals a role for translational control in secretory cell survival. Mol Cell 2001; 7: 1153–1163 [DOI] [PubMed] [Google Scholar]
- 53.Zhang P, McGrath B, Li S, et al. The PERK eukaryotic initiation factor 2α kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol 2002; 22: 3864–3874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rubio‐Cabezas O, Patch AM, Minton JA, et al. Wolcott–Rallison syndrome is the most common genetic cause of permanent neonatal diabetes in consanguineous families. J Clin Endocrinol Metab 2009; 94: 4162–4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kumar S. Wolfram syndrome: important implications for pediatricians and pediatric endocrinologists. Pediatr Diabetes 2010; 11: 28–37 [DOI] [PubMed] [Google Scholar]
- 56.Melloul D, Tsur A, Zangen D. Pancreatic Duodenal Homeobox (PDX‐1) in health and disease. J Pediatr Endocrinol Metab 2002; 15: 1461–1472 [DOI] [PubMed] [Google Scholar]
- 57.Thomas IH, Saini NK, Adhikari A, et al. Neonatal diabetes mellitus with pancreatic agenesis in an infant with homozygous IPF‐1 Pro63fsX60 mutation. Pediatr Diabetes 2009; 10: 492–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nicolino M, Claiborn KC, Senée V, et al. A novel hypomorphic PDX1 mutation responsible for permanent neonatal diabetes with subclinical exocrine deficiency. Diabetes 2010; 59: 733–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sellick GS, Barker KT, Stolte‐Dijkstra I, et al. Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat Genet 2004; 36: 1301–1305 [DOI] [PubMed] [Google Scholar]
- 60.Tutak E, Satar M, Yapicioğlu H, et al. A Turkish newborn infant with cerebellar agenesis/neonatal diabetes mellitus and PTF1A mutation. Genet Couns 2009; 20: 147–152 [PubMed] [Google Scholar]
- 61.Fukuda A, Kawaguchi Y, Furuyama K, et al. Reduction of Ptf1a gene dosage causes pancreatic hypoplasia and diabetes in mice. Diabetes 2008; 57: 2421–2431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Watanabe N, Hiramatsu K, Miyamoto R, et al. A murine model of neonatal diabetes mellitus in Glis3‐deficient mice. FEBS Lett 2009; 583: 2108–2113 [DOI] [PubMed] [Google Scholar]
- 63.Senée V, Chelala C, Duchatelet S, et al. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat Genet 2006; 38: 682–687 [DOI] [PubMed] [Google Scholar]
- 64.Naya FJ, Stellrecht CM, Tsai MJ, et al. Tissue‐specific regulation of the insulin gene by a novel basic helix‐loop‐helix transcription factor. Genes Dev 1995; 9: 1009–1019 [DOI] [PubMed] [Google Scholar]
- 65.Taha D, Bardise J, Hegab A, et al. Neonatal diabetes mellitus because of pancreatic agenesis with dysmorphic features and recurrent bacterial infections. Pediatr Diabetes 2008; 9: 240–244 [DOI] [PubMed] [Google Scholar]
- 66.Chen R, Hussain K, Al‐Ali M, et al. Neonatal and late‐onset diabetes mellitus caused by failure of pancreatic development: report of 4 more cases and a review of the literature. Pediatrics 2008; 121: e1541–e1547 [DOI] [PubMed] [Google Scholar]
- 67.Balasubramanian M, Shield JP, Acerini CL, et al. Pancreatic hypoplasia presenting with neonatal diabetes mellitus in association with congenital heart defect and developmental delay. Am J Med Genet A 2010; 152A: 340–346 [DOI] [PubMed] [Google Scholar]
- 68.Pearson ER, Flechtner I, Njolstad PR, et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med 2006; 355: 467–477 [DOI] [PubMed] [Google Scholar]
- 69.Rafiq M, Flanagan SE, Patch AM, et al. Effective treatment with oral sulfonylureas in patients with diabetes due to sulfonylurea receptor 1 (SUR1) mutations. Diabetes Care 2008; 31: 204–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stanik J, Gasperikova D, Paskova M, et al. Prevalence of permanent neonatal diabetes in Slovakia and successful replacement of insulin with sulfonylurea therapy in KCNJ11 and ABCC8 mutation carriers. J Clin Endocrinol Metab 2007; 92: 1276–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Suzuki S, Makita Y, Mukai T, et al. Molecular basis of neonatal diabetes in Japanese patients. J Clin Endocrinol Metab 2007; 92: 3979–3985 [DOI] [PubMed] [Google Scholar]
- 72.Begum‐Hasan J, Polychronakos C, Brill H. Familial permanent neonatal diabetes with KCNJ11 mutation and the response to glyburide therapy – a three‐year follow‐up. J Pediatr Endocrinol Metab 2008; 21: 895–903 [DOI] [PubMed] [Google Scholar]
- 73.Slingerland AS, Hurkx W, Noordam K, et al. Sulphonylurea therapy improves cognition in a patient with the V59M KCNJ11 mutation. Diabet Med 2008; 25: 277–281 [DOI] [PubMed] [Google Scholar]
- 74.Koster JC, Cadario F, Peruzzi C, et al. The G53D mutation in Kir6.2 (KCNJ11) is associated with neonatal diabetes and motor dysfunction in adulthood that is improved with sulfonylurea therapy. J Clin Endocrinol Metab 2008; 93: 1054–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sumnik Z, Kolouskova S, Wales JK, et al. Sulphonylurea treatment does not improve psychomotor development in children with KCNJ11 mutations causing permanent neonatal diabetes mellitus accompanied by developmental delay and epilepsy (DEND syndrome). Diabet Med 2007; 24: 1176–1178 [DOI] [PubMed] [Google Scholar]
- 76.Mlynarski W, Tarasov AI, Gach A, et al. Sulfonylurea improves CNS function in a case of intermediate DEND syndrome caused by a mutation in KCNJ11. Nat Clin Pract Neurol 2007; 3: 640–645 [DOI] [PubMed] [Google Scholar]
- 77.UK Prospective Diabetes Study (UKPDS) Group . Intensive blood‐glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352: 837–853 [PubMed] [Google Scholar]
- 78.Codner E, Flanagan S, Ellard S, et al. High‐dose glibenclamide can replace insulin therapy despite transitory diarrhea in early‐onset diabetes caused by a novel R201L Kir6.2 mutation. Diabetes Care 2005; 28: 758–759 [DOI] [PubMed] [Google Scholar]
- 79.Kumaraguru J, Flanagan SE, Greeley SA, et al. Tooth discoloration in patients with neonatal diabetes after transfer onto glibenclamide: a previously unreported side effect. Diabetes Care 2009; 32: 1428–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Greeley SA, Tucker SE, Worrell HI, et al. Update in neonatal diabetes. Curr Opin Endocrinol Diabetes Obes 2010; 17: 13–19 [DOI] [PubMed] [Google Scholar]
- 81.Martín‐Frías M, Colino E, Pérez deNanclaresG, et al. Glibenclamide treatment in relapsed transient neonatal diabetes as a result of a KCNJ11 activating mutation (N48D). Diabet Med 2009; 26: 567–569 [DOI] [PubMed] [Google Scholar]
- 82.Gloyn AL, Cummings EA, Edghill EL. Permanent neonatal diabetes due to paternal germline mosaicism for an activating mutation of the KCNJ11 gene encoding the Kir6.2 subunit of the β‐cell potassium adenosine triphosphate channel. J Clin Endocrinol Metab 2004; 89: 3932–3935 [DOI] [PubMed] [Google Scholar]
- 83.Philipson LH, Murphy R, Ellard S, et al. Genetic testing in diabetes mellitus: a clinical guide to monogenic diabetes In: Weiss RE, Refetoff S (eds). Genetic Diagnosis of Endocrine Disorders. Elsevier, London, 2010; 17–25 [Google Scholar]
- 84.Greeley SA, John PM, Winn AN, et al. The cost‐effectiveness of personalized genetic medicine: the case of genetic testing in neonatal diabetes. Diabetes Care 2011. doi: 10.2337/dc10‐1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grulich‐Henn J, Wagner V, Thon A, et al. Entities and frequency of neonatal diabetes: data from the diabetes documentation and quality management system (DPV). Diabet Med 2010; 27: 709–712 [DOI] [PubMed] [Google Scholar]
- 86.Gach A, Malecki MT, Noczynska A. Islet‐specific antibody seroconversion in patients with long duration of permananet neonatal diabetes caused by mutations in the KCNJ11 gene. Diabetes Care 2007; 30: 2080–2082 [DOI] [PubMed] [Google Scholar]
- 87.Jose B, Griffiths U, Barrett T, et al. Glibenclamide controls ketosis‐prone diabetes in a 38‐year‐old woman with Kir6.2 mutation. Pract Diab Int 2009; 26: 244–246 [Google Scholar]