

Summary
Bronchopulmonary dysplasia (BPD), a multifactorial disease of preterm neonates of complex etiology, is a significant problem within very low birth weight infants. Nitric oxide (NO) has been implicated in both the pathogenesis and as a potential therapeutic of this disease. At this time, there is little direct evidence of the changes in NO production and metabolism that occur within BPD in humans. Animal models have implied that reduced nitric oxide synthase (NOS) expression and NO production in the early stages of the disease may be critical factors. However, inflammation and hence iNOS expression, is also thought to play a role. In the present study we have utilized pathological samples to determine changes in the expression of NOS and NO metabolites within late stage BPD. It is our contention that within these samples iNOS expression is increased and associated with increased NO metabolite production. Mild immunostaining of all three nitric oxide synthase (NOS) enzymes (neuronal, inducible and endothelial) is observed in control lung with tight localization to the endothelium and epithelial airway. This tight localization was lost in samples from subjects with BPD. There was also a marked increase in iNOS expression throughout the lung tissue with strong coexistence with an epithelial cell marker cytokeratin. NO reaction products are altered with BPD as evidenced by increased S-nitrosothiol (SNO) and strong nitrotyrosine (NO2Y) imunoreactivity. This study demonstrates a strong correlation between products of NO reactivity and NOS localization in BPD.
Keywords: bronchopulmonary dysplasia, chronic lung disease, inflammation, S-nitrosothiol, nitrotyrosine
INTRODUCTION
Bronchopulmonary dysplasia (BPD) is a disease of the lung in the premature infant defined as a requirement for supplemental oxygen at 36 weeks postmenstrual age.1 This pathophysiology appears to be caused by a combination of factors including inflammatory injury, mechanical ventilation, oxygen therapy and lung prematurity. When first described by Northway in 1967, premature infants who developed the disease were more mature (31–34 weeks gestation) than those seen today.2 Because of advances in mechanical ventilation, surfactant replacement therapy and improvements in neonatal intensive care, the incidence of BPD in infants older than 30 weeks gestation has decreased. However, in more premature infants (gestational age less than 28 weeks) and those of a very low birth weight (less than 1,500 g) BPD continues to be problematic.3–5 Infants described by Northway typically had a history of severe hyaline membrane disease, whereas today, the disease appears to be manifested primarily by arrested alveolar development.6 While treatment induced oxygen injury and volutrauma continues to play a role in BPD, extreme lung immaturity, inflammation and multiple other factors have been implicated in an animal model of BPD.7 NO produced endogenously by the three isoforms of the enzyme nitric oxide synthase (NOS) (neuronal, inducible and endothelial) has multiple functions within many biological systems.8,9 In particular it plays an important role within the functioning airway epithelium.10–12 Nitric oxide has now been recognized to play a key role in several inflammatory airway diseases, including BPD. Although the etiology of BPD is multifactorial, it has been shown that one factor, hyperoxia, results in a loss of NO-mediated airway relaxation, contributing to increased airway contractility under in vivo conditions. In a fetal baboon model of BPD, decreased levels of two of the three NOS isoforms (neuronal and endothelial) and a parallel down regulation of NO production was characteristic in the early postnatal period of the disease.13
Within human studies, it has been suggested that products of NO metabolism such as NO2Y may be biomarkers of the disease,14,15 suggesting that NO, potentially produced by iNOS as a result of inflammatory processes, may play a role. However, such studies have only been able to examine secondary evidence of NO function, namely biomarkers in plasma or urine. It is our proposal that these peripheral changes in NO metabolites are associated with increased iNOS expression and NO metabolite production within the lung. In order to understand how NOS expression is altered, and how this affects NO metabolite production, we have undertaken an immunohistochemical investigation of both NOS expression and the formation of NO metabolites in pathological samples from infants with and without BPD. These studies reveal that not only is NO production apparently altered within BPD, but that the metabolism of NO is also considerably changed.
MATERIALS AND METHODS
Study Samples
This study was carried out under an institutionally approved protocol from the IRB at the Children’s Hospital of Philadelphia. Sixteen autopsy and biopsy lung tissue samples were obtained from a tissue repository and clinical database of a Specialized Center of Research Program between 1995 and 2002. BPD was defined as a requirement for supplemental oxygen or assisted ventilation at 36 weeks postmenstrual age (n = 7). Control subjects (n = 7). The characteristics of the samples are shown in Table 1. It should be noted that the samples from subjects 6 & 7 were prepared from biopsies of the recipient not the donor lung. All of the other samples were from tissue obtained at autopsy. Study sections were paraffin embedded and cut in serial sections.
TABLE 1.
Characteristics of Study Group
| Study no. | Gestational age (weeks) | Primary pulmonary diagnoses | Cause of death | Age at death or transplant |
|---|---|---|---|---|
| 1 | 24 | BPD | BPD, sepsis, arrest | 7 Months |
| 2 | 24 | BPD | BPD | 4 Months |
| 3 | 29 | BPD | Endocarditis | 3 Months |
| 4 | 25 | BPD | Sepsis | 39 Days |
| 5 | 27 | BPD | Respiratory failure, sepsis | 31 Days |
| 6 | NK | Alveolar proteinosis and BPD | Transplant related | 11 Months |
| 7 | 29 | Congenital chylothorax and BPD | Transplant related | 12 Months |
| 8 | Term | Control | NK | Stillborn |
| 9 | Term | Control | Brain Tumor | 1 Day |
| 10 | Term | Control | IVH | 2 Months |
| 11 | Term | Control | Hydrocephalus | 4 Days |
| 12 | Term (donor) | Control | Trauma | NK |
| 13 | 36 | Control | Cerebral infarct | 6 Days |
| 14 | 32 | Control | Gastroschesis | 3 Days |
BPD: bronchopulmonary dysplasia; CLD: chronic lung disease; IVH: intraventricular hemorrhage; NK: unknown.
Detection of nNOS, iNOS and eNOS by Immunohistochemistry
Lung sections were deparafinized and re-hydrated before antigen retrieval via low power microwave exposure for 5 min. To quench background fluorescence, sections were washed in fresh solutions of saturated sodium borohydride which not only removes the excess aldehydes from tissue preparation but it also efficiently reduces processing-related fluorescence in glutaraldehyde-fixed tissues. After blocking non-specific antigen binding, sections were incubated with primary antibodies overnight in 4°C. Pure goat serum (Sigma, St. Louis, MO) was used for all antibody dilutions. The following antibodies and dilutions were used: polyclonal anti-nNOS primary antibody (BD Transduction, San Jose, CA), 1:100; polyclonal anti-iNOS primary antibody (Calbiochem, San Diego, CA), 1:500; polyclonal anti-eNOS primary antibody (BD Transduction), 1:100. Since the antibodies to the three NOS isoforms do not cross react, they each served as controls for the other isoforms. Additional control was achieved by omission of primary antibody in serial sections. A fragment of affinity isolated antigen specific antibody from Sigma (Anti-Rabbit Cy3 conjugate, 1:100 dilution) was used as secondary antibody.
Detection of SNO and NO2Y
Immunohistochemical detection of NO metabolites was carried out as described for NOS isoforms and as previously described.16,17 Rabbit polyclonal anti-SNO, 1:100 dilution (Calbiochem) was used to localize S-nitrosylated proteins in tissues. Mercury chloride pre-treatment of the slides was used as a negative control. Anti-Rabbit IgG, Cy3 conjugate (Sigma) was used as secondary antibody. To detect nitrated proteins, mouse monoclonal anti-NO2Y (a gift from Dr. H. Ischiropoulos) was used. Sodium hydrosulfite which reduces nitrotyrosine to aminotyrosine was used on serial sections as a negative control. Anti-mouse IgG (Fab specific fluorescein isothiocyanate) from Sigmawas used as a secondary antibody.
Double Immunofluorescence Staining
Lung sections were prepared as described above prior to treatment with the primary antibody. Monoclonal mouse anti-human CD68 (Serotec, Raleigh, NC, 1:200 dilution), an inflammatory cell marker combined with anti-nNOS or anti-iNOS primary antibodies. Cytokeratin, a monoclonal pan epithelial cell marker (Zymed, San Francisco, CA 1:250 dilution) was diluted in goat serum and incubated at 4°C over night. The isotype matched fluorescence conjugated secondary antibodies were used to detect the specific proteins.
Microscopy and Image Analysis
An inverted fluorescence microscope (Nikon, Melville, NY) and a Metamorph imaging software (Universal Imaging Corp., PA) were used for analysis. Staining intensity was characterized as follows: 0 = negative or the same level as background staining; 1 = slightly more than background staining; 2 = mild positivity; 3 = moderate; 4 = intense staining. Scoring was performed by different investigators in a blinded fashion.
Statistical Analysis
Reported scores are expressed as mean. Non-parametric median of these scores (three to five areas were examined) are shown. Comparisons between control group and those with BPD as respects to overall immunoreactivity were made using Wilcoxon’s rank test with a significance level of <0.05.
RESULTS
NOS Immunoreactivity
All three NOS isoforms (nNOS, iNOS and eNOS) were detected in both control and BPD lung tissue. Minimal iNOS staining was observed in control lung samples and what could be seen was restricted to large airway epithelium (Fig. 1B). Notably, both the quantity and distribution of iNOS was markedly altered in BPD where intense immunoreactivity was observed throughout the lung (Fig. 1D, Table 2). In control lung, the predominant eNOS staining occurs in the large blood vessels and airway lining. There is a moderate degree of staining in small blood vessels but no eNOS is apparent within the lung parenchyma (Fig. 1F). On the other hand, BPD lungs show moderate staining throughout the lung, although still predominantly in blood vessels (Fig. 1H). In control lung nNOS was predominantly located within the pulmonary epithelium, in contrast to the other isoforms there was no significant change in nNOS staining within the BPD lung samples (Fig. 1J and L, Table 2).
Fig. 1.
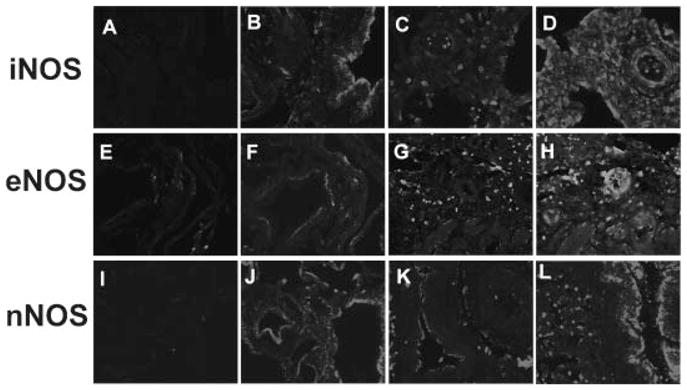
NOS Staining in BPD and control lung. Immunohistochemical staining of NOS isotypes iNOS (A–D), eNOS (E–H) and nNOS (I–L) in control lung (A, B, E, F, I, J) and BPD lung (C, D, G, H, K, L). Stains are shown in panels B, D, F, H, J, and L, corresponding negative controls are shown in panels A, C, E, G, I and K. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
TABLE 2.
Nonparametric Scores of NOS Isoforms and NO Metabolites
| Study group | Primary diagnosis | eNOS | nNOS | iNOS | NO2Y | SNO |
|---|---|---|---|---|---|---|
| 1 | BPD | 2 | 2 | 3 | 4 | 4 |
| 2 | BPD | 3 | 2 | 2 | 4 | 4 |
| 3 | BPD | 3 | 3 | 2 | 3 | 4 |
| 4 | BPD | 0 | 0 | 3 | 3 | 4 |
| 5 | BPD | 0 | 0 | 0 | 2 | 0 |
| 6 | BPD | 2 | 2 | 2 | 1 | 1 |
| 7 | BPD | 1 | 1 | NK | 2 | 4 |
| Median | BPD | 2 | 2 | 2* | 3* | 4* |
| 8 | Control | 0 | 1 | 1 | 3 | 2 |
| 9 | Control | 0 | 2 | 0 | 1 | 0 |
| 10 | Control | 0 | 2 | 0 | 1 | 0 |
| 11 | Control | 1 | 0 | 0 | 1 | 2 |
| 12 | Control | 1 | 3 | 0 | 2 | 0 |
| 13 | Control | 2 | 2 | 0 | 2 | 2 |
| 14 | Control | 1 | 2 | 0 | 1 | 2 |
| Median | Control | 1 | 2 | 0 | 1 | 2 |
The degree of staining for SNO and NO2Yon a zero to four scale (0 = no staining or same level as background staining; 1 = slightly more than background staining; 2 = mild positivity, 3 = moderate; 4 = intense staining).
NK: unknown.
Represents significantly different from control, P <0.05.
NOS in Inflammatory and Epithelial Cells
There is a considerable morphological rearrangement in the lungs of infants with BPD. Therefore, in order to know what cell types express NOS within BPD, we chose to conduct dual-staining of iNOS or nNOS and cell markers. We chose to examine iNOS as it appeared to be the most significantly altered within the BPD lung, whilst nNOS is the predominant form of NOS within the control lung. Figures 2 and 3 show examples of lung sections double stained for iNOS or nNOS and CD68, a marker of inflammatory cells, or cytokeratin, an epithelial marker. The staining patterns shown in these figures are representative of the nine BPD and seven control samples examined. Distinct cytokeratin staining is observed in the lining of the large airway (Fig. 2A and C). To some extent this staining pattern is maintained within the BPD lung, although, in general, the intensity of the staining is reduced (Fig. 2D). In both situations nNOS is associated with cytokeratin expressing cells, although the association appears tighter in the control lung. However, in the BPD lung, iNOS and cytokeratin strongly coexist throughout the lung tissue, even in the smaller airways (Fig. 2B). In the control lungs, iNOS is not seen within cytokeratin positive cells, the epithelium. iNOS positive cells, presumably airway macrophages, can be observed within the large airway (Fig. 2A).
Fig. 2.

NOS with cytokeratin in control and BPD lung. Double label staining showing iNOS immunostaining (red) and cytokeratin, an epithelial marker (green) in control and BPD lung (A, B). Note attached macrophages (arrow in A) with positive iNOS staining but no coexistence with cytokeratin. BPD lung shows strong coexistence of iNOS and cytokeratin immunostaining (yellow in B). nNOS (red), cytokeratin (green) and coexistence (yellow) are shown in control and BPD lung (C, D). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Fig. 3.

NOS with CD68 in BPD and Control Lung. Double label staining showing iNOS (red), CD68-a macrophage marker (green), Coexistence (yellow) in control and BPD lung (A, B). Note cells that stain for iNOS but not for CD68 and vice versa (arrow in A). In BPD (B), all CD68 positive cells stain for iNOS (no green). nNOS (red) and CD68 (green) in control and BPD lung (C, D), with minimal coexistence in BPD lung (D). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
In the control lung, there are a few CD68 and iNOS positive cells (Fig. 3A). In BPD lung, all CD68 positive cells are iNOS positive, indicating that inflammatory cells in BPD are one source for iNOS (Fig. 3B). nNOS was not changed in control and BPD lungs (Fig. 3C and D).
Markers of NO Metabolism
To investigate whether changes in NOS expression are reflected in markers of NO metabolism, we examined lung tissue for biomarkers of NO reactivity, namely S-nitrosothiol (SNO) and nitrotyrosine (NO2Y). Within the control lung the staining pattern for SNO is very similar to that seen with eNOS. SNO staining appears to be localized predominantly in the lining of blood vessels with some minimal formation in airway lining (Fig. 4A). In contrast, BPD lungs show moderate to intense SNO staining in all lung structures, especially in areas of hypercellularity. Immunoreactivity for SNO was notably altered with disease and was statistically significant, P <0.05 (Fig. 4C and Table 2). In order to confirm the specificity of SNO staining two negative and one positive control was performed for each section. Examples of these controls are shown for one of the BPD samples (Fig. 4D–F). Mercuric chloride treatment, which specifically cleaves the SNO bond by forming a mercaptan, significantly abrogated SNO-immunoreactive staining (Fig. 4D). There was no staining when the staining was repeated in the absence of primary antibody (Fig. 4E), indicating there was not significant non-specific secondary antibody mediated staining or background fluorescence. Pretreatment of the samples with acidified nitrite, which nitrosates all available thiol residues, dramatically increased the immunoreactivity of the antibody (Fig. 4F) providing a positive control for SNO staining.
Fig. 4.

S-Nitrosothiol staining in BPD and control lung. SNO staining in control (A, C) and BPD lung (B, D–F). Stains are shown in panels A and C with corresponding negative controls (achieved by treatment with mercury chloride). Note concentrated SNO staining in areas of hypercellularity (C). There was no staining within the BPD samples when the primary antibody was excluded (E). The pre-treatment of samples with acidified nitrite enhanced the SNO staining in BPD samples (F).
Immunoreactivity for NO2Y was also altered in BPD. Whilst the small degree of NO2Y seen in the control lung is restricted to airway epithelium (Fig. 5A), in BPD, moderate to intense staining is seen throughout the lung tissue (Fig. 5C). The tight localization seen within the intima of airway in control lung is lost in BPD. Pretreatment with Sodium Hydrosulfite, which reduces NO2Y to aminotyrosine, significantly reduced NO2Y antibody mediated staining (Fig. 5B and D).
Fig. 5.
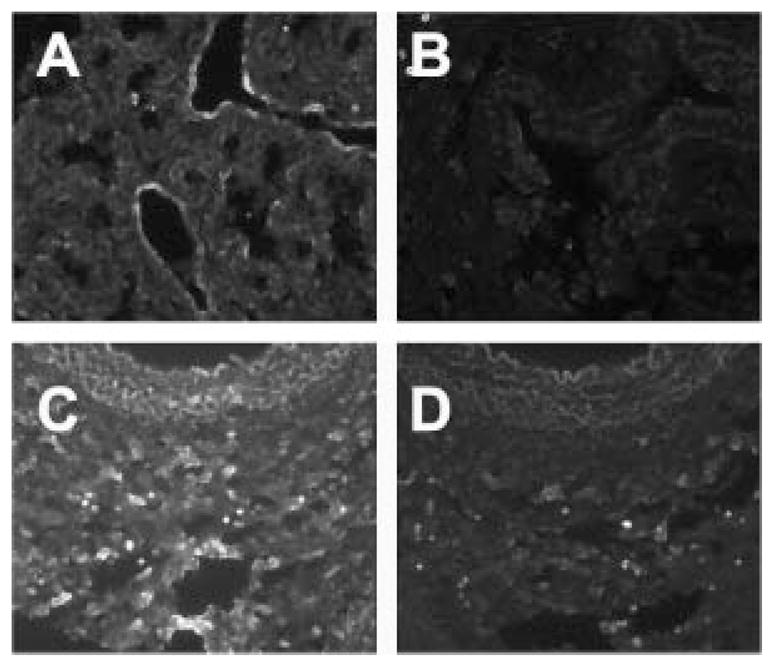
NO2Y staining in control and BPD lung. Control lung had limited NO2Y immunoreactive staining (A) while BPD samples had increased levels of NO2Y (C). Pretreatment of the samples with Sodium hydrosulfite abolished the NO2Y staining in control (B) and BPD (D) samples.
DISCUSSION
In this study we have demonstrated alteration in NOS expression and production of NO metabolites in lung tissue from infants with end stage BPD. Localized expression of all three NOSs was observed in control lung; however, in BPD lung there was increased eNOS and iNOS immunoreactivity throughout a morphologically altered lung. iNOS strongly coexists with the epithelial cell marker cytokeratin and, to a lesser degree, with the inflammatory macrophage marker CD68. NO modified proteins (SNO and NO2Y) are also altered both in quantity and distribution, with immunostaining for NO2Y mirroring that of iNOS in both control and BPD tissue.
Control lungs show a mild to moderate staining for nNOS, iNOS and eNOS with a tight localization in the airway epithelium and endothelium. Potentially, the constitutive NO-producing enzymes (nNOS and eNOS) function primarily to regulate normal blood vessel and airway function, including development, tone and cellular responsiveness. In BPD, immunoreactivity for nNOS was not increased; however, we detected increased eNOS staining in airway, blood vessel and in lung parenchyma. This is in contrast to results observed in the early postnatal period of a baboon model of CLD, where a deficiency in nNOS and eNOS was observed. However, in agreement with our human studies, iNOS expression in the animal model of CLD was increased over the first 2 weeks of postnatal life.13 When considering this comparison it is important to note that the results of our human studies represent NOS expression in end stage BPD.
The most dramatic changes we observed were in iNOS immunoreactivity in all BPD samples. Immunoreactivity for iNOS was rarely detected in control lung and an epithelial focus is strictly maintained (Figs. 1 and 2). No iNOS immunoreactivity was detected in other lung structures. However, in BPD we observed intense iNOS immunoreactivity throughout the lung with the greatest concentration in large airways and blood vessels. iNOS is expressed in both macrophages and epithelial cells.18,19 Double label studies show increased epithelial iNOS in BPD (Fig. 2A and B). Notably only a limited amount of the iNOS detected appeared to colocalize with CD68, a macrophage cell marker (Fig. 3A and B). Colocalization studies with nNOS (immunoreactivity of which did not change in BPD) show no significant difference between control and BPD samples. Thus suggesting some specificity of the increased epithelial iNOS observed in BPD samples. This is of particular interest in regard to the established role of iNOS in mediating acute inflammatory lung injury. Several authors have reported involvement of iNOS in inflammatory diseases of the airway.20–22 The significance and the consequences of elevated epithelial iNOS expression observed in our BPD studies are not clear, however, inappropriate production of NO may lead to the formation of cytotoxic species or inappropriate cellular signaling and contribute to the pathology of the disease.
In order to understand how the changes in iNOS expression may be related to NO production we examined the tissues for NO-modified proteins. NO can interact with ROS to produce higher oxides of nitrogen, which are capable of nitrating tyrosine residues to form NO2Y23,24. In addition in its lower oxide state NO is capable of reacting with thiol residues to produce SNO. Using an immunohistochemical methodology we previously developed,16 we observed both an increase in quantity of SNO and a shift in localization from the control state. Within control lung, SNO is almost exclusively found within the airway epithelium and blood vessel endothelium, regions of lung where one would presume nNOS and eNOS function to predominate. In contrast, within BPD samples, SNO immunoreactivity is seen throughout the lung tissue with the greatest concentration in regions of hypercellularity, where iNOS expression is increased. Interestingly, it has been suggested that SNO formation can operate as a signal for cellular proliferation.25,26 In addition SNO signaling has clearly been shown to be involved in endothelial function.17
In this study we demonstrated significant alteration in the formation of NO2Y. In control samples, there is minimal NO2Y and it is restricted to pulmonary epithelia, however, in BPD there is an increase in NO2Y staining and it is distributed throughout the tissue especially in larger airway walls. In this way, NO2Y staining closely mirrors that of iNOS. NO2Y immunoreactivity has been identified in lungs of patients with acute respiratory distress syndrome and several inflammatory disorders including asthma, pulmonary fibrosis and cystic fibrosis.27–29 NO2Y has also been detected in plasma of infants who develop BPD.14 Inflammatory cells produce cytokines and several other mediators that can activate iNOS expression through endocrine or paracrine pathways.10,30 Mechanistically, NO2Y is more likely to be formed at higher flux rates of NO such as may occur with iNOS. A number of pathways exist for the production of intermediates capable of nitrating or oxidizing proteins.23,31 In many of these pathways there is an oxidant stress component. Therefore, we also conducted inmmunochemistry stain for protein carbonyls. Mild to moderate levels of protein carbonyl were found in lung tissue of samples of both control and BPD tissue (results not shown). No significant difference between the two groups was observed, thus indicating that non-specific oxidation of proteins was not altered in BPD.
There are significant limitations within this study that should be considered when analyzing the results. This study has been conducted using banked and stored samples from a repository within Children’s Hospital of Philadelphia rather than from prospectively collected samples. Hence, other than for two of the samples, the tissues were collected at autopsy. The use of autopsy samples without any special preservation techniques may have affected the levels of labile metabolites such as SNO. However, both control and BPD samples were treated the same way and within the BPD samples robust SNO staining is observed, indicating that this epitope survived sample preparation to a significant extent. A second limitation in the study was the selection of control samples. The ideal control group for this study would have been ventilated premature infants that had not developed BPD, a group that is not available. In the BPD group of premature infants, the long-term ventilation, which allowed them to reach gestational ages equivalent to those of the control infants, did not prevent the high levels of iNOS and NO metabolites. Therefore, it seems unlikely that the low values of the control group are a result of age and maturity alone. As there are a number of variables that are inconsistent between control and BPD infants, this study should be considered as a preliminary examination of NO-related endpoints, which potentially warrant further investigation. In addition, it should also be emphasized that the sample population is from the end-stage of the disease and thus it is possible that the changes observed relate to other factors than those involved in the original development of the pathology, such as long-term ventilation and hospitalization.
In summary, within end-stage BPD, NOS expression is altered both in quantity and distribution and that these changes are matched by alterations in the profile of NO metabolites. There was a significant increase in epithelial iNOS within BPD. These alterations represent the possibility that within BPD there is both a gain of toxic species and a loss of normal NO signaling. It remains unclear whether these shifts are representative of the mechanisms involved in the development of BPD or are consequential upon the establishment of disease. Studies with animal models of BPD will be critical in further understanding how NO is involved in the pathogenesis of the disease. These data demonstrate the importance of NO signaling within BPD, which highlights the necessity of understanding how restoration of NO signaling, such as through the use of inhaled gases which has been performed in both animal models and clinical trial,32,33 may be critical in prevention and treatment of the disease.
Acknowledgments
The authors thank Sree Angampali and Ekaterina Polyakova for assisting in these studies. We also appreciate the generous gift of Dr. H. Ischiropoulos (monoclonal nitrotyrosine antibody). Supported in part by The Crown Foundation (CWD&AJG), NHLBI HL56401 (LWG; RAB; PLB) and HL074115 (AJG).
Grant sponsor: The Crown Foundation; Grant sponsor: NHLBI; Grant numbers: HL56401, HL074115.
References
- 1.Jobe AH, Bancalari E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2001;163:1723–1729. doi: 10.1164/ajrccm.163.7.2011060. [DOI] [PubMed] [Google Scholar]
- 2.Northway WH, Jr, Rosan RC, Porter DY. Pulmonary disease following respirator therapy of hyaline-membrane diseaseBronchopulmonary dysplasia. N Engl J Med. 1967;276:357–368. doi: 10.1056/NEJM196702162760701. [DOI] [PubMed] [Google Scholar]
- 3.Jobe AH. Antenatal factors and the development of bronchopulmonary dysplasia. Semin Neonatol. 2003;8:9–17. doi: 10.1016/s1084-2756(02)00188-4. [DOI] [PubMed] [Google Scholar]
- 4.Ventura SJ, Martin JA, Curtin SC, Mathews TJ. Report of final nationality statistics, 1996. Mon Vital Stat Rep. 1998;46:1–99. [PubMed] [Google Scholar]
- 5.Parker RA, Lindstrom DP, Cotton RB. Evidence from twin study implies possible genetic susceptibility to bronchopulmonary dysplasia. Semin Perinatol. 1996;20:206–209. doi: 10.1016/s0146-0005(96)80049-8. [DOI] [PubMed] [Google Scholar]
- 6.Jobe AJ. The new BPD: an arrest of lung development. Pediatr Res. 1999;46:641–643. doi: 10.1203/00006450-199912000-00007. [DOI] [PubMed] [Google Scholar]
- 7.Coalson JJ, Winter VT, Siler-Khodr T, Yoder BA. Neonatal chronic lung disease in extremely immature baboons. Am J Respir Crit Care Med. 1999;160:1333–1346. doi: 10.1164/ajrccm.160.4.9810071. [DOI] [PubMed] [Google Scholar]
- 8.Murad F. The 1996 Albert Lasker Medical Research Awards. Signal transduction using nitric oxide and cyclic guanosine monophosphate. JAMA. 1996;276:1189–1192. [PubMed] [Google Scholar]
- 9.Schmidt HHHW, Walter U. NO at work. Cell. 1994;78:919–925. doi: 10.1016/0092-8674(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 10.Barnes PJ. Nitric oxide and airway disease. Ann Med. 1995;27:389–393. doi: 10.3109/07853899509002592. [DOI] [PubMed] [Google Scholar]
- 11.Ricciardolo FLM, Sterk PJ, Gaston B, Folkerts G. Nitric oxide in health and disease of the respiratory system. Physiol Rev. 2004;84:731–765. doi: 10.1152/physrev.00034.2003. [DOI] [PubMed] [Google Scholar]
- 12.Nijkamp FP, Folkerts G. Nitric oxide and bronchial hyperresponsiveness. Arch Int Pharmacodyn Ther. 1995;329:81–96. [PubMed] [Google Scholar]
- 13.Afshar S, Gibson LL, Yuhanna IS, Sherman TS, Kerecman JD, Grubb PH, Yoder BA, McCurnin DC, Shaul PW. Pulmonary NO synthase expression is attenuated in a fetal baboon model of chronic lung disease. Am J Physiol Lung Cell Mol Physiol. 2003;284:L749–L758. doi: 10.1152/ajplung.00334.2002. [DOI] [PubMed] [Google Scholar]
- 14.Banks BA, Ischiropoulos H, McClelland M, Ballard PL, Ballard RA. Plasma 3-nitrotyrosine is elevated in premature infants who develop bronchopulmonary dysplasia. Pediatrics. 1998;101:870–874. doi: 10.1542/peds.101.5.870. [DOI] [PubMed] [Google Scholar]
- 15.Farkouh CR, Merrill JD, Ballard PL, Ballard RA, Ischiropoulos H, Lorch SA. Urinary metabolites of oxidative stress and nitric oxide in preterm and term infants. Biol Neonate. 2006;90:233–242. doi: 10.1159/000093633. [DOI] [PubMed] [Google Scholar]
- 16.Gow AJ, Davis CW, Munson D, Ischiropoulos H. Immunohistochemical detection of S-nitrosylated proteins. Methods Mol Biol. 2004;279:167–172. doi: 10.1385/1-59259-807-2:167. [DOI] [PubMed] [Google Scholar]
- 17.Gow AJ, Chen Q, Hess DT, Day BJ, Ischiropoulos H, Stamler JS. Basal and stimulated protein S-nitrosylation in multiple cell types and tissues. J Biol Chem. 2002;277:9637–9640. doi: 10.1074/jbc.C100746200. [DOI] [PubMed] [Google Scholar]
- 18.Kobzik L, Bredt DS, Lowenstein CJ, Drazen J, Gaston B, Sugarbaker D, Stamler JS. Nitric oxide synthase in human and rat lung: immunocytochemical and histochemical localization. Am J Respir Cell Mol Biol. 1993;9:371–377. doi: 10.1165/ajrcmb/9.4.371. [DOI] [PubMed] [Google Scholar]
- 19.Hamid Q, Springall DR, Riveros-Moreno V, Chanez P, Howarth P, Redington A, Bousquet J, Godard P, Holgate S, Polak JM. Induction of nitric oxide synthase in asthma. Lancet. 1993;342:1510–1513. doi: 10.1016/s0140-6736(05)80083-2. [DOI] [PubMed] [Google Scholar]
- 20.Guo FH, Raeve HRD, Rice TW, Stuehr DJ, Thunnissen F, Erzurum SC. Continuous nitric oxide synthesis by inducible nitric oxide synthase in normal human airway epithelium in vivo. Proc Natl Acad Sci. 1995;92:7809–7813. doi: 10.1073/pnas.92.17.7809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiba Y, Arimoto T, Yoshikawa T, Misawa M. Elevated nitric oxide synthase activity concurrent with antigen-induced airway hyperresponsiveness in rats. Exp Lung Res. 2000;26:535–549. doi: 10.1080/019021400750048081. [DOI] [PubMed] [Google Scholar]
- 22.Haddad EB, Liu SF, Salmon M, Robichaud A, Barnes PJ, Chung KF. Expression of inducible nitric oxide synthase mRNA in Brown Norway rats exposed to ozone: effect of dexamethasone. Eur J Pharmacol. 1995;293:287–290. doi: 10.1016/0926-6917(95)00032-1. [DOI] [PubMed] [Google Scholar]
- 23.Ischiropoulos H. Biological tyrosine nitration: a pathophysiological function of nitric oxide and reactive oxygen species. Arch Biochem Biophys. 1998;356:1–11. doi: 10.1006/abbi.1998.0755. [DOI] [PubMed] [Google Scholar]
- 24.Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin JC, Smith CD, Beckman JS. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochem Biophys. 1992;298:431–437. doi: 10.1016/0003-9861(92)90431-u. [DOI] [PubMed] [Google Scholar]
- 25.Stamler JS, Lamas S, Fang FC. Nitrosylation. the prototypic redox-based signaling mechanism. Cell. 2001;106:675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 26.Nelson EJ, Connolly J, McArthur P. Nitric oxide and S-nitrosylation: excitotoxic and cell signaling mechanism. Biol Cell. 2003;95:3–8. doi: 10.1016/s0248-4900(03)00004-2. [DOI] [PubMed] [Google Scholar]
- 27.Andreadis AA, Hazen SL, Comhair SA, Erzurum SC. Oxidative and nitrosative events in asthma. Free Radic Biol Med. 2003;35:213–225. doi: 10.1016/s0891-5849(03)00278-8. [DOI] [PubMed] [Google Scholar]
- 28.Lamb NJ, Gutteridge JM, Baker C, Evans TW, Quinlan GJ. Oxidative damage to proteins of bronchoalveolar lavage fluid in patients with acute respiratory distress syndrome: evidence for neutrophil-mediated hydroxylation, nitration, and chlorination. Crit Care Med. 1999;27:1738–1744. doi: 10.1097/00003246-199909000-00007. [DOI] [PubMed] [Google Scholar]
- 29.Morrissey BM, Schilling K, Weil JV, Silkoff PE, Rodman DM. Nitric oxide and protein nitration in the cystic fibrosis airway. Arch Biochem Biophys. 2002;406:33–39. doi: 10.1016/s0003-9861(02)00427-7. [DOI] [PubMed] [Google Scholar]
- 30.Dugas B, Kolb JP. Nitric oxide production in human inflammatory processes. Res Immunol. 1995;146:661–663. doi: 10.1016/0923-2494(96)84913-3. [DOI] [PubMed] [Google Scholar]
- 31.Eiserich JP, Hristova M, Cross CE, Jones AD, Freeman BA, Halliwell B, van der Vliet A. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature. 1998;391:393–397. doi: 10.1038/34923. [DOI] [PubMed] [Google Scholar]
- 32.Auten RL, Mason SN, Whorton MH, Lampe WR, Foster WM, Goldberg RN, Li B, Stamler JS, Auten KM. Inhaled ethyl nitrite prevents hyperoxia-impaired postnatal alveolar development in newborn rats. Am J Respir Crit Care Med. 2007;176:291–299. doi: 10.1164/rccm.200605-662OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ballard RA, Truog WE, Cnaan A, Martin RJ, Ballard PL, Merrill JD, Walsh MC, Durand DJ, Mayock DE, Eichenwald EC, Null DR, Hudak ML, Puri AR, Golombek SG, Courtney SE, Stewart DL, Welty SE, Phibbs RH, Hibbs AM, Luan X, Wadlinger SR, Asselin JM, Coburn CE the NOCLDSG. Inhaled nitric oxide in preterm infants undergoing mechanical ventilation. N Engl J Med. 2006;355:343–353. doi: 10.1056/NEJMoa061088. [DOI] [PubMed] [Google Scholar]