

Abstract
Increased hepatic lipid content is associated with hepatic as well as whole‐body insulin resistance and is typical for individuals with type 2 diabetes mellitus. However, whether insulin resistance causes hepatic steatosis or whether hepatic steatosis per se reduces insulin sensitivity remains unclear. Multiple metabolic pathways lead to the development of hepatic steatosis, including enhanced free fatty acid release from adipose tissues (lipolysis), increased de novo fatty acid synthesis (lipogenesis), decreased mitochondrial β‐oxidation and decreased very low‐density lipoprotein secretion. Although the molecular mechanisms leading to the development of hepatic steatosis in the pathogenesis of type 2 diabetes mellitus are complex, several recent animal models have shown that modulating important enzymes involved in hepatic fatty acid and glycerolipid synthesis might be a key for treating hepatic insulin resistance. We highlight recent advances in the understanding of the molecular mechanisms leading to the development of hepatic steatosis and insulin resistance. (J Diabetes Invest, doi: 10.1111/j.2040‐1124.2011.00111.x, 2011)
Keywords: Liver, Fatty acids, Diabetes
Introduction
In the past few decades, the prevalence of obesity and type 2 diabetes mellitus has grown to epidemic proportions1. Hepatic insulin resistance is a principal component of type 2 diabetes2. Decreased hepatic insulin sensitivity leads to elevated hepatic glucose production, hyperinsulinemia, increased β‐cell mass and hyperglycemia.
Hepatic steatosis is a common finding in patients with type 2 diabetes. Approximately 70% of patients who are obese or who have type 2 diabetes develop non‐alcoholic fatty liver disease (NAFLD), and 20% of these patients show progression to non‐alcoholic steatohepatitis3,4. Despite the existing correlation between hepatic steatosis and insulin resistance, whether insulin resistance causes hepatic steatosis or whether the increase in triacylglycerol (TAG) and/or lipid metabolites might play a causal role in the development of hepatic or systemic insulin resistance remains unclear. Recent studies have favored the concept that hepatic TAG itself is not toxic and might in fact protect the liver from lipotoxicity by buffering the accumulation of toxic fatty acids (FA).
Hepatic Lipid Metabolism is involved in Fatty Liver Development
Hepatic FA are derived from several different sources, including peripheral fats stored in white adipose tissue that flow to the liver as plasma free FA (FFA), dietary FA (mainly through the uptake of intestinally derived chylomicron remnants) and FA newly synthesized in the liver through de novo lipogenesis (DNL). After the esterification step, TAG can be stored as lipid droplets within hepatocytes or secreted into the blood as very low‐density lipoprotein (VLDL), but they can also be hydrolyzed and FA can be channeled toward the β‐oxidation pathway. Therefore, excessive hepatic lipid accumulation can be caused by the following four different metabolic perturbations: (i) increased FFA flux to the liver from lipolyzed adipose TAG or dietary lipids; (ii) increased DNL; (iii) reduced FA oxidation; and (iv) reduced lipid export in the form of VLDL5,6 (Figure 1). Studies in humans and rodents have shown that the mechanisms leading to excessive hepatic lipid accumulation are mainly linked to increased FFA delivery from peripheral expanded adipose tissue to the liver and enhanced hepatic DNL, whereas lipid disposal through β‐oxidation and VLDL export are only moderately affected7.
Figure 1.
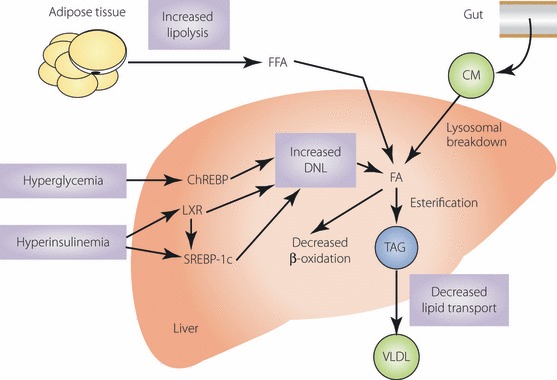
Potential sources and mechanisms leading to the development of hepatic steatosis. Different sources of fatty acids (FA) contribute to the development of hepatic steatosis. Under conditions of insulin resistance, the ability of insulin to suppress hormone sensitive lipase activity is impaired, leading to an elevated rate of triacylglycerol (TAG) lipolysis and release of increased free‐FA (FFA) into the liver. Dietary FA are also taken up by the liver through the uptake of intestinally derived chylomicron remnants. In the liver, hyperglycemia and hyperinsulinemia induce sterol regulatory element‐binding protein (SREBP)‐1c expression and activate liver X receptors (LXR), leading to the transcriptional activation of all lipogenic genes. In addition, hyperglycemia activates carbohydrate response element‐binding protein (ChREBP), which transcriptionally activates liver pyruvate kinase (L‐PK) and all lipogenic genes. The combined actions of SREBP‐1c and ChREBP promote de novo lipogenesis (DNL). A consequence of increased DNL is increased production of malonyl‐CoA, which inhibits carnitine palmitoyltransferase‐1 (CPT‐1), resulting in impaired mitochondrial β‐oxidation. After the esterification step (conversion of FA into TAG), TAG can then be stored as lipid droplets in hepatocytes or secreted into the blood as very low‐density lipoprotein (adapted from Postic and Girard5). Copyright 2008 by American Society For Clinical Investigation. Reproduced with permission of American Society For Clinical Investigation.
FA Delivery and Uptake
The rate of hepatic FFA uptake depends on FFA delivery to the liver and the capacity of the liver for FFA transport. In the postabsorptive state, the major source of FFA delivered to the liver is derived from FFA released from white adipose tissue. FFA and glycerol hydrolyzed from visceral adipocytes are transported to the liver, because the blood stream of visceral organs flows directly into the liver through the portal vein. The inflow of FA into the liver by lipolysis in adipose tissue increases in obesity and insulin resistant states8. In addition, expression of genes encoding hepatic lipase and hepatic lipoprotein lipase is higher in obese subjects with NAFLD than subjects without NAFLD9, suggesting that FFA released by lipolysis of circulating TAG also contribute to hepatic FA accumulation and steatosis. These increases in hepatic lipase and hepatic lipoprotein lipase, along with higher postprandial lipidemia and FFA concentrations in NAFLD subjects, might be responsible for the increased postprandial incorporation of dietary FA into intrahepatic TAG observed in obese subjects with type 2 diabetes. Furthermore, membrane proteins that direct trafficking of FFA from plasma into tissues are also likely to be involved in increased hepatic FFA uptake. Gene expression of FAT/CD36, which is an important regulator of tissue FFA uptake from plasma, increases in the liver of obese subjects with hepatic steatosis10. Therefore, these data suggest that alterations in adipose tissue lipolytic activity, regional hepatic lipolysis of circulating TAG and tissue FFA transport proteins could be involved in the pathogenesis of hepatic steatosis.
DNL
Synthesis of hepatic FA and TAG is nutritionally regulated5. When a high‐carbohydrate diet is ingested, carbohydrate is converted into hepatic TAG by key enzymes involved in the glycolytic and lipogenic pathways, including glucokinase and liver pyruvate kinase (L‐PK) for glycolysis; adenosine triphosphate (ATP) citrate lyase, acetyl‐CoA carboxylase (ACC) and FA synthase (FAS) for lipogenesis; ELOVL6 and stearoyl‐CoA desaturase 1 (SCD1) for catalyzing FA elongation and desaturation steps; and mitochondrial glycerol‐3‐phosphate acyltransferase (GPAT) and diacylglycerol acyltransferase (DGAT) for TAG synthesis (Figure 2). Under normal conditions, the contribution of DNL to FA, TAG, and VLDL synthesis is small in humans and estimated to be <5% in the postabsorptive state11. Conditions associated with a high rate of lipogenesis, such as ingestion of a high‐carbohydrate diet, hyperglycemia and hyperinsulinemia, are associated with a shift in cellular metabolism from lipid oxidation to lipid synthesis. Using a stable isotope approach, nearly 60% of hepatic TAG in subjects with NAFLD, has been shown to be derived from circulating FFA, a little more than 10% from the diet, and close to 30% from DNL11. This suggests the importance of DNL in the pathology of hepatic steatosis observed in obese subjects with type 2 diabetes.
Figure 2.
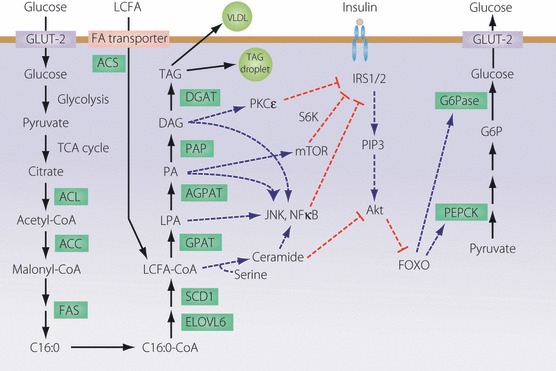
Metabolic pathways of fatty acid (FA) and triacylglycerol (TAG) synthesis, and mechanisms by which lipid intermediates affect hepatic insulin sensitivity. Excess lipid intermediates in the TAG biosynthetic pathway, such as diacylglycerol (DAG), phosphatidic acid (PA) and lysophosphatidic acid (LPA), cause insulin resistance by activating several serine/threonine kinases, including protein kinase C (PKC), mammalian target of rapamycin (mTOR), c‐Jun N‐terminal kinase 1 (JNK1), IκB kinase (IKK), which inhibit insulin signaling either directly through insulin receptor substrate (IRS)‐1 and IRS‐2 serine phosphorylation or indirectly through a series of transcriptional events mediated by nuclear factor (NF)‐κB. This, in turn, results in reduced insulin activation of phosphatidylinositol (PI) 3‐kinase and Akt. Ceramide can also impair insulin action through interactions with Akt. Reduced Akt activation resulting in lower fork‐head box (FOX)O phosphorylation leads to increases in the transcription of phosphoenolpyruvate carboxycarboxykinase (PEPCK) and glucose‐6‐phosphatase (G6Pase). This enhances gluconeogenesis and hepatic glucose output (adapted from Nagle et al.6). Copyright 2009 The American Society for Biochemistry and Molecular Biology.
Transcriptional Regulation of DNL
Enzymes involved in DNL are acutely regulated by post‐translational and allosteric mechanisms, whereas they are mainly controlled on a long‐term basis by modulating their transcription rate. The fact that the transcription factor, sterol regulatory element‐binding protein (SREBP)‐1c, which is itself stimulated by glucose and insulin, activates the transcription of genes involved in FA (e.g. ACC, FAS, ELOVL6 and SCD1) and TAG synthesis (e.g. GPAT and DGAT)12 is now well accepted (Figure 2). SREBP‐1c overexpression in livers of transgenic mice leads to the development of fatty liver as a result of increased lipogenesis13. Inactivation of the SREBP‐1c gene in livers of ob/ob mice, a genetic model of leptin deficiency that develops obesity and hepatic steatosis, results in an approximately 50% reduction in hepatic TAG14. We and others have shown that increased rates of hepatic FA synthesis contribute to the development of fatty livers in rodent models of insulin resistant diabetes and obesity14,15. In addition, SREBP directly repress the transcription of insulin receptor substrate (IRS)‐2, the main insulin signaling molecule in the liver16. IRS‐2 repression by hepatic SREBP‐1c leads to the inhibition of the process regulating insulin signaling. Thus, chronic activation of hepatic SREBP‐1c causes hepatic steatosis, hypertriglyceridemia, hyperglycemia and insulin resistance, leading to the development of type 2 diabetes.
In addition to SREBP‐1c, liver X receptors (LXR) are also important regulators of lipid synthesis (Figure 2). Hepatic LXR activation by endogeneous oxysterol ligands results in the upregulation of genes involved in lipid and carbohydrate metabolism. Oral administration of LXR agonists to mice results in elevated hepatic FA synthesis and steatosis17. This increased hepatic lipogenesis is largely attributed to a LXR‐dependent upregulation of SREBP‐1c expression. Insulin and glucose can stimulate the transcriptional activity of LXR18,19. Thus, hyperinsulinemia and hyperglycemia can activate LXR and SREBP‐1c, resulting in increased lipogenesis, hepatic lipid accumulation and worsening of hepatic insulin resistance.
Carbohydrate‐mediated DNL stimulation is transcriptionally mediated by the carbohydrate response element‐binding protein (ChREBP)20. Glucose activates ChREBP by regulating its entry from the cytosol into the nucleus and by activating the binding of this transcription factor to DNA (Figure 2). ChREBP regulates hepatic lipid synthesis through the transcriptional control of L‐PK, a key regulatory enzyme in glycolysis, and the lipogenic genes ACC and FAS in response to glucose. Hepatic TAG content decreases in ChREBP knockout mice compared with wild‐type mice21. Furthermore, deletion of ChREBP in ob/ob mice results in decreased hepatic steatosis and obesity, and ameliorates glucose intolerance and insulin resistance22. Thus, clarification of the role of ChREBP and its target genes in glucose and lipid metabolism will be useful for preventing and treating type 2 diabetes.
The Role of Lipid Metabolites in the Development of Hepatic Insulin Resistance
The cellular mechanisms responsible for FA‐induced insulin resistance are not fully understood. As discussed earlier, an association between hepatic steatosis and insulin resistance exists, and several rodent models have shown that decrease in hepatic TAG content correlates with improvement in insulin sensitivity. However, whether a causal relationship exists remains uncertain. Several lines of evidence suggest that lipid intermediates, such as diacylglycerol (DAG), long chain fatty acyl‐CoA, ceramide, lysophosphatidic acid (LPA) and phosphatidic acid (PA), rather than TAG itself, are important for the development of hepatic insulin resistance.
TAG
Hepatic TAG accumulation was once believed to impair insulin signaling and cause insulin resistance. However, recent data show that hepatic TAG accumulation is a parallel phenomenon and not the direct cause for impaired insulin signaling. Dissociation between hepatic steatosis and insulin resistance has been observed in some genetically altered or pharmacologically manipulated animal models. Overexpression of hepatic DGAT223, blockade of hepatic VLDL secretion24 and pharmacological blockade of β‐oxidation in mice25 cause significant hepatic steatosis, but not hepatic insulin resistance. In contrast, inhibiting hepatic TAG synthesis by decreasing DGAT2 in diet‐induced and genetically obese mice decreases hepatic steatosis, but does not improve insulin sensitivity26. These data support the notion that hepatic TAG accumulation is not necessary for causing insulin resistance, and that excessive accumulation of certain FA‐derived lipid metabolites is lipotoxic and is a cause of hepatic insulin resistance. In fact, TAG synthesis is beneficial for removing potentially lipotoxic FA derivatives, such as FFA, DAG and ceramide, by channeling FA substrates into a storage form of TAG27.
However, TAG in excess might obstruct hepatic blood flow by compressing the sinusoids28. Furthermore, storage of TAG as lipid droplets is only a temporary measure; FA in TAG can be released at any time, and if the cell is still unable to handle FA appropriately through other metabolic pathways, the stored TAG can serve as a source of toxic intermediates.
DAG
Recent studies implicate DAG as an inducer of insulin resistance29. When excess DAG is formed, protein kinase C (PKC)ε is activated, and IRS‐1 and IRS‐2 are phosphorylated on tyrosine residues, which diminishes phosphatidylinositol 3‐kinase and Akt activation2. This is not entirely surprising, because lipogenic diets normally increase SREBP‐1c‐mediated glycerolipid synthesis.
Both GPAT1 knockout mice30 and adenovirus‐mediated GPAT1 overexpression in the liver31 provide evidence for an important role of this enzyme in the development of hepatic steatosis and insulin resistance. GPAT1 knockout mice show reduced levels of both hepatic DAG and TAG, and are protected against high‐fat, diet‐induced hepatic insulin resistance, possibly as a result of low DAG‐mediated PKCε activation30. Interestingly, although GPAT1 inhibition leads to a significant accumulation of hepatic acyl‐CoA content, GPAT knockout mice do not show hepatic insulin resistance, suggesting that DAG is most likely the better candidate to account for insulin resistance. GPAT1 overexpression in livers of rats also supports the importance of DAG in the development of hepatic insulin resistance. Hepatic GPAT1 overexpression causes hepatic steatosis and insulin resistance. Hepatic insulin resistance observed in GPAT1‐overexpressing rats is associated with elevated DAG, LPA and TAG concentrations, as well as activated PKCε.
Other Potential Mediators
In addition to DAG, glycerolipid intermediates, such as LPA and PA, can also initiate signaling pathways. LPA is a ligand for peroxisome proliferator‐activated receptor (PPAR)γ and might be responsible for the upregulation of PPARγ target genes observed in fatty liver32. Intracellular PA activates the mammalian target of rapamycin, which downregulates the insulin signal by promoting serine phosphorylation of IRS‐1 when nutrients are present in excess33.
Recent studies have also shown that FA composition of the lipid species (the length of carbon chain and/or the number and position of unsaturated bonds) could be another determinant of the development of hepatic insulin resistance accompanying intracellular lipid accumulation. An unexpected phenotype has been observed while analyzing mice deficient for ELOVL6, a microsomal enzyme involved in the elongation of saturated and monounsaturated FA with 12, 14 and 16 carbons. Loss of ELOVL6 function increases the levels of palmitate (C16:0) and palmitoleate (C16:1), but reduces the levels of stearate (C18:0) and oleate (C18:1). Elovl6 knockout mice are protected against developing hepatic insulin resistance when fed a high‐fat/high‐sucrose diet, despite that hepatic steatosis and obesity observed in these mice are similar to those observed in wild‐type mice34, suggesting that hepatic FA composition, particularly the conversion of palmitate to stearate, is crucial for insulin sensitivity rather than only lipid accumulation. In addition, some DAG and sphingolipid species might be poor inhibitors of insulin signaling6. FA composition of glycerolipid intermediates might influence their signaling capacities and importance as contributors to hepatic insulin resistance.
Relationship between Hepatic Lipid Metabolism, Inflammation and Insulin Resistance
Although hepatic lipid accumulation is associated with and might be sufficient to explain the development of insulin resistance, systemic chronic inflammation has been proposed to play an important role in the development of insulin resistance and type 2 diabetes.
Several signaling pathways link the inflammatory mechanisms of insulin resistance. An important kinase likely to mediate the cross‐talk between inflammatory and metabolic signaling is c‐Jun N‐terminal kinase 1 (JNK1), a serine/threonine protein kinase that is regulated by FFA and many inflammatory stimuli, such as tumor necrosis factor‐α and interleukin‐1β. In both genetic and dietary animal models of obesity, JNK1 activity increases in the liver, muscle and adipose tissue, and loss of JNK1 function prevents insulin resistance35. The target of JNK1 action is serine phosphorylation of IRS‐1, which impairs insulin action36.
Furthermore, FA‐induced insulin resistance might be mediated by phosphorylating inhibitor of NF‐κB (IκB) kinase β (IKKβ). Transgenic mice expressing constitutively low levels of IKKβ in hepatocytes activate NF‐κB, which leads to hepatic and systemic insulin resistance37. Furthermore, pharmacological inhibition of IKKβ activity with high‐dose aspirin treatment reduces fasting hyperglycemia and basal hepatic glucose production, as well as improves peripheral glucose uptake in a mouse model and in patients with type 2 diabetes38,39. IKKβ inhibits insulin signaling by directly phosphorylating IRS‐1 serine residues and by activating IκB, which leads to NF‐κB translocation and stimulates the production of multiple inflammatory mediators that can cause insulin resistance40. Once activated, this process might trigger a vicious positive‐feedback loop of inflammatory responses that negatively regulate insulin signaling.
In the liver, insulin resistance seems to be somewhat paradoxically associated with a reduced ability of insulin signaling to inhibit glucose production, whereas insulin‐stimulated lipogenesis is enhanced. Despite a number of arguments, whether insulin resistance leads to hepatic steatosis and whether steatosis or rather the presence of FA derivatives involved in TAG synthesis enhances insulin resistance are still under debate. In addition, not only gross lipid content, but also intracellular FA composition is crucial for insulin sensitivity. Identification of the molecular mechanisms leading to disordered lipid metabolism and insulin resistance in steatotic livers using genomic, proteomic and lipidomic approaches involving tissues from patients with type 2 diabetes are expected to improve both diagnostic and therapeutic approaches. Further studies will be required to identify the molecular mechanisms leading to disordered lipid metabolism and insulin resistance in steatotic livers. These studies are also expected to develop novel therapeutic approaches for the treatment and prevention of insulin resistance and type 2 diabetes.
Acknowledgements
The authors are grateful to current and former colleagues in the laboratory of Shimano. The authors were supported by a grant‐in‐aid for Scientific Research 21689025 (to TM) and 21390275 (to HS) from the Ministry of Science, Education, Culture, and Technology of Japan. The authors have no conflict of interest.
References
- 1.James WP. The epidemiology of obesity: the size of the problem. J Intern Med 2008; 263: 336–352 [DOI] [PubMed] [Google Scholar]
- 2.Savage DB, Petersen KF, Shulman GI. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol Rev 2007; 87: 507–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roden M. Mechanisms of disease: hepatic steatosis in type 2 diabetes‐pathogenesis and clinical relevance. Nat Clin Pract Endocrinol Metab 2006; 2: 335–348 [DOI] [PubMed] [Google Scholar]
- 4.Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology 2006; 43: S99–S112 [DOI] [PubMed] [Google Scholar]
- 5.Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest 2008; 118: 829–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagle CA, Klett EL, Coleman RA. Hepatic triacylglycerol accumulation and insulin resistance. J Lipid Res 2009; 50: S74–S79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewis GF, Carpentier A, Adeli K, et al. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev 2002; 23: 201–229 [DOI] [PubMed] [Google Scholar]
- 8.Mittendorfer B, Magkos F, Fabbrini E, et al. Relationship between body fat mass and free fatty acid kinetics in men and women. Obesity (Silver Spring) 2009; 17: 1872–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Westerbacka J, Kolak M, Kiviluoto T, et al. Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin‐resistant subjects. Diabetes 2007; 56: 2759–2765 [DOI] [PubMed] [Google Scholar]
- 10.Greco D, Kotronen A, Westerbacka J, et al. Gene expression in human NAFLD. Am J Physiol Gastrointest Liver Physiol 2008; 294: G1281–G1287 [DOI] [PubMed] [Google Scholar]
- 11.Donnelly KL, Smith CI, Schwarzenberg SJ, et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005; 115: 1343–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shimano H. SREBPs: physiology and pathophysiology of the SREBP family. FEBS J 2009; 276: 616–621 [DOI] [PubMed] [Google Scholar]
- 13.Shimano H, Horton JD, Shimomura I, et al. Isoform 1c of sterol regulatory element‐binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest 1997; 99: 846–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yahagi N, Shimano H, Hasty AH, et al. Absence of sterol regulatory element‐binding protein‐1 (SREBP‐1) ameliorates fatty livers but not obesity or insulin resistance in Lep(ob)/Lep(ob) mice. J Biol Chem 2002; 277: 19353–19357 [DOI] [PubMed] [Google Scholar]
- 15.Shimomura I, Bashmakov Y, Horton JD. Increased levels of nuclear SREBP‐1c associated with fatty livers in two mouse models of diabetes mellitus. J Biol Chem 1999; 274: 30028–30032 [DOI] [PubMed] [Google Scholar]
- 16.Ide T, Shimano H, Yahagi N, et al. SREBPs suppress IRS‐2‐mediated insulin signalling in the liver. Nat Cell Biol 2004; 6: 351–357 [DOI] [PubMed] [Google Scholar]
- 17.Grefhorst A, Elzinga BM, Voshol PJ, et al. Stimulation of lipogenesis by pharmacological activation of the liver X receptor leads to production of large, triglyceride‐rich very low density lipoprotein particles. J Biol Chem 2002; 277: 34182–34190 [DOI] [PubMed] [Google Scholar]
- 18.Chen G, Liang G, Ou J, et al. Central role for liver X receptor in insulin‐mediated activation of Srebp‐1c transcription and stimulation of fatty acid synthesis in liver. Proc Natl Acad Sci USA 2004; 101: 11245–11250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitro N, Mak PA, Vargas L, et al. The nuclear receptor LXR is a glucose sensor. Nature 2007; 445: 219–223 [DOI] [PubMed] [Google Scholar]
- 20.Uyeda K, Repa JJ. Carbohydrate response element‐binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab 2006; 4: 107–110 [DOI] [PubMed] [Google Scholar]
- 21.Iizuka K, Bruick RK, Liang G, et al. Deficiency of carbohydrate response element‐binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci USA 2004; 101: 7281–7286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iizuka K, Miller B, Uyeda K. Deficiency of carbohydrate‐activated transcription factor ChREBP prevents obesity and improves plasma glucose control in leptin‐deficient (ob/ob) mice. Am J Physiol Endocrinol Metab 2006; 291: E358–E364 [DOI] [PubMed] [Google Scholar]
- 23.Monetti M, Levin MC, Watt MJ, et al. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab 2007; 6: 69–78 [DOI] [PubMed] [Google Scholar]
- 24.Minehira K, Young SG, Villanueva CJ, et al. Blocking VLDL secretion causes hepatic steatosis but does not affect peripheral lipid stores or insulin sensitivity in mice. J Lipid Res 2008; 49: 2038–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grefhorst A, Hoekstra J, Derks TG, et al. Acute hepatic steatosis in mice by blocking beta‐oxidation does not reduce insulin sensitivity of very‐low‐density lipoprotein production. Am J Physiol Gastrointest Liver Physiol 2005; 289: G592–G598 [DOI] [PubMed] [Google Scholar]
- 26.Yu XX, Murray SF, Pandey SK, et al. Antisense oligonucleotide reduction of DGAT2 expression improves hepatic steatosis and hyperlipidemia in obese mice. Hepatology 2005; 42: 362–371 [DOI] [PubMed] [Google Scholar]
- 27.Listenberger LL, Han X, Lewis SE, et al. Triglyceride accumulation protects against fatty acid‐induced lipotoxicity. Proc Natl Acad Sci USA 2003; 100: 3077–3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schaffer JE. Lipotoxicity: when tissues overeat. Curr Opin Lipidol 2003; 14: 281–287 [DOI] [PubMed] [Google Scholar]
- 29.Samuel VT, Liu ZX, Qu X, et al. Mechanism of hepatic insulin resistance in non‐alcoholic fatty liver disease. J Biol Chem 2004; 279: 32345–32353 [DOI] [PubMed] [Google Scholar]
- 30.Neschen S, Morino K, Hammond LE, et al. Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl‐CoA:glycerol‐sn‐3‐phosphate acyltransferase 1 knockout mice. Cell Metab 2005; 2: 55–65 [DOI] [PubMed] [Google Scholar]
- 31.Nagle CA, An J, Shiota M, et al. Hepatic overexpression of glycerol‐sn‐3‐phosphate acyltransferase 1 in rats causes insulin resistance. J Biol Chem 2007; 282: 14807–14815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McIntyre TM, Pontsler AV, Silva AR, et al. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARgamma agonist. Proc Natl Acad Sci USA 2003; 100: 131–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X, Devaiah SP, Zhang W, et al. Signaling functions of phosphatidic acid. Prog Lipid Res 2006; 45: 250–278 [DOI] [PubMed] [Google Scholar]
- 34.Matsuzaka T, Shimano H, Yahagi N, et al. Crucial role of a long‐chain fatty acid elongase, Elovl6, in obesity‐induced insulin resistance. Nat Med 2007; 13: 1193–1202 [DOI] [PubMed] [Google Scholar]
- 35.Hirosumi J, Tuncman G, Chang L, et al. A central role for JNK in obesity and insulin resistance. Nature 2002; 420: 333–336 [DOI] [PubMed] [Google Scholar]
- 36.Aguirre V, Uchida T, Yenush L, et al. The c‐Jun NH(2)‐terminal kinase promotes insulin resistance during association with insulin receptor substrate‐1 and phosphorylation of Ser(307). J Biol Chem 2000; 275: 9047–9054 [DOI] [PubMed] [Google Scholar]
- 37.Cai D, Yuan M, Frantz DF, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK‐beta and NF‐kappaB. Nat Med 2005; 11: 183–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yuan M, Konstantopoulos N, Lee J, et al. Reversal of obesity‐ and diet‐induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 2001; 293: 1673–1677 [DOI] [PubMed] [Google Scholar]
- 39.Hundal RS, Petersen KF, Mayerson AB, et al. Mechanism by which high‐dose aspirin improves glucose metabolism in type 2 diabetes. J Clin Invest 2002; 109: 1321–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shoelson SE, Lee J, Yuan M. Inflammation and the IKK beta/I kappa B/NF‐kappa B axis in obesity‐ and diet‐induced insulin resistance. Int J Obes Relat Metab Disord 2003; 27: S49–S52 [DOI] [PubMed] [Google Scholar]