

Abstract
Aims/Introduction: To evaluate the efficacy and safety of the glucagon‐like peptide‐1 receptor agonist, exenatide, in Japanese patients with type 2 diabetes mellitus suboptimally controlled despite therapeutic doses of a sulfonylurea alone or combined with a biguanide or thiazolidinedione.
Materials and Methods: Patients were randomized to a placebo or exenatide, either 5 or 10 μg, given subcutaneously b.i.d. in addition to oral therapy. Patients randomized to 10 μg exenatide received 5 μg b.i.d. for the first 4 weeks, followed by 10 μg b.i.d. for the last 20 weeks.
Results: A total of 179 patients received the study drug and composed the full analysis set (n = 35, placebo; n = 72, exenatide 5 μg; n = 72, exenatide 10 μg; 68% male; 58 ± 10 years; body mass index 25.5 ± 4.1 kg/m2; HbA1c 8.2 ± 0.9%; means ± standard deviations). Baseline to end‐point (least‐squares means ± standard errors) HbA1c changes (%) were −0.28 ± 0.15 (placebo), −1.34 ± 0.11 (exenatide 5 μg) and −1.62 ± 0.11 (exenatide 10 μg) (both P < 0.001, exenatide vs placebo). Baseline to end‐point bodyweight changes (kg) were −0.47 ± 0.39 (placebo), −0.39 ± 0.28 (exenatide 5 μg) and −1.54 ± 0.27 (exenatide 10 μg; P = 0.026, exenatide 10 μg vs placebo). Nausea, generally mild to moderate, was reported in 8.6% (placebo), 25.0% (exenatide 5 μg) and 36.1% (exenatide 10 μg) of patients. Mild to moderate hypoglycemia was reported in 22.9% (placebo), 51.4% (exenatide 5 μg) and 58.3% (exenatide 10 μg) of patients.
Conclusions: Over 24 weeks, exenatide vs the placebo improved glycemic control, reduced bodyweight (10 μg) and was well tolerated in Japanese patients with type 2 diabetes mellitus suboptimally controlled, despite oral therapy including a sulfonylurea. This trial was registered with ClinicalTrials.gov (no. NCT00577824). (J Diabetes Invest, doi: 10.1111/j.2040‐1124.2010.00084.x, 2011)
Keywords: Exenatide, Glycemic control, Japanese
Introduction
The number of diabetes patients in Japan is growing. According to an ‘Outline of the National Health and Nutrition Survey, Japan 2006’, which was carried out by The Health Service Bureau, Ministry of Health, Labor and Welfare, approximately 8.2 million people are strongly suspected of having diabetes (HbA1c ≥ 6.1%), the estimated population reaching approximately 18.7 million when those who cannot deny the possibility of having diabetes (6.0% ≥ HbA1c ≥ 5.6%) are included1. Japanese type 2 diabetes mellitus patients are less obese with the ‘thrifty’ genotype, which causes more insulin secretion deficiency and less insulin resistance than Westerners2.
Exenatide is the first in a class of anti‐diabetic agents known as glucagon‐like peptide‐1 (GLP‐1) receptor agonists. Exenatide, which is a 39‐amino acid peptide, shares several metabolic effects with endogenous GLP‐1, including glucose‐dependent stimulation of insulin secretion3, suppression of glucagon secretion4, slowing of gastric emptying5, and increasing satiety resulting in reduction of food intake and improving β‐cell function6,7. Because abnormal β‐cell function has been recognized as a major factor for the development of type 2 diabetes mellitus in Japanese patients8, exenatide might be beneficial for those Japanese patients.
A randomized, double‐blind, placebo‐controlled, parallel phase 2 study has previously evaluated the dose‐dependent effects on glycemic control and safety of 2.5, 5 and 10 μg exenatide over a period of 12 weeks in 153 Japanese patients whose type 2 diabetes mellitus was suboptimally controlled despite therapeutic doses of sulfonylurea (SU), biguanide (BG) or thiazolidine derivative (TZD). That study showed that, at doses up to 10 μg b.i.d., exenatide reduced plasma glucose in a dose‐dependent manner and was generally well tolerated9.
In the present study, we investigated safety and efficacy of exenatide in Japanese patients with suboptimally controlled type 2 diabetes mellitus over 24 weeks. This is the first phase 3 study of exenatide in Japanese patients with type 2 diabetes mellitus.
Subjects and Methods
Subjects
Japanese patients were included if they were between 20 and 75 years‐of‐age and had been diagnosed with type 2 diabetes mellitus according to the classifications by the Japan Diabetes Society and World Health Organization, and with bodyweight ≥ 50 kg. Patients were required to have been treated with SU monotherapy, combination therapy with SU and BG, or SU and TZD without any dose change for 90 days before screening. Patients on α‐glucosidase inhibitors (α‐GI) or short‐acting insulin secretion inducers at the time of screening could be included in the present study, but had to be discontinued and washed‐out for a period of 2–3 weeks.
Patients had inadequate glycemic control, as shown by HbA1c ≥ 7.0% and ≤10.0% at screening. Exclusion criteria included treatment with any exogenous insulin or drug directly affecting gastrointestinal motility within 90 days before screening, a clinically significant gastrointestinal disorder or hepatic disorder, serum creatinine ≥ 1.5 mg/dL in men or ≥1.4 mg/dL in women, and fasting plasma glucose (FPG) ≥ 250 mg/dL or casual blood glucose ≥ 350 mg/dL or at least one episode of severe hypoglycemia. Female patients of childbearing age were excluded if they were pregnant at the time of enrolment, intended to become pregnant during the study, had not practiced a reliable method of birth control for 90 days before screening or did not agree to continue practicing a reliable method of birth control during the study.
This study was carried out in 23 centers in Japan. Patients, investigators and the sponsor were unblinded to the injection volume, but blinded to the identity of exenatide and the placebo. Of 211 screened patients, 181 fulfilled inclusion/exclusion criteria (one patient decided to withdraw and 29 patients didn’t meet inclusion/exclusion criteria) and were randomly assigned (1:2:2) to subcutaneous injection of placebo, 5 μg exenatide or 10 μg exenatide b.i.d. using a dynamic allocation algorithm that involved stratification factors including HbA1c value and prior use of α‐GI. Patients randomized to exenatide 10 μg b.i.d. received 5 μg b.i.d. for the first 4 weeks, followed by 10 μg twice daily for the last 20 weeks. Following randomization, all patients were instructed to self‐administer the study drug into the abdomen within 60 min before their morning and evening meals.
Institutional review boards provided written approval of the study protocol and the informed consent document. The study was carried out in accordance with the ethical principles of the Declaration of Helsinki and was consistent with good clinical practices and applicable laws and regulations. Investigators obtained written informed consent from patients before carrying out protocol procedures. The study also included an open‐label extension period to 52 weeks, which will be reported separately.
Study End‐points
The primary end‐point was a change in HbA1c from baseline to end‐point (week 24 or last available observation on the treatment). All HbA1c values used in the present study were Japan Diabetes Society values. Secondary end‐points included: the percentage of patients who achieved HbA1c < 7.0% or <6.5% at end‐point (only patients with a baseline HbA1c ≥ 7% or ≥6.5% were eligible for the analysis), changes from baseline to end‐point of FPG, bodyweight, serum lipids (total cholesterol, low‐density lipoprotein [LDL] cholesterol, high‐density lipoprotein [HDL] cholesterol, triglycerides), 7‐point self‐monitored blood glucose (SMBG) levels (before breakfast, lunch and dinner, at 2 h after starting each meal and before bedtime), markers of insulin secretion, resistance and glycemic control (homeostasis model assessment beta cell function [HOMA‐B], homeostatis model assessment insulin resistance [HOMA‐R], 1,5‐anhydroglucitol), and safety. Safety measures included treatment‐emergent adverse events (TEAE) including hypoglycemia and antibodies to exenatide. Hypoglycemia was defined as the presence of any signs or symptoms of hypoglycemia, regardless of blood glucose concentration. Hypoglycemia events that showed a blood glucose level of <70 mg/mL without signs and symptoms were collected with the designation of ‘blood glucose decreased’. Hypoglycemia that required the assistance of another person was considered as severe hypoglycemia. If hypoglycemia symptoms occurred, the investigators instructed the subjects to measure blood glucose using the blood glucose meter for self‐monitoring. In addition, patients were instructed to notify the principal investigator if any hypoglycemia symptoms occurred or if their blood glucose level was <70 mg/dL. If a dosage of SU reduction was deemed appropriate by the investigator, the dosage was reduced to 50% or less of the dose at the time of the reported events.
Assays
Antibodies to exenatide were measured using a solid‐phase enzyme‐linked immunosorbent assay as previously described10.
Statistical Analyses
Data are presented for the full analysis set (FAS), which includes all randomized patients who received at least one dose of study drug and who had post‐baseline data available. All tests of treatment effects were carried out at a two‐sided significance level of 5%, unless otherwise stated. Unless otherwise stated, data are presented as mean ± standard deviation (SD).
The primary end‐point was change in HbA1c from baseline to end‐point. Change in HbA1c was evaluated by analysis of covariance with treatment group as a factor and baseline HbA1c as the covariate, and comparison with the placebo group was carried out with a t‐test using least square (LS) means. Here, in consideration of the multiplicity of tests between the placebo and the exenatide groups, the significance level was set to 2.5% (two‐sided) with Bonferroni’s method.
A dose–response relationship in the proportion of patients achieving the HbA1c target value was evaluated using the Cochran–Armitage test.
We set the sample size to be 175 patients (70 patients for each exenatide group) for the FAS in consideration of the requirement in the viewpoint of long safety evaluation11. This sample size was confirmed to ensure enough statistical power on the comparison of change in HbA1c from baseline to end‐point between the placebo and exenatide 10 μg groups.
Results
Patient Disposition and Baseline Characteristics
Patient disposition is shown in Figure 1. Of 181 randomized patients, 179 were included in FAS. Two patients (one in the placebo group, one in the exenatide 10 μg group) decided to discontinue before administration of the study drug. Overall, 152 patients (84%) in the FAS completed the study. The percentages of patients who discontinued after randomization were 5.6% (2/36) in the placebo group, 9.7% (7/72) in the exenatide 5 μg group and 27.4% (20/73) in the exenatide 10 μg group. Adverse events were the most common reason for discontinuation among exenatide patients, with nausea being the most frequent reason for discontinuation.
Figure 1.

Patients’ dispositions.
Patients in the FAS were generally balanced among treatment groups with respect to baseline characteristics (Table 1). Using a two‐sided significance level of 15%, no significant differences were observed. Oral anti‐diabetic drugs (OAD) at the time of informed consent are shown in Table 1. The most frequent combinations were SU combined with BG, and SU combined with BG and α‐GI.
Table 1. Baseline characteristics.
| Placebo | Exenatide 5 μg | Exenatide 10 μg | P† | |
|---|---|---|---|---|
| n = 35 | n = 72 | n = 72 | ||
| Sex (male), % | 24 (68.6) | 49 (68.1) | 49 (68.1) | 0.998 |
| Age (years) | 56.3 ± 11.4 | 58.5 ± 9.3 | 59.4 ± 9.8 | 0.321 |
| Weight (kg) | 70.3 ± 13.3 | 67.0 ± 11.5 | 69.1 ± 11.2 | 0.348 |
| Body mass index (kg/m2) | 25.8 ± 4.2 | 25.0 ± 4.1 | 25.8 ± 3.9 | 0.391 |
| Duration of type 2 diabetes (years) | 12.4 ± 6.5 | 12.2 ± 6.3 | 11.6 ± 7.0 | 0.764 |
| HbA1c (%) | 8.1 ± 0.9 | 8.3 ± 0.8 | 8.2 ± 1.0 | 0.679 |
| FPG (mg/dL) | 160 ± 31.4 | 164 ± 41.5 | 164 ± 39.0 | 0.818 |
| Total cholesterol (mg/dL) | 201 ± 25.7 | 204 ± 35.7 | 202 ± 30.8 | 0.880 |
| HDL cholesterol (mg/dL) | 56 ± 12.5 | 57 ± 14.9 | 55 ± 10.6 | 0.556 |
| LDL cholesterol (mg/dL) | 123 ± 24.0 | 124 ± 28.3 | 125 ± 27.1 | 0.917 |
| Triglycerides (mg/dL) | 126 ± 79.2 | 133 ± 95.1 | 131 ± 69.8 | 0.914 |
| Oral anti‐diabetic agents at informed consent | ||||
| SU alone | 3 (8.6) | 4 (5.6) | 8 (11.1) | |
| SU + α‐GI | 3 (8.6) | 1 (1.4) | 4 (5.6) | |
| SU + BG | 14 (40.0) | 33 (45.8) | 27 (37.5) | |
| SU + BG + α‐GI | 9 (25.7) | 22 (30.6) | 13 (18.1) | |
| SU + BG + meglitinide derivative | 0 (0.0) | 0 (0.0) | 1 (1.4) | |
| SU + TZD | 4 (11.4) | 6 (8.3) | 12 (16.7) | |
| SU + TZD + α‐GI | 2 (5.7) | 6 (8.3) | 7 (9.7) | |
Data are n (%) or means ± SD for the full analysis set. α‐GI, α‐glucosidase inhibitors; BG, biguanide; FPG, fasting blood glucose; SU, sulfonylurea; TZD, thiazolidine derivative. †P for comparison among the treatment groups.
Changes in Glycemic Control and Bodyweight
The mean change in HbA1c level from baseline to each visit is shown in Figure 2. Reduction became apparent at week 4 in both exenatide groups and continued until week 16. Reduction was maintained at all visits after week 16 up to week 24 in both exenatide groups. The LS mean change ± standard error (SE) in HbA1c level observed from baseline to end‐point were −0.28 ± 0.15% in the placebo group, −1.34 ± 0.11% in the 5 μg exenatide group and −1.62 ± 0.11% in the 10 μg exenatide group. The changes in HbA1c level were significantly greater (P < 0.001) in both exenatide treatment groups than in the placebo group.
Figure 2.
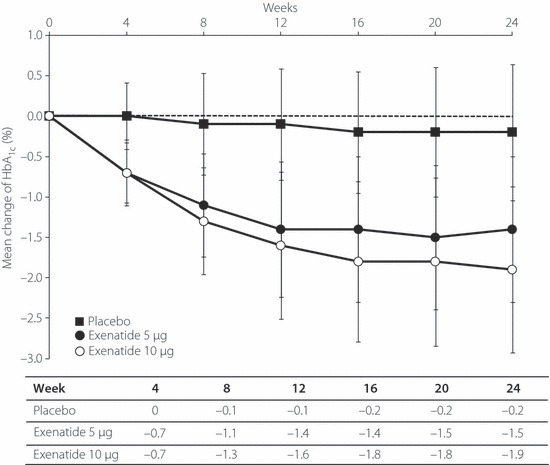
Mean changes of HbA1c from baseline to each visit. Data are mean ± SD for the full analysis set. The dotted line represents the baseline value.
The percentage of patients achieving the HbA1c target value of <7.0% at end‐point was 15.2% (5/33) in the placebo group, 67.1% (47/70) in the exenatide 5 μg group and 71.0% (49/69) in the exenatide 10 μg group (Figure 3). The percentage of patients achieving the HbA1c target value of <6.5% at end‐point was 8.6% (3/35) in the placebo group, 36.6% (26/71) in the exenatide 5 μg group and 47.2% (34/72) in the exenatide 10 μg group (Figure 3). There was a statistically significant dose–response relationship in the proportion of patients achieving the HbA1c target value of both <7.0% and <6.5% at end‐point (both P < 0.001).
Figure 3.
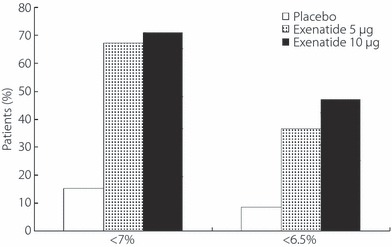
Percentage of patients who received placebo, exenatide 5 μg or exenatide 10 μg achieving HbA1c <7.0% or <6.5% at end‐point.
Figure 4 shows the mean change in fasting blood glucose concentration from baseline to each visit. Marked reductions were observed at 4 weeks in both the exenatide 5 and 10 μg groups, and these reductions in mean fasting blood glucose concentration were sustained at all visits after week 4 up to the study end in each exenatide group. The LS mean change ± SE in the fasting blood glucose from baseline to end‐point was −7.59 ± 5.46 mg/dL in the placebo group, −25.15 ± 3.83 mg/dL in the exenatide 5 μg group and −29.00 ± 3.81 mg/dL in the exenatide 10 μg group based on the same ancova model as that used for the primary analysis. Changes in fasting blood glucose were significantly greater in the exenatide treatment groups than in the placebo group (5 μg group, P = 0.009; 10 μg group, P = 0.002).
Figure 4.

Mean changes of fasting blood glucose from baseline to each visit. Data are means ± SD for full analysis set. The dotted line represents the baseline value.
Figure 5 shows a summary of 7‐point SMBG concentration by treatment group. Reductions from baseline were observed in both exenatide treatment groups and were especially marked in the postbreakfast and postdinner blood glucose levels for patients who were in the exenatide 10 μg group.
Figure 5.

Effects of (a) placebo, (b) exenatide 5 μg and (c) exenatide 10 μg on 7‐point self‐monitored blood glucose at end‐point. Data are means ± SD for the full analysis set.
The mean ± SE changes in 1,5‐anhydroglucitol from baseline to end‐point were 0.66 ± 0.30 μg/mL in the placebo group, 5.29 ± 0.55 μg/mL in the exenatide 5 μg group and 4.52 ± 0.54 μg/mL in the exenatide 10 μg group. The increase in 1,5‐anhydroglucitol was significantly greater in both exenatide treatment groups than in the placebo group (exenatide 5 μg, P < 0.001; exenatide 10 μg, P < 0.001).
The LS means change ± SE in bodyweight from baseline to end‐point were −0.47 ± 0.39 kg in the placebo group, −0.39 ± 0.28 kg in the exenatide 5 μg group and −1.54 ± 0.27 kg in the exenatide 10 μg group based on the ancova model. Changes in bodyweight were significantly greater (P = 0.026) in the exenatide 10 μg group than in the placebo group (Figure 6).
Figure 6.
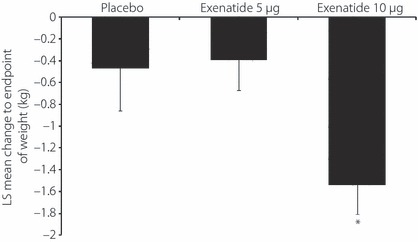
Change in bodyweight from baseline to end‐point. Data are least square (LS) means − SE for the full analysis set. *P = 0.026 vs placebo.
Reductions in total cholesterol, LDL‐cholesterol and HDL‐cholesterol from baseline were observed in both exenatide treatment groups. The mean changes in HDL‐cholesterol were −1 ± 7.1 mg/dL in the placebo group, −5 ± 7.6 mg/dL in the exenatide 5 μg group and −4 ± 5.9 mg/dL in the exenatide 10 μg group. The reduction in HDL‐cholesterol was statistically significantly greater in both exenatide groups (exenatide 5 μg, P = 0.020; exenatide 10 μg, P = 0.014) than in the placebo group. No statistically significant difference was observed in total cholesterol, LDL‐cholesterol and triglycerides. All changes in total cholesterol, LDL‐cholesterol, HDL‐cholesterol and triglycerides were within the normal range.
HOMA‐B and HOMA‐R were calculated using serum insulin and fasting blood glucose. The mean in HOMA‐B values at baseline were similar: 27.55 ± 24.44 (placebo), 30.87 ± 38.04 (exenatide 5 μg) and 27.96 ± 26.49 (exenatide 10 μg). The mean changes from baseline to end‐point on HOMA‐B were −0.71 ± 20.33 (placebo), 2.48 ± 88.40 (exenatide 5 μg) and 6.84 ± 117.59 (exenatide 10 μg). Mean HOMA‐R values at baseline were 2.48 ± 1.76 (placebo), 2.86 ± 3.04 (exenatide 5 μg) and 2.58 ± 1.71 (exenatide 10 μg). The mean changes in HOMA‐R at end‐point were −0.38 ± 0.97 (placebo), −0.54 ± 3.37 (exenatide 5 μg) and −0.37 ± 1.59 (exenatide 10 μg). No statistically significant differences in mean HOMA‐B and HOMA‐R were observed between the treatment groups.
Safety
Serious adverse events (SAE) were reported in six patients: four patients in the placebo group and two patients in the exenatide 5 μg group. SAE in the exenatide 5 μg group were prostate cancer, major depression and diabetic ketoacidosis. No deaths occurred during this study. There were no SAE reported in the exenatide 10 μg group. The incidences of adverse events other than SAE leading to withdrawal from the study were 0.0% (0/35) in the placebo group, 5.6% (4/72) in the exenatide 5 μg group and 25.0% (18/72) in the exenatide 10 μg group.
TEAE accounted for 74.3% (26/35) in the placebo group, 97.2% (70/72) in the exenatide 5 μg group and 94.4% (68/72) in the exenatide 10 μg group. TEAE reported by >10% of patients either in the exenatide 5 μg or the exenatide 10 μg groups are shown in Table 2. Hypoglycemia, nausea and vomiting diminished over time with continued use of exenatide.
Table 2. Treatment‐emergent adverse events with incidence >10% either in exenatide 5 μg or 10 μg group.
| Placebo n = 35 n (%) | Exenatide | ||
|---|---|---|---|
| 5 μg n = 72 n (%) | 10 μg n = 72 n (%) | ||
| Hypoglycemia | 8 (22.9) | 37 (51.4) | 42 (58.3) |
| Nausea | 3 (8.6) | 18 (25.0) | 26 (36.1) |
| Blood glucose decreased | 4 (11.4) | 10 (13.9) | 18 (25.0) |
| Vomiting | 1 (2.9) | 3 (4.2) | 12 (16.7) |
| Constipation | 1 (2.9) | 10 (13.9) | 11 (15.3) |
| Nasopharyngitis | 8 (22.9) | 8 (11.1) | 9 (12.5) |
| Stomach discomfort | 1 (2.9) | 7 (9.7) | 9 (12.5) |
| Appetite loss | 1 (2.9) | 7 (9.7) | 9 (12.5) |
| Anorexia | 1 (2.9) | 2 (2.8) | 8 (11.1) |
| Diarrhea | 2 (5.7) | 8 (11.1) | 4 (5.6) |
During the study, 22.9% (8/35), 51.4% (37/72) and 58.3% (42/72) of patients in the placebo, exenatide 5 μg and exenatide 10 μg groups reported hypoglycemia, respectively. With the one exception in the exenatide 5 μg group, all reported hypoglycemia were judged to be related to the study drug by the study investigators. Also, all cases of hypoglycemia were mild in severity with the exception of one case of moderate hypoglycemia in the exenatide 10 μg group. No severe hypoglycemia occurred during the study.
At end‐point, 59.7% (43/72) and 44.4% (32/72) of patients in the exenatide 5 μg and exenatide 10 μg groups, respectively, had detectable antibodies to exenatide (≥1/25 dilution). No relationship between anti‐exenatide antibody status and overall TEAE incidence was observed, nor were any dose‐dependent trends observed.
Discussion
This is the first phase 3 study to evaluate exenatide efficacy and safety in Japanese patients with type 2 diabetes mellitus. The study shows that exenatide in combination with OAD including SU improved glycemic control, as evidenced by improvements in HbA1c, fasting blood glucose levels and postprandial blood glucose, and also reduced bodyweight over 24 weeks in patients with type 2 diabetes mellitus who were suboptimally controlled with OAD. The most commonly reported adverse events associated with exenatide treatment were nausea and hypoglycemia. No severe hypoglycemia was reported during the study.
Patients in the present study had been diagnosed with type 2 diabetes mellitus on average for 10–15 years, with mean HbA1c values at baseline of 8.1–8.3%, indicating a moderately advanced stage of type 2 diabetes mellitus. The LS mean change ± SE in HbA1c (−1.34 ± 0.11%, −1.62 ± 0.11%) and FPG (−25.15 ± 3.83 mg/dL, −29.00 ± 3.81 mg/dL) observed from baseline to end‐point for the exenatide 5 μg treatment group and the exenatide 10 μg group, respectively, were clinically meaningful and consistent with those from previous studies of exenatide b.i.d. in primarily Caucasian, Asian and Japanese populations9,10,12–17.
In the present study, both the exenatide 5 and 10 μg groups reduced postprandial glucose (PPG) compared with the placebo group. The importance of PPG concentration in patients with type 2 diabetes mellitus has been recognized. The Diabetes Epidemiology Collaborative Analysis of Diagnostic Criteria in Europe study group reported that 2‐h oral glucose tolerance test levels were a better predictor of on‐study death from all causes and from cardiovascular disease than FPG levels18. The guideline for Management of Postmeal glucose, recently issued by the International Diabetes Federation, stated that postmeal and postchallenge hyperglycemia are independent risk factors for macrovascular disease as a major evidence statement. The guideline highlights the importance of PPG control in the context of an overall strategy to improve glycemic control as measured by HbA1c levels19.
In the current study, only the exenatide 10 μg group significantly reduced bodyweight compared with the placebo. This apparent weight loss as a result of exenatide is important, because other anti‐diabetic therapies, including SU, TZD and insulins, can cause weight gain in type 2 diabetes mellitus patients20. In some previous placebo‐controlled studies of exenatide 5 and 10 μg combined with OAD, both doses of exenatide were associated with a reduction in bodyweight13,15. This difference might be related to the difference of baseline body mass index, which was greater in the predominantly Caucasian patient population of the earlier studies (33–34 kg/m2) compared with the Japanese population of the current study (26 kg/m2). Not all exenatide‐induced reductions in bodyweight are explained by gastrointestinal side‐effects, as weight loss was observed in subjects who never experienced these adverse events. There was essentially no correlation between weight change and a subject’s total days of nausea21.
A significant dose‐dependent increase in the incidence of hypoglycemia was observed in the current study. It is reported that exenatide enhances insulin secretion in a glucose‐dependent manner, wherein insulin secretion decreases as glucose levels normalize7,22. This mechanism would reduce the potential for exenatide to cause hypoglycemia. However, up to 54.9% of exenatide‐treated patients reported hypoglycemia during the present study. This observation is likely because all patients were taking a concomitant SU with or without additional concomitant oral agent(s) (BG or TZD). In previous placebo‐controlled studies of exenatide combined with OAD carried out in primarily Caucasian populations, concomitant SU have been implicated in increasing the incidence of hypoglycemia when coupled with lower ambient glycemia and increasing exenatide dose12,13. Patients in the placebo arms of the present study were treated with background SU therapy (with or without additional OAD); hypoglycemia was reported in 22.9% of placebo‐treated patients. Hypoglycemia was reported in 38.5% of the glibenclamide‐treated patients in the study of a GLP‐1 receptor agonist compared with SU for monotherapy (liraglutide and glibenclamide) in Japanese patients23. A proactive approach to SU dose management has been suggested in order to limit the incidence of hypoglycemia in exenatide‐treated patients13. However, in the present study, SU was discontinued or the dose was reduced after a documented hypoglycemic episode instead of proactive SU dose adjustment. Established dose adjustment guidelines for the concomitant use of exenatide and SU in Japanese patients would be expected.
Consistent with the potentially immunogenic properties of protein and peptide pharmaceuticals, antibodies to exenatide were measured. In the present study, the presence of antibodies to exenatide had no clinically relevant effect.
In the present study, the average duration of diabetes was more than 10 years, which is consistent with that reported in a previous study of Japanese patients9. However, the duration of diabetes in the present study was longer than those from previous studies of exenatide b.i.d. in patient populations that were primarily Caucasian and from other Asian countries12–14,17. The difference was caused by different inclusion criteria. In the current study, patients who were receiving treatment with from one to up to three concomitant OAD were included. In contrast, in other studies, patients who were medicated with only one concurrent OAD were included.
The present study had several limitations. Patients, investigators and the sponsor were blinded to the distinction between exenatide and the placebo, but were not blinded to injection volume. A fully double‐blind design might have been more robust. Also, there were no standardized diet and exercise recommendations in the current study, and we do not know what effect this additional treatment had on bodyweight changes.
In conclusion, both the 5‐ and 10‐μg b.i.d. doses of exenatide improved glycemic control and study results suggest that 10 μg b.i.d. might provide the additional benefit of weight loss for Japanese patients who are suboptimally controlled with OAD. The safety profile is similar to previous studies. The current study has shown that exenatide appears to provide glycemic control benefits for Japanese patients with type 2 diabetes mellitus similar to those reported in Caucasian and other Asian patients.
Acknowledgements
This study was supported by Amylin Pharmaceuticals (San Diego, CA, USA) and Eli Lilly and Company (Indianapolis, IN, USA). T Kadowaki has participated in advisory boards for Novo Nordisk, Novartis, and Eli Lilly and Company; has received consulting fees from Novo Nordisk, Novartis and Merck; and has received financial support or grants to carry out research from Novo Nordisk and Eli Lilly and Company. M Namba has participated in advisory boards for Novo Nordisk, Novartis and Banyu Pharmaceutical Company, Ltd; has received consulting fees from Novo Nordisk and Banyu Pharmaceutical Company; and has received financial support or grants to carry out research from Novo Nordisk, Eli Lilly and Company, Banyu Pharmaceutical Company, Sanwa Kagaku Kenkyusho and Ono Pharmaceutical. A Yamamura, T Imaoka, W Goto and H Sowa are employees and shareholders of Eli Lilly Japan KK (Kobe, Japan). MK Boardman is an employee and shareholder of Eli Lilly and Company (Indianapolis, IN, USA).
References
- 1.Health, Labor and Welfare Association, Ministry of Health . Outline of the National Health and Nutrition Survey. Japan 2006. [In Japanese]. http://www.mhlw.go.jp/houdou/2008/04/dl/h0430‐2c.pdf
- 2.Kawamori R. Diabetes trends in Japan. Diabetes Metab Res Rev 2002; 18: S9–S13 [DOI] [PubMed] [Google Scholar]
- 3.Parkes DG, Pittner R, Jodka C, et al. Insulinotropic actions of exendin‐4 and glucagon‐like peptide‐1 in vivo and in vitro. Metabolism 2001; 50: 583–589 [DOI] [PubMed] [Google Scholar]
- 4.Kolterman OG, Buse JB, Fineman MS, et al. Synthetic exendin‐4 (exenatide) significantly reduces postprandial and fasting plasma glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab 2003; 88: 3082–3089 [DOI] [PubMed] [Google Scholar]
- 5.Nauck MA, Wollschläger D, Werner J, et al. Effects of subcutaneous glucagon‐like peptide 1 (GLP‐1 [7‐36 amide]) in patients with NIDDM. Diabetologia 1996; 12: 1546–1553 [DOI] [PubMed] [Google Scholar]
- 6.Li Y, Hansotia T, Yusta B, et al. Glucagon‐like peptide‐1 receptor signaling modulates beta cell apoptosis. J Biol Chem 2003; 278: 471–478 [DOI] [PubMed] [Google Scholar]
- 7.Triplitt CL. New technologies and therapeies in the management of diabetes. Am J Manag Care 2007; 13: S47–S54 [PubMed] [Google Scholar]
- 8.Kuroe A, Fukushima M, Usami M, et al. Impaired beta‐cell function and insulin sensitivity in Japanese subjects with normal glucose tolerance. Diabetes Res Clin Pract 2003; 59: 71–77 [DOI] [PubMed] [Google Scholar]
- 9.Kadowaki T, Namba M, Yamamura A, et al. Exenatide exhibits dose‐dependent effects on glycemic control over 12 weeks in Japanese patients with suboptimally controlled type 2 diabetes. Endocr J 2009; 56: 415–424 [DOI] [PubMed] [Google Scholar]
- 10.Fineman MS, Bicsak TA, Shen LZ, et al. Effect on glycemic control of exenatide (synthetic exendin‐4) additive to existing metformin and/or sulfonylurea treatment in patients with type 2 diabetes. Diabetes Care 2003; 26: 2370–2377 [DOI] [PubMed] [Google Scholar]
- 11.Health, Labor and Welfare Association, Ministry of Health . Extent of population exposure to assess clinical safety for drugs intended for long‐term treatment of non‐life‐threatening conditions. Notification No.592 of the Pharmaceuticals and Cosmetic Division 1995. [In Japanese].
- 12.Buse JB, Henry RR, Han J, et al. Effects of exenatide (exendin‐4) on glycemic control over 30 weeks in sulfonylurea‐treated patients with type 2 diabetes. Diabetes Care 2004; 27: 2628–2635 [DOI] [PubMed] [Google Scholar]
- 13.Kendall DM, Riddle MC, Rosenstock J, et al. Effects of exenatide (exendin‐4) on glycemic control over 30 weeks in patients with type 2 diabetes treated with metformin and a sulfonylurea. Diabetes Care 2005; 28: 1083–1091 [DOI] [PubMed] [Google Scholar]
- 14.Zinman B, Hoogwerf BJ, Durán García S, et al. The effect of adding exenatide to a thiazolidinedione in suboptimally controlled type 2 diabetes: a randomized trial. Ann Intern Med 2007; 146: 477–485 [DOI] [PubMed] [Google Scholar]
- 15.DeFronzo RA, Ratner RE, Han J, et al. Effects of exenatide (exendin‐4) on glycemic control and weight over 30 weeks in metformin‐treated patients with type 2 diabetes. Diabetes Care 2005; 28: 1092–1100 [DOI] [PubMed] [Google Scholar]
- 16.Poon T, Nelson P, Shen L, et al. Exenatide improves glycemic control and reduces body weight in subjects with type 2 diabetes: a dose‐ranging study. Diabetes Technol Ther 2005; 7: 467–477 [DOI] [PubMed] [Google Scholar]
- 17.Gao Y, Yoon KH, Chuang LM, et al. Efficacy and safety of exenatide in patients of Asian descent with type 2 diabetes inadequately controlled with metformin or metformin and a sulphonylurea. Diabetes Res Clin Pract 2009; 83: 69–76 [DOI] [PubMed] [Google Scholar]
- 18.The DECODE study group, the European Diabetes Epidemiology Group . Glucose tolerance and cardiovascular mortality: comparison of fasting and 2‐hour diagnostic criteria. Arch Intern Med 2001; 161: 397–405 [DOI] [PubMed] [Google Scholar]
- 19.International Diabetes Federation: Guideline for Management of Postmeal Glucose. 2005; http://www.idf.org/webdata/docs/Guideline_PMG_final.pdf . [DOI] [PMC free article] [PubMed]
- 20.Riddle MC. Glycemic management of type 2 diabetes: an emerging strategy with oral agents, insulins, and combinations. Endocrinol Metab Clin North Am 2005; 34: 77–98 [DOI] [PubMed] [Google Scholar]
- 21.Blonde L, Klein EJ, Han J, et al. Interim analysis of the effects of exenatide treatment on A1C, weight and cardiovascular risk factors over 82 weeks in 314 overweight patients with type 2 diabetes. Diabetes Obes Metab 2006; 8: 436–447 [DOI] [PubMed] [Google Scholar]
- 22.Drucker DJ. Enhancing incretin action for the treatment of type 2 diabetes. Diabetes Care 2003; 26: 2929–2940 [DOI] [PubMed] [Google Scholar]
- 23.Seino Y, Rasmussen MF, Nishida T, et al. Efficacy and safety of the once‐daily human GLP‐1 analogue, liraglutide, vs glibenclamide monotherapy in Japanese patients with type 2 diabetes. Curr Med Res Opin 2010; 26: 1013–1022 [DOI] [PubMed] [Google Scholar]