

Abstract
Aims/Introduction: 2‐Methoxyestradiol (2ME) is an estradiol metabolite with little estrogenic activity. Previous data identified its anti‐carcinogenic properties and possible cardiovascular benefits. However, its effect on diabetes mellitus has not been fully elucidated. The aim of the present study was to determine the effects of 2ME on glucose metabolism in the diabetic state.
Materials and Methods: To evaluate the effects of 2ME, pellets of two different doses of the drug were implanted into female db/db mice at the age of 5 weeks. Intraperitoneal glucose tolerance test and insulin tolerance test were carried out at the age of 8 weeks. The pancreas was harvested for morphological analysis and β‐cell function at the age of 9 weeks.
Results: 2ME improved random blood glucose levels and glucose tolerance with increases in insulin levels during an intraperitoneal glucose tolerance test. Insulin sensitivity judged by an insulin tolerance test was comparable in the low‐ and high‐dose 2ME groups and the control group. Although glucose‐stimulated insulin secretion in isolated islets was comparable among the three groups, β‐cell mass in 2ME‐treated groups was higher than the control group. In the 2ME‐treated groups, the number of Ki67‐positive cells in islets was higher, whereas the number of cleaved caspase‐3‐positive cells was comparable with the control.
Conclusions: 2ME ameliorates glucose tolerance by promoting the proliferation of β‐cell mass in db/db mice. Our data suggests its potential clinical usefulness as a disease‐modifying drug for type 2 diabetes mellitus. (J Diabetes Invest, doi: 10.1111/j.2040‐1124.2010.00087.x, 2011)
Keywords: Diabetes mellitus, Insulin, Pancreatic β‐cells
Introduction
The prevalence of type 2 diabetes mellitus is lower in premenopausal women, but the trend is reversed after menopause1. This change might be related to the anti‐diabetic effects of the ovarian hormones. In particular, the anti‐diabetic actions of estrogens were confirmed in two large clinical trials. The Women’s Health Initiative (WHI) Hormone Trial was a randomized, double‐blind trial that investigated the effect of estrogen plus progesterone during a 5.6‐year follow‐up period. The participants were over 15,000 postmenopausal women. The cumulative incidence of diabetes was 20% lower in women on hormone replacement therapy than those on a placebo2. The Heart and Estrogen/Progestin Replacement Study (HERS) was also a randomized, double‐blind trial. The participants were approximately 3000 postmenopausal women with coronary heart disease who received daily estrogen plus progestin or a placebo. Over a period of 4 years, the incidence of diabetes decreased by 35% in the estrogen plus progestin group3. Hyperglycemia cannot develop without β‐cell failure, thus, the aforementioned studies suggest that estrogen or its metabolites has a preferential effect on β‐cell function. This conclusion is supported by basic experiments that had shown the preferential role of 17β‐estradiol (E2) on β‐cell function4. It should be cautioned, however, that both the WHI and HERS found that the hormone replacement therapy increased the risk of cardiovascular disease after menopause5,6.
Several effects of E2 are known to be mediated by its metabolites rather than by the compound itself7. Among them is 2‐methoxyestradiol (2ME), which lacks the estrogen receptor binding ability. 2ME is produced from 17β‐estradiol by sequential hepatic hydration (cytochrome P450, family 1, subfamily A polypeptide 2 [p450 CYP1A2]) and methylation (catechol‐O‐methyltransferase). The plasma concentrations of 2ME are higher in women than men, and increase in the luteal phase compared with the follicular phase. Under non‐pregnant conditions, plasma 2ME levels are in the picomolar range; however, during late pregnancy, the levels increase to the tens of nanomolar8. Although estrogen enhances cell proliferation in some tissues, 2ME at pharmacological doses blocks angiogenesis, induces apoptosis and disrupts microtubules. 2ME is the first steroid with antitumor effects. It appears to target only active proliferating cells, not cytotoxic to quiescent cells8. 2ME is currently being used clinically in patients with various types of cancers9–13. In addition, 2ME has protective effects on the kidney, cardiovascular system and also on osteoporosis in postmenopausal women14–17.
In the present study, as a first step to understanding the role of 2ME in glucose metabolism, we investigated the effects of 2ME on glucose metabolism in female db/db mice, which are obese diabetic mice with hypo‐ovarian function18.
Materials and methods
Animals and Experimental Procedures
The study protocol was reviewed and approved by the Animal Care and Use Committee of Juntendo University. The db/db female mice were purchased from CLEA Japan (Tokyo, Japan) at the age of 5 weeks. All mice were housed in specific pathogen‐free barrier facilities, maintained under 12‐h light/dark cycle, and fed a standard rodent food (Oriental Yeast, Tokyo, Japan) and water ad libitum. At the age of 5 weeks, mice were implanted subcutaneously with small pellets (Innovative Research of America, Sarasota, FL, USA) that released 0.5 mg (n = 11) or 5 mg (n = 14) of 2ME or a placebo (n = 12) gradually over 50 days.
Measurement of Blood Glucose and Insulin Levels
At the age of 8 weeks, mice were injected with glucose at 1.0 g/kg for an intraperitoneal glucose tolerant test (IPGTT), after an overnight fast and 2.0 U/kg of insulin for an insulin tolerance test (ITT), as described previously19. Glucose levels were measured using a glucose analyzer (Free Style; KISSEI Pharma, Nagano, Japan) at the indicated time‐points and insulin levels were measured by an enzyme‐linked immunosorbent assay (ELISA) kit (Morinaga, Kanagawa, Japan). ITT data were represented by percentage reduction of basal blood glucose level.
Assay of Insulin Secretion From Isolated Islets and Islet Insulin Content
Pancreatic islets were isolated from 9‐week‐old mice by collagenase digestion, as described previously19. Briefly, five size‐matched islets were incubated in 400 μL Krebs–Ringer solution (129 mmol/L NaCl, 5 mmol/L NaHCO3, 4.8 mmol/L KCl, 1.2 mmol/L KH2PO4, 1.2 mmol/L MgSO4, 0.2% bovine serum albumin, 10 mmol/L 4‐[2‐hydroxyethyl]‐1‐piperazineethanesulfonic acid and 2.5 mmol/L CaCl2 at pH 7.4), with 2.8 or 16.7 mmol/L glucose. To measure islet insulin content, islets were solubilized in acid‐ethanol solution overnight at 4°C and insulin concentration was analyzed by an ELISA kit (Morinaga). Insulin secretion was expressed as percentage secretion (serum insulin divided by insulin content) divided by islet DNA content.
In Vitro Analysis of Cell Proliferation and Apoptosis
In these studies, we analyzed the pancreatic tissue immunohistochemically for cell proliferation (using Ki67 as a marker protein) and cell apoptosis (using cleaved caspase 3 as a protein marker). The pancreas was removed from anesthetized mice after cardiac perfusion with 4% zinc formalin fixative. The pancreatic tissue was fixed by immersion at 4°C overnight, paraffin embedded and then cut at 5‐μm thick sections before mounting on slides. Immunohistochemical analysis was carried out using guinea pig anti‐human insulin antibody (dilution 1:2,000; Linco Research, St Charles, MO, USA), mouse antihuman Ki67 antibody (dilution 1:1000; Pharmingen, San Jose, CA, USA) and rabbit anticleaved caspase 3 antibody (1:400; Cell Signaling Technology, Danvers, MA, USA). All primary antibody reactions were visualized using the appropriate biotinylated secondary antibody and incubated with streptavidin conjugated with horseradish peroxidase regent (Dako, Glostrup, Denmark) and peroxidase substrate solution containing 3.3′‐diaminobenzidine tetrahydrochloride. Sections were counterstained with methylene blue. The islet β‐cell mass and the percentages of areas immunopositive for Ki67 and cleaved caspase 3 were determined as described previously20. Briefly, an insulin positive area was determined on five of each immunostained sections from each mouse, each separated by at least 200 μm. The areas of positive staining were automatically measured using Image analysis software (Image Pro4.5J; Planetron, Tokyo, Japan). Non‐specific staining was excluded from the quantification. β‐cell mass was calculated using the following equation:
The number of Ki67‐positive cells and caspase 3 positive cells per islet were counted using five immunostained sections each from five mice, with each section separated by at least 200 μm.
Statistical Analysis
Results were expressed as mean ± SEM. Student’s unpaired t‐test was used to compare the laboratory data of two groups. Differences among three or more groups were analyzed by anova. A P‐value <0.05 was considered significant.
Results
2ME Treatment Improves Glucose Tolerance in db/db Mice
To investigate the effect of 2ME on blood glucose level, we divided db/db mice into three groups: (i) the high‐dose 2ME‐treated group (2ME‐H); (ii) the low‐dose 2ME‐treated group (2ME‐L); and (iii) the placebo group (control). 2ME and a placebo were provided by implanted pellets and the estimated administered dose of 2ME was 0.1 and 0.01 mg/day in the 2ME‐H and 2ME‐L group, respectively.
After implantation of the pellets at the age of 5 weeks, we measured serial changes in bodyweight and food intake. 2ME‐treated mice showed lower food intake at the age of 7 weeks, but higher food intake at the age of 8 weeks. Total food intake was comparable among the groups In contrast, 2ME‐treated mice gained more weight than the control mice (Figure 1a,b). The randomly‐measured glucose level was significantly lower in 2ME‐treated mice than the control (Figure 1c). Whereas fasting blood glucose level was comparable among the groups, IPGTT carried out at the age of 8 weeks showed a marked improvement in glucose tolerance of 2ME‐treated mice (Figure 2a). Although blood glucose levels during IPGTT were lower in 2ME‐treated mice, simultaneously measured insulin level in the 2ME‐L group was comparable to the control, though that of 2ME‐H mice was significantly higher than the control at the age of 8 weeks (Figure 2b). ITT showed comparable insulin sensitivity in the three groups (Figure 2c). These results suggest that 2ME treatment improves glucose tolerance and enhances glucose‐stimulated insulin secretion in db/db mice.
Figure 1.
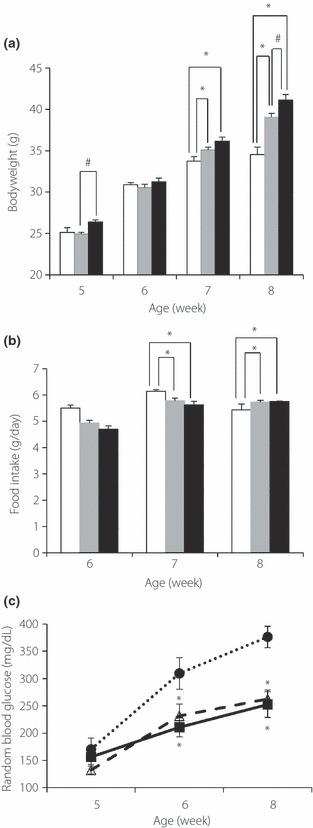
Treatment with 2‐methoxyestradiol (2ME) reduces random glucose levels in db/db mice. Serial changes in (a) bodyweight, (b) food intake and (c) random blood glucose level in placebo treated (control) mice, low‐dose 2ME‐treated mice (0.01 mg/day; 2ME‐L), and high‐dose 2ME‐treated mice (0.1 mg/day; 2ME‐H) between 5 and 8 weeks‐of‐age. Solid circles and open bars, control (n = 12); open triangles and grey bars, 2ME‐L (n = 14); solid squares and solid bars, 2ME‐H (n = 11). Data are mean ± SEM. *P < 0.05 vs control, #P < 0.05 vs 2ME‐L.
Figure 2.
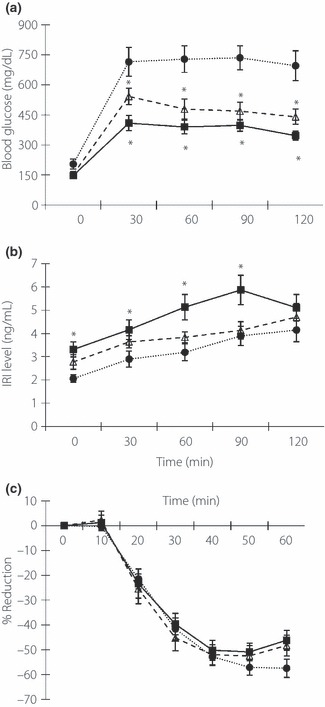
Treatment with 2‐methoxyestradiol (2ME) improves glucose tolerance in db/db mice. (a) Blood glucose concentrations and (b) serum insulin levels during intraperitoneal glucose tolerant test in placebo (control) mice (n = 12), low‐dose 2ME‐treated mice (n = 14, 0.01 mg/day; 2ME‐L), high‐dose 2ME‐treated mice (n = 11, 0.1 mg/day; 2ME‐H) at the age of 8 weeks. (c) Results of insulin tolerance test at the age of 9 weeks in each group (control, n = 12; 2ME‐L, n = 14; 2ME‐H, n = 11). Solid circles, control; open triangles, 2ME‐L; solid squares, 2ME‐H. Data are mean ± SEM. *P < 0.05 vs control, #P < 0.05 vs 2ME‐L. IRI, immunoreactive insulin.
2ME Increases β‐cell Mass
To determine the mechanism of improved glucose‐stimulated insulin secretion in 2ME‐treated mice, we investigated islet morphology by immunohistochemical staining for insulin. As shown in Figure 3a, treatment with 2ME increased β‐cell mass, compared with control mice. Quantitative analysis showed significant increases in β‐cell mass in both 2ME‐H and ‐L groups (Figure 3b).
Figure 3.
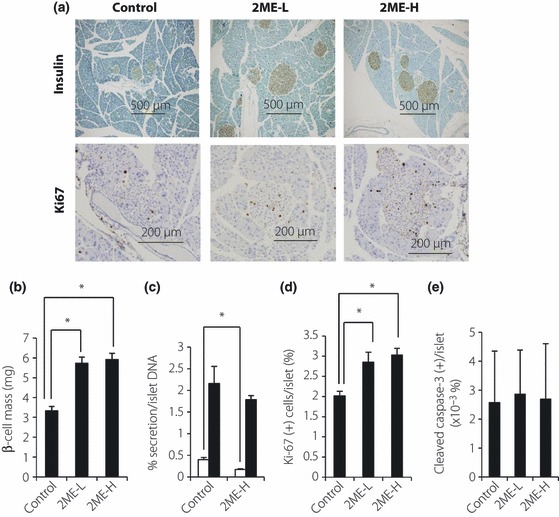
Treatment with 2‐methoxyestradiol (2ME) increases β‐cell mass with enhanced cell proliferation in db/db mice. (a) Representative images of immunohistochemical staining with insulin and Ki67 in control mice, low‐dose 2ME‐treated mice and high‐dose 2ME‐treated mice. (b) β‐cell mass in placebo (control) mice (n = 5), low‐dose 2ME‐treated mice (n = 5; 0.01 mg/day; 2ME‐L), and high‐dose 2ME‐treated mice. Data are mean ± SEM. *P < 0.05 vs control. (c) Insulin secretion of batch‐incubated pancreatic islets of each group at the age of 8 weeks (control, n = 10 and 2ME‐H, n = 10). Open bars, under 2.8 mmol/L glucose concentration; solid bars, under 16.7 mmol/L glucose concentration. Data are mean ± SEM. *P < 0.05 vs control. (d) The number of Ki67‐positive cells in islets in each group (n = 5 each). Data are mean ± SEM. *P < 0.05 vs control. (e) The number of cleaved‐caspase 3‐positive cells in islets in each group (n = 5 each). Data are mean ± SEM.
Using isolated islets, we further studied the effect of 2ME on islet function. Insulin secretion by islets incubated under a low concentration of glucose was lower than by islets of 2ME‐H, but insulin secretion responded to high glucose in a manner comparable between islets from 2ME‐H and control mice (Figure 3c). It was noteworthy that these results were after normalization by islet DNA content.
Through what mechanism does 2ME increase β‐cell mass? To answer this question, we quantitated the proliferation and apoptotic rate of islets by Ki67 and cleaved‐caspase 3 staining, respectively. The number of Ki67‐positive cells in the islets was higher in islets of 2ME‐treated mice than control mice (Figure 3a,d). In contrast, the number of cleaved‐caspase 3‐positive cells was similar among the three groups (Figure 3e). Thus, the increase in β‐cell mass induced by 2ME was mainly a result of increased rate of cell proliferation in islets, not by suppression of the apoptotic rate.
Discussion
In the present study, we investigated the effects of 2ME on glucose metabolism. The results suggested that 2ME ameliorated glucose intolerance in db/db mice at least in part through the expansion of β‐cell mass by increasing the proliferation rate of these cells. Zang et al.21 investigated the effect of 2ME on glucose metabolism in diabetic ZSF1 rats. They found that 2ME significantly improved HbA1c level without changes in insulin sensitivity, based on ITT. Our data are in agreement with those of the aforementioned study.
We found that 2ME enhanced proliferation of β‐cell in db/db mice. This is in contrast to the reported antiproliferative effect of 2ME on various cells. For example, 2ME is reported to inhibit proliferation of endothelial cells17,22, smooth muscle cells23 and fibroblasts24. In addition, several studies showed that 2ME inhibits the proliferation of various types of tumor cells8. However, it is plausible that sensitivity of cells to 2ME depends on the cell type and 2ME concentration. In methylnitrosourea‐induced mammary carcinoma, low‐dose 2ME stimulated tumor growth, whereas at a high dose, it inhibited tumor growth25. In this regard, the dose of 2ME used in the present study and the cell‐specific response of β‐cells might be key factors for the unique cell proliferating effect of 2ME.
Several mechanisms have been proposed for the effects of 2ME. The antiproliferative effects of 2ME are considered to a result of the induction of cell cycle arrest and apoptosis through the activation of p5326,27 or JNK‐Bcl228,29. This effect is also caused by the inhibition of tubulin polymerization to disturb cytoskeletal function30. Furthermore, the anti‐angiogenic effect of 2ME is mediated by the inhibition of hypoxia‐inducible factor‐1α, a key angiogenic transcription factor30. In addition, it has been reported that 2ME has a calcium antagonistic effect8 and oxidation inhibiting effects31. However, the exact signal transduction pathway of 2ME has not been yet elucidated. Further studies are required to determine the exact mechanisms on various actions of 2ME, including the proliferation of β‐cells.
In the present study, IPGTT data showed that 2ME treatment improves β‐cell function in a dose‐dependent manner (Figure 2a). In contrast, the batch incubation data using isolated islets shows that 2ME treatment did not improve insulin secretion from each islet (Figure 3c). The measurement of β‐cell mass showed that low and high doses of 2ME treatment comparably increase β‐cell mass in vivo (Figure 3b). These results suggest that the increase of β‐cell mass might be implicated in the amelioration of β‐cell function. However, the effect of 2ME on β‐cell mass cannot explain why the 2ME‐H group showed better β‐cell function judged by IPGTT than the 2ME‐L group. If 2ME improved islet endothelial function or altered the environment of extracellular matrix in islets in a dose‐dependent manner, 2ME could improve insulin secretion in vivo, but could not improve insulin secretion using isolated islets. Further studies are required to elucidate its mechanism in detail.
In conclusion, the present study showed that 2ME increases β‐cell mass, which is relatively decreased in female db/db mice, without major adverse effects. The effect of 2ME on male db/db mice is under our investigation. Until now, we have found that 2ME treatment decreases fasting blood glucose, with the tendency to increase immunoreactive insulin levels in male db/db mice (Uchida and Watada, unpublished observation). 2ME might be useful not only in a female diabetic model, but also in a male diabetic model. Considering that 2ME has already been used in a clinical setting, this drug is a promising agent to prevent any decrease in β‐cell mass, which occurs during the natural history of type 2 diabetes.
Acknowledgements
We thank Naoko Daimaru, Eriko Magoshi and Kiyomi Nakamura for their excellent technical assistance. This study was supported by grants from the Institute for Environmental and Gender‐Specific Medicine, Juntendo University Graduate School of Medicine. There is no conflict of interest.
References
- 1.Wild S, Roglic G, Green A, et al. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 2004; 27: 1047–1053 [DOI] [PubMed] [Google Scholar]
- 2.Margolis KL, Bonds DE, Rodabough RJ, et al. Effect of oestrogen plus progestin on the incidence of diabetes in postmenopausal women: results from the Women’s Health Initiative Hormone Trial. Diabetologia 2004; 47: 1175–1187 [DOI] [PubMed] [Google Scholar]
- 3.Kanaya AM, Herrington D, Vittinghoff E, et al. Glycemic effects of postmenopausal hormone therapy: the Heart and Estrogen/progestin Replacement Study. A randomized, double‐blind, placebo‐controlled trial. Ann Intern Med 2003; 138: 1–9 [DOI] [PubMed] [Google Scholar]
- 4.Liu S, Mauvais‐Jarvis F. Minireview: estrogenic protection of beta‐cell failure in metabolic diseases. Endocrinology 2010; 151: 859–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hulley S, Grady D, Bush T, et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA 1998; 280: 605–613 [DOI] [PubMed] [Google Scholar]
- 6.Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288: 321–333 [DOI] [PubMed] [Google Scholar]
- 7.Dubey RK, Tofovic SP, Jackson EK. Cardiovascular pharmacology of estradiol metabolites. J Pharmacol Exp Ther 2004; 308: 403–409 [DOI] [PubMed] [Google Scholar]
- 8.Mueck AO, Seeger H. 2‐Methoxyestradiol – biology and mechanism of action. Steroids 2010; 75: 625–631 [DOI] [PubMed] [Google Scholar]
- 9.Rajkumar SV, Richardson PG, Lacy MQ, et al. Novel therapy with 2‐methoxyestradiol for the treatment of relapsed and plateau phase multiple myeloma. Clin Cancer Res 2007; 13: 6162–6167 [DOI] [PubMed] [Google Scholar]
- 10.Matei D, Schilder J, Sutton G, et al. Activity of 2 methoxyestradiol (Panzem NCD) in advanced, platinum‐resistant ovarian cancer and primary peritoneal carcinomatosis: a Hoosier Oncology Group trial. Gynecol Oncol 2009; 115: 90–96 [DOI] [PubMed] [Google Scholar]
- 11.James J, Murry DJ, Treston AM, et al. Phase I safety, pharmacokinetic and pharmacodynamic studies of 2‐methoxyestradiol alone or in combination with docetaxel in patients with locally recurrent or metastatic breast cancer. Invest New Drugs 2007; 25: 41–48 [DOI] [PubMed] [Google Scholar]
- 12.Tevaarwerk AJ, Holen KD, Alberti DB, et al. Phase I trial of 2‐methoxyestradiol NanoCrystal dispersion in advanced solid malignancies. Clin Cancer Res 2009; 15: 1460–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sweeney C, Liu G, Yiannoutsos C, et al. A phase II multicenter, randomized, double‐blind, safety trial assessing the pharmacokinetics, pharmacodynamics, and efficacy of oral 2‐methoxyestradiol capsules in hormone‐refractory prostate cancer. Clin Cancer Res 2005; 11: 6625–6633 [DOI] [PubMed] [Google Scholar]
- 14.Sibonga JD, Lotinun S, Evans GL, et al. Dose‐response effects of 2‐methoxyestradiol on estrogen target tissues in the ovariectomized rat. Endocrinology 2003; 144: 785–792 [DOI] [PubMed] [Google Scholar]
- 15.Zacharia LC, Jackson EK, Gillespie DG, et al. Increased 2‐methoxyestradiol production in human coronary versus aortic vascular cells. Hypertension 2001; 37(2 Part 2): 658–662 [DOI] [PubMed] [Google Scholar]
- 16.Kurokawa A, Azuma K, Mita T, et al. 2‐Methoxyestradiol reduces monocyte adhesion to aortic endothelial cells in ovariectomized rats. Endocr J 2007; 54: 1027–1031 [DOI] [PubMed] [Google Scholar]
- 17.Tsukamoto A, Kaneko Y, Yoshida T, et al. 2‐Methoxyestradiol, an endogenous metabolite of estrogen, enhances apoptosis and beta‐galactosidase expression in vascular endothelial cells. Biochem Biophys Res Commun 1998; 248: 9–12 [DOI] [PubMed] [Google Scholar]
- 18.Campfield LA, Smith FJ, Guisez Y, et al. OB protein: a peripheral signal linking adiposity and central neural networks. Appetite 1996; 26: 302 [DOI] [PubMed] [Google Scholar]
- 19.Uchida T, Iwashita N, Ohara‐Imaizumi M, et al. Protein kinase Cdelta plays a non‐redundant role in insulin secretion in pancreatic beta cells. J Biol Chem 2007; 282: 2707–2716 [DOI] [PubMed] [Google Scholar]
- 20.Ebato C, Uchida T, Arakawa M, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high‐fat diet. Cell Metab 2008; 8: 325–332 [DOI] [PubMed] [Google Scholar]
- 21.Zhang X, Jia Y, Jackson EK, et al. 2‐Methoxyestradiol and 2‐ethoxyestradiol retard the progression of renal disease in aged, obese, diabetic ZSF1 rats. J Cardiovasc Pharmacol 2007; 49: 56–63 [DOI] [PubMed] [Google Scholar]
- 22.Fotsis T, Zhang Y, Pepper MS, et al. The endogenous oestrogen metabolite 2‐methoxyoestradiol inhibits angiogenesis and suppresses tumour growth. Nature 1994; 368: 237–239 [DOI] [PubMed] [Google Scholar]
- 23.Nishigaki I, Sasaguri Y, Yagi K. Anti‐proliferative effect of 2‐methoxyestradiol on cultured smooth muscle cells from rabbit aorta. Atherosclerosis 1995; 113: 167–170 [DOI] [PubMed] [Google Scholar]
- 24.Dubey RK, Gillespie DG, Jackson EK, et al. 17Beta‐estradiol, its metabolites, and progesterone inhibit cardiac fibroblast growth. Hypertension 1998;31(1 Pt 2): 522–528 [DOI] [PubMed] [Google Scholar]
- 25.Lippert TH, Adlercreutz H, Berger MR, et al. Effect of 2‐methoxyestradiol on the growth of methyl‐nitroso‐urea (MNU)‐induced rat mammary carcinoma. J Steroid Biochem Mol Biol 2003; 84: 51–56 [DOI] [PubMed] [Google Scholar]
- 26.LaVallee TM, Zhan XH, Johnson MS, et al. 2‐methoxyestradiol up‐regulates death receptor 5 and induces apoptosis through activation of the extrinsic pathway. Cancer Res 2003; 63: 468–475 [PubMed] [Google Scholar]
- 27.Yue TL, Wang X, Louden CS, et al. 2‐Methoxyestradiol, an endogenous estrogen metabolite, induces apoptosis in endothelial cells and inhibits angiogenesis: possible role for stress‐activated protein kinase signaling pathway and Fas expression. Mol Pharmacol 1997; 51: 951–962 [DOI] [PubMed] [Google Scholar]
- 28.Bu S, Blaukat A, Fu X, et al. Mechanisms for 2‐methoxyestradiol‐induced apoptosis of prostate cancer cells. FEBS Lett 2002; 531: 141–151 [DOI] [PubMed] [Google Scholar]
- 29.Shimada K, Nakamura M, Ishida E, et al. Roles of p38‐ and c‐jun NH2‐terminal kinase‐mediated pathways in 2‐methoxyestradiol‐induced p53 induction and apoptosis. Carcinogenesis 2003; 24: 1067–1075 [DOI] [PubMed] [Google Scholar]
- 30.Mabjeesh NJ, Escuin D, LaVallee TM, et al. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell 2003; 3: 363–375 [DOI] [PubMed] [Google Scholar]
- 31.Seeger H, Mueck AO, Lippert TH. The inhibitory effect of endogenous estrogen metabolites on copper‐mediated in vitro oxidation of LDL. Int J Clin Pharmacol Ther 1998; 36: 383–385 [PubMed] [Google Scholar]