

Abstract
Hyperglycaemia occurs frequently in the critically ill, even in those patients without a history of diabetes. The mechanisms underlying hyperglycaemia in this group are complex and incompletely defined. In health, the gastrointestinal tract is an important modulator of postprandial glycaemic excursions and both the rate of gastric emptying and the so-called incretin hormones, glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide, are pivotal determinants of postprandial glycaemia. Incretin-based therapies (that is, glucagon-like peptide-1 agonists and dipeptidyl-peptidase-4 inhibitors) have recently been incorporated into standard algorithms for the management of hyperglycaemia in ambulant patients with type 2 diabetes and, inevitably, an increasing number of patients who were receiving these classes of drugs prior to their acute illness will present to ICUs. This paper summarises current knowledge of the incretin effect as well as the incretin-based therapies that are available for the management of type 2 diabetes, and provides suggestions for the potential relevance of these agents in the management of dysglycaemia in the critically ill, particularly to normalise elevated blood glucose levels.
Hyperglycaemia and critical illness: prevalence and association with adverse outcomes
Both type 1 and type 2 diabetes mellitus (T2DM) increase the propensity for macrovascular and microvascular disease as well as infection, all of which are likely to predispose individuals to critical illness necessitating intensive care admission [1]. While the worldwide prevalence of formally diagnosed T2DM is about 3% [2], the prevalence in ICU patients is variably reported as 15 to 20% or even higher [3].
In the critically ill, hyperglycaemia also occurs frequently in patients without known diabetes; this group includes patients with undiagnosed type 2 diabetes and those with so-called stress hyperglycaemia. We propose that the latter should be referred to as critical illness induced hyperglycaemia (CIIH), given that this describes the pathogenesis more appropriately. Several hormonal mechanisms appear to be important pathophysiological mediators of hyperglycaemia during critical illness. These mechanisms include increases in counter-regulatory hormones, such as endogenous glucagon, catecholamines and glucocorticoids, inadequate insulin secretion for the degree of hyperglycaemia and insulin resistance [4, 5]. The exogenous administration of catecholamines, dextrose, corticosteroids and nutritional support has the potential to further exacerbate the elevation in blood glucose [4, 6].
CIIH is therefore characterised by hyperglycaemia (fasting blood glucose ≥7 mmol/l or random blood glucose ≥11.1 mmol/l) in critically ill patients, who were glucose tolerant prior to their acute illness, and are shown in the longer term not to have diabetes [1]. The true prevalence of CIIH is unknown, at least in part because the vast majority of studies have failed to discriminate it from undiagnosed T2DM [3]. Nevertheless, CIIH is clearly common – for example, in the NICE-SUGAR study >60% of patients without known diabetes in the control arm had blood glucose concentrations >10 mmol/l requiring exogenous insulin [7].
Marked acute hyperglycaemia is associated with increased morbidity and mortality in the critically ill [8]. The magnitude of the elevation in blood glucose required to cause harm remains uncertain and is likely to differ between T2DM and CIIH. In observational studies, a robust association between increased mortality and hyperglycaemia has been reported consistently in patients with CIIH, but not in those with pre-existing T2DM [9–11]. Moreover, preliminary – albeit retrospective and observational – data suggest that the adverse impact of hyperglycaemia is attenuated or abolished by pre-existing or chronic hyperglycaemia, and that hyperglycaemia during acute illness may, in fact, be protective [12]. Accordingly, glucose concentrations that are considered safe in patients with CIIH may well be harmful in patients with type 1 or (more frequently) type 2 diabetes and chronic hyperglycaemia [12]. Furthermore, it is intuitively logical to tailor strategies for glycaemic control based on pre-existing glycaemia, rather than managing all critically ill patients as a homogeneous group. Prospective studies are urgently required in this area to clarify these important issues.
The incretin effect – the historical context
The characterisation of the incretin effect arguably began in 1902 when Bayliss and Starling, with the characterisation of secretin, speculated that the gastrointestinal tract could communicate with the pancreas via a messenger(s) in the blood [13]. Until that time, primarily as a result of Pavlov’s influence, it was thought that the functions of the body were regulated exclusively by nerves. However, it was not until the early 1960s that the insulin response to enteral glucose was demonstrated to be much greater than an intravenous glucose load, despite the latter resulting in substantially higher blood glucose concentrations [14, 15], suggesting that a hormone (or hormones) secreted from the gastrointestinal tract stimulates insulin secretion. When blood glucose concentrations resulting from oral and intravenous glucose load were matched, Perley and Kipnis observed that the plasma insulin response to intravenous glucose was approximately 40% of that resulting from the oral glucose load in health, quantifying the magnitude of the incretin effect for the first time [16] (Figure 1). The hormones responsible for the incretin effect were subsequently shown to be glucose-dependent insulinotropic peptide (GIP) (in 1973) and glucagon-like peptide (GLP)-1 (in 1985).
Figure 1.
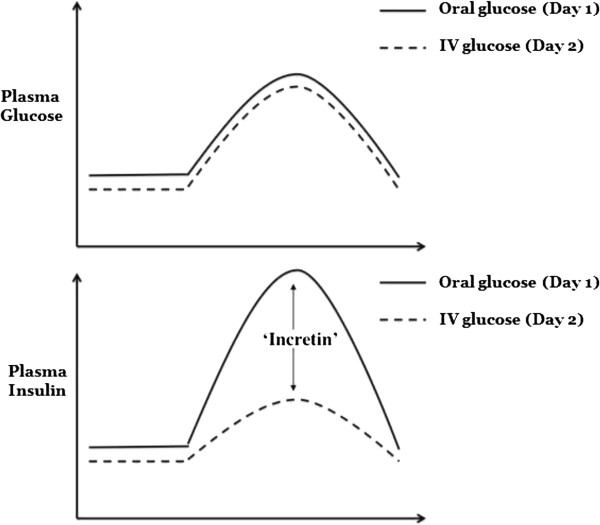
The incretin effect. There is a much greater release of insulin in response to oral glucose administration as compared with administering the same amount of glucose by intravenous (IV) infusion. Subjects were given oral glucose on day 1 with plasma insulin levels recorded. The same volunteers returned on a second day and an IV glucose infusion was titrated to match the plasma glucose excursion achieved with the oral load. The difference in the measured plasma insulin is the incretin effect, mediated by the hormones glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide. Adapted from [17].
GIP is secreted predominantly from duodenal K cells in response to luminal fat and carbohydrate [17], while GLP-1 is secreted from intestinal L cells, located primarily in the distal ileum and colon, in response to luminal fat, carbohydrate, protein and bile acids [18, 19]. At least in health, both GIP and GLP-1 are insulinotropic and, in the case of GLP-1, glucagonostatic in a strictly glucose-dependent fashion, so that below a blood glucose threshold of ~8 mmol/l even pharmacological doses of these hormones have little or no effect on fasting blood glucose [20]. In addition to its insulinotropic and glucagonostatic properties, GLP-1 is a physiological modulator to slow gastric emptying [17, 21], while GIP probably has no effect on gastric emptying and is glucagonotropic during euglycaemia [22].
Attenuation of the incretin effect and pathogenesis of hyperglycaemia in type 2 diabetes
In 1986 Nauck and colleagues reported that the magnitude of the incretin effect was markedly diminished in longstanding T2DM [23], and they went on to show that the insulinotropic effect of GIP is also attenuated in this group, while that of exogenous GLP-1 is relatively preserved [24]. Whether the reduced incretin effect is a primary phenomenon or epi-phenomenon of β-cell failure is still uncertain [25], but it is apparent that the reduced insulinotropic effect of GIP is, in part, an effect of hyperglycaemia and, hence, reversible [25]. Nevertheless, these observations stimulated the development of GLP-1-based pharmacological therapy for the management of T2DM. One should recognise that a limitation of these early studies is that gastric emptying was not quantified. This is important because the rate of nutrient entry into the small intestine is a major determinant of both GLP-1 and GIP secretion [26]. Studies employing direct, intraduodenal infusion of nutrients indicate that the secretion of GIP and GLP-1 is maintained in both T2DM [26, 27] and the critically ill [28].
Drug development of incretin therapies for the management of type 2 diabetes mellitus
Initial proof-of-principle studies were performed using pharmacological doses of GLP-1 administered by intravenous or subcutaneous infusion. As well as stimulating insulin [29] and inhibiting glucagon secretion, both in a glucose-dependent manner [30], GLP-1 has been shown to have several important and unique pharmacodynamic properties that enhance its therapeutic potential. GLP-1 dose-dependently slows gastric emptying, thereby attenuating postprandial glycaemic excursions [31]. Exogenous GLP-1 also inhibits appetite, resulting in reduced energy intake and sustained weight loss in obese subjects, features that are beneficial in patients with T2DM [32]. In vitro and animal studies have shown that GLP-1 exerts trophic effects on the pancreatic β cells, stimulating proliferation and differentiation [33], as well as reducing β-cell apoptosis [34]. Hence, GLP-1 may have the potential to preserve β-cell mass in patients with type 2 diabetes. Pharmacological administration of GLP-1 has significant extra-gastrointestinal effects with the potential cardioprotective properties of particular relevance to the critically ill. For example: in animal models, GLP-1 attenuates ischaemia-induced myocardial damage [35]; in patients with heart failure, exogenous GLP-1 has been associated with improvements in left ventricular ejection fraction, myocardial oxygen uptake and 6-minute walking distance [36]; and when administered post-coronary artery bypass grafting, GLP-1 is associated with less use of sympathomimetic drugs and fewer arrhythmias [37]. There is evidence, hitherto derived from animal studies, that GLP-1 may possess neurotropic effects [38] which have the potential to be beneficial in the management of neurodegenerative conditions such as Alzheimer’s disease [39]. The implications in the neurocritically ill remain uncertain [40, 41].
Currently available incretin-based agents for use in type II diabetes
The administration of GLP-1 to ambulant patients with diabetes proved clinically impractical. Endogenous GLP-1 (and its synthetic peptide) has a plasma half-life of only 1 to 2 minutes [42] due to rapid metabolism by the ubiquitous, nonspecific enzyme dipeptidyl-peptidase 4 (DPP-4), which forms an inactive metabolite that is rapidly cleared by the kidneys [43], necessitating continuous infusion. The pharmaceutical industry has therefore focused on the development of stable DPP-4-resistant GLP-1 receptor agonists, as well as DPP-4 inhibitors that protect endogenously secreted GLP-1, GLP-2 and GIP from degradation. DPP-4 inhibitors act as incretin enhancers, increasing systemic and, perhaps of more pertinence to their effect, intestinal concentrations of endogenously secreted GLP-1, GLP-2 and GIP [44]. GLP-1 agonists are incretin mimetics, acting as agonists at the GLP-1 receptor and, in contrast to DPP-4 inhibitors, do not stimulate GIP or GLP-2 and may reduce endogenous GLP-1 concentrations [45], possibly because of its effect on gastric emptying [21].
Incretin-based therapies are frequently prescribed and their use is expected to become even more common, with recent international guidelines recommending that GLP-1 agonists and DPP-4 inhibitors be used as part of standard regimens for patients with T2DM [46].
Glucagon-like peptide-1 receptor agonists
A GLP-1 agonist candidate was fortuitously identified in the peptide exendin-4 from the salivary venom of the Gila monster (Heloderma suspectum). The Gila monster is a slow-moving venomous lizard native to North America that ingests food 5 to 10 times per year, and ingestion results in a substantial increase in plasma exendin-4 concentrations that may have endocrine functions related to metabolic control in the lizard [47]. Exendin-4 is a peptide with ~50% amino acid sequence homology to GLP-1 and acts as a full agonist at the GLP-1 receptor. Exendin-4 is also resistant to metabolism by DPP-4, and is eliminated only by glomerular filtration, with a resultant plasma half-life of 30 minutes [48]. Exenatide was developed as the synthetic replica of exendin-4 and in 2005 was the first incretin-based therapy approved by the US Food and Drug Administration. There are now at least eight GLP-1 agonists undergoing clinical trials, but twice-daily exenatide, once-daily lixisenatide, once-daily liraglutide and once-weekly extended-release exenatide are the only agents to have hitherto reached the market.
GLP-1 agonists can be categorised as short-acting or long-acting compounds. All are given subcutaneously and stimulate fasting insulin secretion and reduce glucagon secretion in a glucose-dependent manner [49]. Dose-dependent nausea, vomiting and diarrhoea occur frequently, and while it is recognised as a far more prominent feature of the synthetic agonists when compared with the native peptide, it does decline with time and is attenuated by dose titration [50, 51]. Short-acting and long-acting compounds do, however, differ in their effects on fasting and postprandial glucose.
Short-acting glucagon-like peptide-1 agonists
Exenatide and lixisenatide are considered short-acting GLP-1 agonists and their short half-lives result in large fluctuations in circulating plasma concentrations [49]. The dominant effect of these drugs on glycaemia is mediated by the slowing of gastric emptying, thereby markedly blunting postprandial glycaemic excursions [52]. Patients with diabetes with significant postprandial hyperglycaemia and relatively well-controlled fasting glycaemia therefore achieve the greatest benefit from these short-acting compounds. Additionally, the combination of short-acting GLP-1 agonist and basal insulin confers complementary effects, with control of fasting hyperglycaemia by the basal insulin, control of postprandial glycaemic excursions with the GLP-1 agonists, a reduction in glycaemic variability and a low risk of hypoglycaemia [49].
Exenatide is approved in Europe as adjunctive therapy in patients with inadequately controlled T2DM who are taking oral hypoglycaemic agents or basal insulin, or both. Exenatide has also been approved for use as monotherapy in the United States. In February 2013, lixisenatide was approved in Europe as adjunctive therapy in patients with inadequately controlled T2DM, who are taking oral hypoglycaemic agents or basal insulin, or both. The pharmacological and pharmacodynamic properties of these therapies are summarised in Tables 1 and 2, respectively.
Table 1.
Marketed incretin-based agents
| Therapy | Approval | Dose | Route | Precautions |
|---|---|---|---|---|
| GLP-1 receptor agonists | ||||
| Exantide (Byetta®) | EMA, US FDA | 5 μg, 10 μg | SC, BD | Renal impairmenta |
| Lixisenatide (Lyxumia®) | EMA | 10 μg, 20 μg | SC, OD | Renal impairmenta |
| Extended-release exenatide (Bydureon®) | EMA, US FDA | 2 mg | SC, QW | Renal impairmenta |
| Liraglutide (Victoza®) | EMA, US FDA | 0.6 mg, 1.2 mg, 1.8 mg | SC, OD | MTC of MEN2 |
| DPP-4 inhibitors | ||||
| Sitagliptin (Januvia®)b | EMA,US FDA | 25 mg, 50 mg, 100 mg | Oral OD | Renal iAAmpairmentc |
| Vildagliptin (Galvus®)b | EMA | 50 mg | Oral BD | Renal impairmenta |
| Hepatic impairment | ||||
| Alogliptin (Nesina®)b | US FDA | 25 mg | Oral OD | Renal impairmentc |
| Saxagliptin (Onglyza®) | EMA, US FDA | 2.5 mg, 5 mg | Oral OD | Renal impairmentc |
| Linagliptin (Trajenta®) | EMA, US FDA | 5 mg | Oral OD | – |
BD twice daily, DPP-4 dipeptidyl-peptidase-4, EMA European Medicines Agency, MEN2 multiple endocrine neoplasia type 2, MTC medullary thyroid carcinoma, OD once daily, SC subcutaneous, QW once weekly, US FDA US Food and Drug Administration. aContraindicated in patients with severe renal impairment (creatinine clearance <30 ml/minute) and end-stage renal disease. bAvailable in a fixed-dose combination with metformin. cDose adjustment required in moderate (creatinine clearance ≥30 to <50 ml/minute), severe and end-stage renal disease.
Table 2.
Pharmacodynamics: GLP-1 agonists versus DPP-4 inhibitors
| GLP-1 agonists | DPP-4 inhibitors | |
|---|---|---|
| Plasma GLP-1 concentrations | Pharmacological concentrations of the GLP-1 analogue (sixfold to tenfold greater than endogenous GLP-1) | Twofold to threefold increase |
| Glucose-dependent insulin secretion | Yes | Yes |
| Glucose-dependent inhibition of glucagon secretion | Yes | Yes |
| HbA1c reduction (%) | 0.8 to 1.8 | 0.5 to 1.1 |
| Inhibition of gastric emptying | Short acting only | No |
| Postprandial glucose reduction | Yes: short acting > long acting | Yes (weaker) |
| Fasting plasma glucose reduction | Yes: long acting > short acting | Yes (weaker) |
| Effects on weight | Significant weight loss | Weight neutral |
| Inhibition of food intake | Yes | No |
| Gastrointestinal adverse effects: nausea, vomiting and diarrhoea | Common | Uncommon |
DPP-4 dipeptidyl-peptidase-4, GLP glucagon-like peptide, HbA 1c glycated haemoglobin.
Long-acting glucagon-like peptide-1 agonists
Extended-release exenatide and liraglutide are long-acting GLP-1 receptor agonists that have continuously elevated plasma concentrations between their recommended dosing intervals. These agonists are approved in Europe and the United States to be used in the treatment of T2DM as monotherapy or as second-line therapy in combination with oral hypoglycaemic agents. They achieve higher fasting insulin levels and greater reductions in fasting glucose concentrations [49]. The continuous activation of the GLP-1 receptor is thought to induce tachyphylaxis to the deceleration of gastric emptying, such that the slowing of gastric emptying diminishes markedly with long-term use, and consequently the lowering of postprandial glycaemia is only modest [53]. Long-acting compounds therefore seem to be advantageous when clinicians wish to target fasting hyperglycaemia in patients with T2DM.
Dipeptidyl-peptidase-4 inhibitors
The DPP-4 inhibitors are orally administered agents and are approved as monotherapy or in combination with other oral hypoglycaemic agents for the management of T2DM. The predominant mechanism of DPP-4 inhibitor action is thought to be via a gut effect involving local inhibition of intestinal DPP-4 activity, stimulation of gut incretin receptors and activation of gut-to-pancreas neural pathways, rather than a systemic increase in incretin plasma concentrations [44]. DPP-4 is expressed widely throughout the body, including on T lymphocytes and macrophages, posing a theoretical risk of immune dysfunction with enzyme inhibition [54]. However, a meta-analysis reported a very minor increase in all-cause infection during sitagliptin treatment that was not evident in studies involving other DPP-4 inhibitors [55]. The reliability and significance of this association remains uncertain. The presentation, dose, contraindications and pharmacodynamics of the DPP-4 inhibitors are outlined in Tables 1 and 2.
Extra-pancreatic effects of incretin-based therapies
The cardiovascular outcomes of the incretin-based drugs are currently undergoing assessment in large, multicentre clinical trials [56]. Data from preclinical studies, short-term single-centre human studies and retrospective reviews of healthcare claims support a cardioprotective role for both GLP-1 receptor agonists and DPP-4 inhibitors [57]. The majority of clinical trials have reported reductions in body weight and blood pressure, albeit with a mean increase in heart rate of 2 to 4 beats/minute [57].
Finally, there are postmarketing reports of acute pancreatitis during use of GLP-1 agonists and DPP-4 inhibitors [58]. However, when compared with health, the risk of acute pancreatitis is more than doubled in T2DM, independent of treatment modality [59], and the incidence of pancreatitis appears to be no greater when starting these agents than in patients who are commenced on metformin or a sulfonylurea [60]. For these reasons we consider the risk of GLP-1-induced or DPP-4 inhibitor-induced pancreatitis to be negligible. There have also been concerns, principally from rodent studies, that the use of incretin-based therapies may be associated with chronic pancreatitis [61], particularly as this would intuitively also increase the risk of pancreatic cancer [62]. Hitherto, these potential risks have not been substantiated [63] and while ongoing postmarketing surveillance is important, any increase in risk is likely to be extremely small and is unlikely to be relevant to short-term use in critically ill patients.
Rationale for the use of glucagon-like peptide-1 as a novel therapy in the critically ill
Administration of intravenous insulin guided by validated algorithms is the current standard of care for hyperglycaemic critically ill patients [64]. While the optimal blood glucose for the critically ill remains contentious, concentrations >10 mmol/l should be avoided, at least for those without pre-existing diabetes and chronic hyperglycaemia [7]. Several studies have demonstrated that insulin-induced hypoglycaemia (blood glucose <2.2 mmol/l) occurs more frequently with intensive insulin therapy and that it is an independent risk factor for mortality [7, 65, 66]. Moreover, in the NICE-SUGAR study, moderate hypoglycaemia (≥2.3 to ≤3.9 mmol/l) and severe hypoglycaemia (<2.3 mmol/l) were both associated with insulin use and increased mortality, with the lowest nadirs in glucose concentrations having the strongest association with death [67]. This observation may be of particular relevance in neurocritical care, particularly as there is evidence that systemic glucose concentrations even within the normal physiological range may be associated with cerebral hypoglycaemia and consequent adverse outcomes [68]. These data, accordingly, strongly suggest that treatment-induced hypoglycaemia should be avoided.
Regardless of target blood glucose, intravenous insulin therapy also carries the logistic limitations of frequent monitoring and training of nursing staff in complicated protocols. Additionally, increased glycaemic variability appears to be an independent risk factor for ICU mortality [69, 70] and, although speculative, a therapy that reduces glycaemic variability may lead to improved outcomes.
By inhibiting glucagon secretion as well as increasing insulin secretion, GLP-1 or its agonists provide a plausible pharmacodynamic mechanism to counter the hyperglucagonaemia and relative insulin resistance that typifies stress hyperglycaemia. Furthermore, the cardioprotective properties hitherto demonstrated in incretin-based therapies are particularly attractive in the intensive care setting. In animal models, GLP-1 decreases ischaemia-induced myocardial damage [35]; in patients with heart failure, exogenous GLP-1 has been associated with improvements in left ventricular ejection fraction, myocardial oxygen uptake and 6-minute walking distance [36]; and when administered post-coronary artery bypass grafting, GLP-1 is associated with less use of sympathomimetic drugs and fewer arrhythmias [37].
For the reasons outlined in Table 3, a simplified therapy for the treatment of hyperglycaemia that avoided iatrogenic hypoglycaemia and limited glycaemic variability would be desirable.
Table 3.
Potential benefits of glucagon-like peptide-1 based therapies in the critically ill
| Potential benefit | |
|---|---|
| Negligible risk of severe hypoglycaemia | |
| Reduction in insulin requirement | |
| Cardiovascular protection | |
| Attenuated glycaemic variability | |
| Amenable to continuous infusion without dose titration | |
| Decreased blood glucose testing | |
| Simplified protocol for medical and nursing staff |
Recommendations for incretin-based therapies in critically ill patients
Critical illness may markedly affect pharmacokinetics, particularly for the oral and subcutaneous routes [71]. Furthermore, multiorgan failure with evolving hepatic and renal dysfunction and the consequent derangements in metabolism and elimination may result in unpredictable half-lives and drug clearance [71]. For these reasons, and also because of limited experience with these incretin-based agents, it is appropriate to discontinue these drugs when critically ill patients are admitted to the ICU.
In hyperglycaemic patients transitioning from intensive care to ward care, current recommended practice is to commence scheduled subcutaneous basal-bolus insulin regimens [72], although the American Diabetes Association consensus statement concedes that noninsulin agents may be appropriate in selected, stable patients who are expected to consume meals at regular intervals [72]. There have been no randomised controlled trials to compare subcutaneous basal-bolus insulin with the incretin-based agents in this patient group. However, because of the limitations inherent in basal-bolus insulin administration, in patients who were on incretin-based therapies prior to their acute illness and who are improving clinically and transitioning from the ICU to the hospital ward, recommencement of the established incretin regimen would be a reasonable therapeutic option. However, there is limited information available about the use of incretin-based agents in patients discharged from the ICU. Until such time that the strategy is formally evaluated, we recommend that intensivists using such an approach involve an endocrinologist to provide ongoing care following ICU discharge. A specialist in the field may also assist by advising dose reduction due to renal or hepatic impairment (Table 1), managing dose escalation on recommencing these agents to limit adverse effects, and being aware of potential interactions with drugs that have been commenced in the current hospital admission. Finally, because the glucose-lowering effects of GLP-1 agonists will be diminished in patients with pancreatitis, we recommend avoiding GLP-1 agonists in patients with a history of pancreatitis or surgical manipulation of the pancreas.
Experience using incretin-based therapy in the critically ill
Hitherto, the use of GLP-1 in the critically ill is limited to small studies to establish proof of principle [73]. A limitation of all of these studies has been the short duration of GLP-1 administration (<72 hours); however, in patients with T2DM, glucose lowering was maintained for up to 6 weeks during continuous subcutaneous GLP-1 infusion [74], suggesting that tachyphylaxis, at least to the glucose-lowering effect, does not occur. One should also recognise that while synthetic GLP-1 is currently prohibitively expensive, there may be a substantial reduction in cost should a market become available.
With intravenous GLP-1 infusions at doses ranging from 1.2 to 3.6 pmol/kg/minute, marked glucose lowering has been observed in patients with T2DM post-major surgery [75], post-coronary artery bypass grafting [37, 76] and post-acute myocardial infarction with primary angioplasty [77]. Our group has evaluated the effects of GLP-1 infusions in heterogeneous cohorts of mechanically ventilated patients, both with T2DM and CIIH [78–80]. GLP-1 infusions at 1.2 pmol/kg/minute were shown to reduce the glycaemic response to small intestinal nutrient in mechanically ventilated patients with chronic hyperglycaemia [80] and to intragastric and small intestinal nutrient delivery in patients not known to have T2DM (Figure 2) [78, 79]. At this dose, nutrient-induced hyperglycaemia was attenuated but not suppressed completely, and the effect appears to be more prominent in patients without a history of T2DM. We also reported that GLP-1 slows gastric emptying when emptying is relatively normal, but has minimal effect when emptying is already delayed, consistent with studies in ambulant type 2 diabetics [79]. The infusions were well tolerated in all patient groups and there were no reported episodes of hypoglycaemia.
Figure 2.
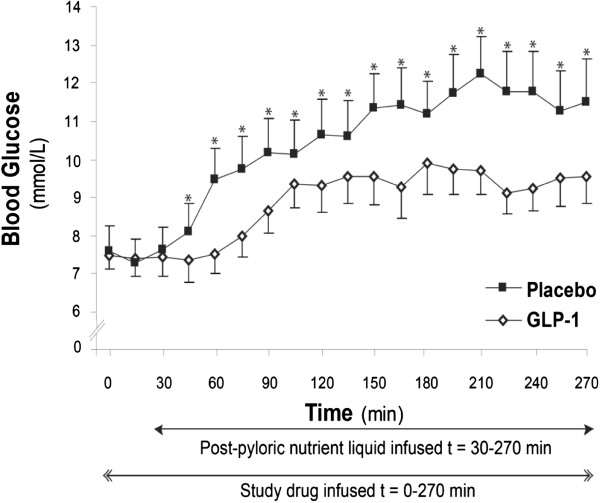
Glucose-lowering effect of glucagon-like peptide-1 in critical illness. In critically ill patients without known diabetes the blood glucose excursion in response to small intestinal feeding is markedly attenuated by 1.2 pmol/kg/minute glucagon-like peptide-1 (GLP-1) (area under the curve at 240 minutes: GLP-1, 2,077 ± 145 mmol/l/minute vs. placebo, 2,568 ± 208 mmol/l/minute; P <0.01). Data are mean ± standard error of the mean (n = 7). Reproduced with permission from [81].
Administration of intravenous exenatide is also being explored, as currently these drugs are substantially cheaper than the synthetic peptide. As outlined (Table 1), exenatide is only approved for subcutaneous administration in the ambulant type 2 patient, but there is experience with its intravenous use in trial settings [81, 82]. Abuannadi and colleagues administered intravenous exenatide for 48 hours in an open-label, nonrandomised, pilot study in 40 cardiac ICU patients [83]. The effectiveness of exenatide was benchmarked to glucose concentrations previously recorded during intensive and moderate insulin therapies [83]. Mean blood glucose concentrations with intravenous exenatide were noninferior to the NICE-SUGAR-endorsed insulin protocol (that is, moderate), and glycaemic variability was reduced [83]. No episodes of severe hypoglycaemia were reported in the exenatide group, but nausea (40%) and vomiting (5%) occurred frequently [83]. Whether these agents could be used as standalone therapy or as insulin-sparing agents with the benefits of reduced hypoglycaemia and diminished glycaemic variability remains to be determined. Further studies using the commercially marketed GLP-1 agonists are warranted [84].
While studies in the critically ill have focused on GLP-1 and its agonists, the role of GIP and DPP-4 inhibitors also warrants consideration [84]. While it is known that the insulinotropic effect of GIP is markedly reduced in T2DM, this may be primarily due to hyperglycaemia rather than an inherent cellular defect [85]. In the critically ill, our group demonstrated that exogenously administered GIP with concurrent GLP-1 does not lower blood glucose concentrations, either in the fasted state or in response to small intestinal nutrient, more than GLP-1 alone [86]. However this was a small, single-centre pilot study and further research to clarify whether GIP retains its insulinotropic effect in critically ill patients, in whom blood glucose concentrations have only been transiently elevated, merits further investigation.
The DPP-4 inhibitors have not yet been investigated as a therapeutic strategy for CIIH. Delayed gastric emptying occurs frequently in critical illness, with absolute gastroparesis not infrequent [87], and also, at least in health, hyperglycaemia per se slows gastric emptying [88]. Accordingly, intragastric administration of an oral glucose-lowering drug may not be the most effective route. Nevertheless, in patients with known normal gastric emptying or with post-pyloric administration in patients with delayed gastric emptying, the local intestinal mechanism of action supports the study of the renally safe DPP-4 inhibitor linagliptin as a potential novel oral agent for glycaemic control in the critically ill.
Conclusions
The discovery of the incretin hormones has encouraged a burgeoning pharmaceutical industry that exploits the entero-insular axis. As the prevalence of T2DM and the use of incretin-based regimens continue to rise, the intensivist is likely to encounter more patients who were taking these drugs prior to their acute illness. Presently, there is little experience with continuing these agents in acute illness and more study is urgently required.
Nevertheless, the pharmacologic strategy of administering incretin mimetics to manage acute hyperglycaemia in the critically ill, while minimising the risk of hypoglycaemia and mitigating glycaemic variability, is appealing. Future studies should focus on the identification of the patient groups most likely to benefit from the administration of incretin receptor agonists, which of the agents is most useful in the critically ill, and at what dose, the relevance of the route of feeding with concurrent incretin therapy and, finally, whether these agents should be used in combination with insulin or as single-agent therapy to provide a safer, more effective and simpler method of achieving glycaemic control.
Acknowledgements
MPP is financially supported by a Dawes Scholarship, co-funded by the University of Adelaide and the Royal Adelaide Hospital. This work was supported by NHMRC Project Grant No. 1025648.
Abbreviations
- CIIH
Critical illness induced hyperglycaemia
- DPP-4
Dipeptidyl-peptidase-4
- GIP
Glucose-dependent insulinotropic polypeptide
- GLP
Glucagon-like peptide
- T2DM
Type 2 diabetes mellitus.
Footnotes
Competing interests
MH has participated in advisory boards and/or symposia for Novo/Nordisk, Sanofi-aventis, Novartis, Eli-Lily, Boehringer Ingelheim, AstraZeneca, Satlogen and Meyer Nutraceuticals. The remaining authors declare that they have no competing interests.
Contributor Information
Mark P Plummer, Email: mark.philip.plummer@gmail.com.
Marianne J Chapman, Email: marianne.chapman@health.sa.gov.au.
Michael Horowitz, Email: michael.horowitz@adelaide.edu.au.
Adam M Deane, Email: adam.m.deane@gmail.com.
References
- 1.Clement S, Braithwaite SS, Magee MF, Ahmann A, Smith EP, Schafer RG, Hirsch IB, American Diabetes Association Diabetes in Hospitals Writing Committee Management of diabetes and hyperglycemia in hospitals. Diabetes Care. 2004;27:553–591. doi: 10.2337/diacare.27.2.553. [DOI] [PubMed] [Google Scholar]
- 2.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 3.Deane AM, Horowitz M. Dysglycaemia in the critically ill – significance and management. Diabetes Obes Metab. 2013;15:792–801. doi: 10.1111/dom.12078. [DOI] [PubMed] [Google Scholar]
- 4.Marik PE, Raghavan M. Stress-hyperglycemia, insulin and immunomodulation in sepsis. Intensive Care Med. 2004;30:748–756. doi: 10.1007/s00134-004-2167-y. [DOI] [PubMed] [Google Scholar]
- 5.Mizock BA. Alterations in fuel metabolism in critical illness: hyperglycaemia. Best Pract Res Clin Endocrinol Metab. 2001;15:533–551. doi: 10.1053/beem.2001.0168. [DOI] [PubMed] [Google Scholar]
- 6.Gearhart MM, Parbhoo SK. Hyperglycemia in the critically ill patient. AACN Clin Issues. 2006;17:50–55. doi: 10.1097/00044067-200601000-00007. [DOI] [PubMed] [Google Scholar]
- 7.Finfer S, Chittock DR, Su SY, Blair D, Foster D, Dhingra V, Bellomo R, Cook D, Dodek P, Henderson WR, Hébert PC, Heritier S, Heyland DK, McArthur C, McDonald E, Mitchell I, Myburgh JA, Norton R, Potter J, Robinson BG, Ronco JJ, NICE-SUGAR Study Investigators Intensive versus conventional glucose control in critically ill patients. N Engl J Med. 2009;360:1283–1297. doi: 10.1056/NEJMoa0810625. [DOI] [PubMed] [Google Scholar]
- 8.van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P, Bouillon R. Intensive insulin therapy in critically ill patients. N Engl J Med. 2001;345:1359–1367. doi: 10.1056/NEJMoa011300. [DOI] [PubMed] [Google Scholar]
- 9.Umpierrez GE, Isaacs SD, Bazargan N, You X, Thaler LM, Kitabchi AE. Hyperglycemia: an independent marker of in-hospital mortality in patients with undiagnosed diabetes. J Clin Endocrinol Metab. 2002;87:978–982. doi: 10.1210/jcem.87.3.8341. [DOI] [PubMed] [Google Scholar]
- 10.Rady MY, Johnson DJ, Patel BM, Larson JS, Helmers RA. Influence of individual characteristics on outcome of glycemic control in intensive care unit patients with or without diabetes mellitus. Mayo Clin Proc. 2005;80:1558–1567. doi: 10.4065/80.12.1558. [DOI] [PubMed] [Google Scholar]
- 11.Egi M, Bellomo R, Stachowski E, French CJ, Hart GK, Hegarty C, Bailey M. Blood glucose concentration and outcome of critical illness: the impact of diabetes. Crit Care Med. 2008;36:2249–2255. doi: 10.1097/CCM.0b013e318181039a. [DOI] [PubMed] [Google Scholar]
- 12.Egi M, Bellomo R, Stachowski E, French CJ, Hart GK, Taori G, Hegarty C, Bailey M. The interaction of chronic and acute glycemia with mortality in critically ill patients with diabetes. Crit Care Med. 2011;39:105–111. doi: 10.1097/CCM.0b013e3181feb5ea. [DOI] [PubMed] [Google Scholar]
- 13.Bayliss WM, Starling EH. The mechanism of pancreatic secretion. J Physiol. 1902;28:325–353. doi: 10.1113/jphysiol.1902.sp000920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McIntyre N, Holdsworth CD, Turner DS. New interpretation of oral glucose tolerance. Lancet. 1964;2:20–21. doi: 10.1016/S0140-6736(64)90011-X. [DOI] [PubMed] [Google Scholar]
- 15.Elrick H, Stimmler L, Hlad CJ, Jr, Arai Y. Plasma insulin response to oral and intravenous glucose administration. J Clin Endocrinol Metab. 1964;24:1076–1082. doi: 10.1210/jcem-24-10-1076. [DOI] [PubMed] [Google Scholar]
- 16.Perley MJ, Kipnis DM. Plasma insulin responses to oral and intravenous glucose: studies in normal and diabetic subjects. J Clin Invest. 1967;46:1954–1962. doi: 10.1172/JCI105685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deane A, Chapman MJ, Fraser RJ, Horowitz M. Bench-to-bedside review: The gut as an endocrine organ in the critically ill. Crit Care. 2010;14:228. doi: 10.1186/cc9039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schirra J, Katschinski M, Weidmann C, Schafer T, Wank U, Arnold R, Goke B. Gastric emptying and release of incretin hormones after glucose ingestion in humans. J Clin Invest. 1996;97:92–103. doi: 10.1172/JCI118411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parker HE, Wallis K, le Roux CW, Wong KY, Reimann F, Gribble FM. Molecular mechanisms underlying bile acid-stimulated glucagon-like peptide-1 secretion. Br J Pharmacol. 2012;165:414–423. doi: 10.1111/j.1476-5381.2011.01561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Combes J, Borot S, Mougel F, Penfornis A. The potential role of glucagon-like peptide-1 or its analogues in enhancing glycaemic control in critically ill adult patients. Diabetes Obes Metab. 2011;13:118–129. doi: 10.1111/j.1463-1326.2010.01311.x. [DOI] [PubMed] [Google Scholar]
- 21.Deane AM, Nguyen NQ, Stevens JE, Fraser RJ, Holloway RH, Besanko LK, Burgstad C, Jones KL, Chapman MJ, Rayner CK, Horowitz M. Endogenous glucagon-like peptide-1 slows gastric emptying in healthy subjects, attenuating postprandial glycemia. J Clin Endocrinol Metab. 2010;95:215–221. doi: 10.1210/jc.2009-1503. [DOI] [PubMed] [Google Scholar]
- 22.Meier JJ, Nauck MA, Schmidt WE, Gallwitz B. Gastric inhibitory polypeptide: the neglected incretin revisited. Regul Pept. 2002;107:1–13. doi: 10.1016/S0167-0115(02)00039-3. [DOI] [PubMed] [Google Scholar]
- 23.Nauck M, Stockmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia. 1986;29:46–52. doi: 10.1007/BF02427280. [DOI] [PubMed] [Google Scholar]
- 24.Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7–36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest. 1993;91:301–307. doi: 10.1172/JCI116186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meier JJ, Nauck MA. Is the diminished incretin effect in type 2 diabetes just an epi-phenomenon of impaired beta-cell function? Diabetes. 2010;59:1117–1125. doi: 10.2337/db09-1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woerle HJ, Carneiro L, Derani A, Goke B, Schirra J. The role of endogenous incretin secretion as amplifier of glucose-stimulated insulin secretion in healthy subjects and patients with type 2 diabetes. Diabetes. 2012;61:2349–2358. doi: 10.2337/db11-1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma J, Pilichiewicz AN, Feinle-Bisset C, Wishart JM, Jones KL, Horowitz M, Rayner CK. Effects of variations in duodenal glucose load on glycaemic, insulin, and incretin responses in type 2 diabetes. Diabet Med. 2012;29:604–608. doi: 10.1111/j.1464-5491.2011.03496.x. [DOI] [PubMed] [Google Scholar]
- 28.Deane AM RC, Keeshan A, Cvijanovic N, Marino Z, Nguyen NQ, Chia B, Summers MJ, Sim JA, Van Beek T, Chapman MJ, Horowitz M, Young RL. Crit Care Med. 2013. Disordered regulation of intestinal glucose sensor, transporters and absorption in critically ill humans and mice. [DOI] [PubMed] [Google Scholar]
- 29.Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1 7–36: a physiological incretin in man. Lancet. 1987;2:1300–1304. doi: 10.1016/S0140-6736(87)91194-9. [DOI] [PubMed] [Google Scholar]
- 30.Nauck MA, Heimesaat MM, Behle K, Holst JJ, Nauck MS, Ritzel R, Hufner M, Schmiegel WH. Effects of glucagon-like peptide 1 on counterregulatory hormone responses, cognitive functions, and insulin secretion during hyperinsulinemic, stepped hypoglycemic clamp experiments in healthy volunteers. J Clin Endocrinol Metab. 2002;87:1239–1246. doi: 10.1210/jcem.87.3.8355. [DOI] [PubMed] [Google Scholar]
- 31.Nauck MA, Niedereichholz U, Ettler R, Holst JJ, Orskov C, Ritzel R, Schmiegel WH. Glucagon-like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. Am J Physiol. 1997;273:E981–E988. doi: 10.1152/ajpendo.1997.273.5.E981. [DOI] [PubMed] [Google Scholar]
- 32.Verdich C, Flint A, Gutzwiller JP, Naslund E, Beglinger C, Hellstrom PM, Long SJ, Morgan LM, Holst JJ, Astrup A. A meta-analysis of the effect of glucagon-like peptide-1 (7–36) amide on ad libitum energy intake in humans. J Clin Endocrinol Metab. 2001;86:4382–4389. doi: 10.1210/jcem.86.9.7877. [DOI] [PubMed] [Google Scholar]
- 33.Xu G, Stoffers DA, Habener JF, Bonner-Weir S. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes. 1999;48:2270–2276. doi: 10.2337/diabetes.48.12.2270. [DOI] [PubMed] [Google Scholar]
- 34.Farilla L, Bulotta A, Hirshberg B, Li Calzi S, Khoury N, Noushmehr H, Bertolotto C, Di Mario U, Harlan DM, Perfetti R. Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology. 2003;144:5149–5158. doi: 10.1210/en.2003-0323. [DOI] [PubMed] [Google Scholar]
- 35.Bose AK, Mocanu MM, Carr RD, Brand CL, Yellon DM. Glucagon-like peptide 1 can directly protect the heart against ischemia/reperfusion injury. Diabetes. 2005;54:146–151. doi: 10.2337/diabetes.54.1.146. [DOI] [PubMed] [Google Scholar]
- 36.Sokos GG, Nikolaidis LA, Mankad S, Elahi D, Shannon RP. Glucagon-like peptide-1 infusion improves left ventricular ejection fraction and functional status in patients with chronic heart failure. J Card Fail. 2006;12:694–699. doi: 10.1016/j.cardfail.2006.08.211. [DOI] [PubMed] [Google Scholar]
- 37.Sokos GG, Bolukoglu H, German J, Hentosz T, Magovern GJ, Jr, Maher TD, Dean DA, Bailey SH, Marrone G, Benckart DH, Elahi D, Shannon RP. Effect of glucagon-like peptide-1 (GLP-1) on glycemic control and left ventricular function in patients undergoing coronary artery bypass grafting. Am J Cardiol. 2007;100:824–829. doi: 10.1016/j.amjcard.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 38.Perry T, Haughey NJ, Mattson MP, Egan JM, Greig NH. Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J Pharmacol Exp Ther. 2002;302:881–888. doi: 10.1124/jpet.102.037481. [DOI] [PubMed] [Google Scholar]
- 39.Perry TA, Greig NH. A new Alzheimer's disease interventive strategy: GLP-1. Curr Drug Targets. 2004;5:565–571. doi: 10.2174/1389450043345245. [DOI] [PubMed] [Google Scholar]
- 40.Bakiner O, Bozkirli E, Giray S, Arlier Z, Kozanoglu I, Sezgin N, Sariturk C, Ertorer E. Impact of early versus late enteral nutrition on cell mediated immunity and its relationship with glucagon like peptide-1 in intensive care unit patients: a prospective study. Crit Care. 2013;17:R123. doi: 10.1186/cc12795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Plummer MP, Meier JJ, Deane AM. The gut–brain axis in the critically ill: is glucagon-like peptide-1 protective in neurocritical care? Crit Care. 2013;17:163. doi: 10.1186/cc12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vilsboll T, Agerso H, Krarup T, Holst JJ. Similar elimination rates of glucagon-like peptide-1 in obese type 2 diabetic patients and healthy subjects. J Clin Endocrinol Metab. 2003;88:220–224. doi: 10.1210/jc.2002-021053. [DOI] [PubMed] [Google Scholar]
- 43.Deacon CF, Johnsen AH, Holst JJ. Degradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo. J Clin Endocrinol Metab. 1995;80:952–957. doi: 10.1210/jcem.80.3.7883856. [DOI] [PubMed] [Google Scholar]
- 44.Waget A, Cabou C, Masseboeuf M, Cattan P, Armanet M, Karaca M, Castel J, Garret C, Payros G, Maida A, Sulpice T, Holst JJ, Drucker DJ, Magnan C, Burcelin R. Physiological and pharmacological mechanisms through which the DPP-4 inhibitor sitagliptin regulates glycemia in mice. Endocrinology. 2011;152:3018–3029. doi: 10.1210/en.2011-0286. [DOI] [PubMed] [Google Scholar]
- 45.Ionut V, Zheng D, Stefanovski D, Bergman RN. Exenatide can reduce glucose independent of islet hormones or gastric emptying. Am J Physiol Endocrinol Metab. 2008;295:E269–E277. doi: 10.1152/ajpendo.90222.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Inzucchi SE, Bergenstal RM, Buse JB, Diamant M, Ferrannini E, Nauck M, Peters AL, Tsapas A, Wender R, Matthews DR. Management of hyperglycaemia in type 2 diabetes: a patient-centered approach. Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) Diabetologia. 2012;55:1577–1596. doi: 10.1007/s00125-012-2534-0. [DOI] [PubMed] [Google Scholar]
- 47.Kolterman OG, Buse JB, Fineman MS, Gaines E, Heintz S, Bicsak TA, Taylor K, Kim D, Aisporna M, Wang Y, Baron AD. Synthetic exendin-4 (exenatide) significantly reduces postprandial and fasting plasma glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab. 2003;88:3082–3089. doi: 10.1210/jc.2002-021545. [DOI] [PubMed] [Google Scholar]
- 48.Edwards CM, Stanley SA, Davis R, Brynes AE, Frost GS, Seal LJ, Ghatei MA, Bloom SR. Exendin-4 reduces fasting and postprandial glucose and decreases energy intake in healthy volunteers. Am J Physiol Endocrinol Metab. 2001;281:E155–E161. doi: 10.1152/ajpendo.2001.281.1.E155. [DOI] [PubMed] [Google Scholar]
- 49.Meier JJ. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. 2012;8:728–742. doi: 10.1038/nrendo.2012.140. [DOI] [PubMed] [Google Scholar]
- 50.Holst JJ, Vilsboll T, Deacon CF. The incretin system and its role in type 2 diabetes mellitus. Mol Cell Endocrinol. 2009;297:127–136. doi: 10.1016/j.mce.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 51.Neumiller JJ. Clinical pharmacology of incretin therapies for type 2 diabetes mellitus: implications for treatment. Clin Ther. 2011;33:528–576. doi: 10.1016/j.clinthera.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 52.Linnebjerg H, Park S, Kothare PA, Trautmann ME, Mace K, Fineman M, Wilding I, Nauck M, Horowitz M. Effect of exenatide on gastric emptying and relationship to postprandial glycemia in type 2 diabetes. Regul Pept. 2008;151:123–129. doi: 10.1016/j.regpep.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 53.Nauck MA, Kemmeries G, Holst JJ, Meier JJ. Rapid tachyphylaxis of the glucagon-like peptide 1-induced deceleration of gastric emptying in humans. Diabetes. 2011;60:1561–1565. doi: 10.2337/db10-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mentlein R. Dipeptidyl-peptidase IV (CD26) – role in the inactivation of regulatory peptides. Regul Pept. 1999;85:9–24. doi: 10.1016/S0167-0115(99)00089-0. [DOI] [PubMed] [Google Scholar]
- 55.Richter B, Bandeira-Echtler E, Bergerhoff K, Lerch CL. Dipeptidyl peptidase-4 (DPP-4) inhibitors for type 2 diabetes mellitus. Cochrane Database Syst Rev. 2008;2:CD006739. doi: 10.1002/14651858.CD006739.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fonseca VA. Ongoing clinical trials evaluating the cardiovascular safety and efficacy of therapeutic approaches to diabetes mellitus. Am J Cardiol. 2011;108:52B–58B. doi: 10.1016/j.amjcard.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 57.Ussher JR, Drucker DJ. Cardiovascular biology of the incretin system. Endocr Rev. 2012;33:187–215. doi: 10.1210/er.2011-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bain SC, Stephens JW. Exenatide and pancreatitis: an update. Expert Opin Drug Saf. 2008;7:643–644. doi: 10.1517/14740330802432003. [DOI] [PubMed] [Google Scholar]
- 59.Noel RA, Braun DK, Patterson RE, Bloomgren GL. Increased risk of acute pancreatitis and biliary disease observed in patients with type 2 diabetes: a retrospective cohort study. Diabetes Care. 2009;32:834–838. doi: 10.2337/dc08-1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dore DD, Seeger JD, Arnold Chan K. Use of a claims-based active drug safety surveillance system to assess the risk of acute pancreatitis with exenatide or sitagliptin compared to metformin or glyburide. Curr Med Res Opin. 2009;25:1019–1027. doi: 10.1185/03007990902820519. [DOI] [PubMed] [Google Scholar]
- 61.Gier B, Matveyenko AV, Kirakossian D, Dawson D, Dry SM, Butler PC. Chronic GLP-1 receptor activation by exendin-4 induces expansion of pancreatic duct glands in rats and accelerates formation of dysplastic lesions and chronic pancreatitis in the Kras(G12D) mouse model. Diabetes. 2012;61:1250–1262. doi: 10.2337/db11-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Butler PC, Matveyenko AV, Dry S, Bhushan A, Elashoff R. Glucagon-like peptide-1 therapy and the exocrine pancreas: innocent bystander or friendly fire? Diabetologia. 2010;53:1–6. doi: 10.1007/s00125-009-1591-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nauck MA. A critical analysis of the clinical use of incretin-based therapies: the benefits by far outweigh the potential risks. Diabetes Care. 2013;36:2126–2132. doi: 10.2337/dc12-2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moghissi ES, Korytkowski MT, DiNardo M, Einhorn D, Hellman R, Hirsch IB, Inzucchi SE, Ismail-Beigi F, Kirkman MS, Umpierrez GE. American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Diabetes Care. 2009;32:1119–1131. doi: 10.2337/dc09-9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Egi M, Bellomo R, Stachowski E, French CJ, Hart GK, Taori G, Hegarty C, Bailey M. Hypoglycemia and outcome in critically ill patients. Mayo Clin Proc. 2010;85:217–224. doi: 10.4065/mcp.2009.0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Van den Berghe G, Wilmer A, Hermans G, Meersseman W, Wouters PJ, Milants I, Van Wijngaerden E, Bobbaers H, Bouillon R. Intensive insulin therapy in the medical ICU. N Engl J Med. 2006;354:449–461. doi: 10.1056/NEJMoa052521. [DOI] [PubMed] [Google Scholar]
- 67.Finfer S, Liu B, Chittock DR, Norton R, Myburgh JA, McArthur C, Mitchell I, Foster D, Dhingra V, Henderson WR, Ronco JJ, Bellomo R, Cook D, McDonald E, Dodek P, Hébert PC, Heyland DK, Robinson BG. Hypoglycemia and risk of death in critically ill patients. N Engl J Med. 2012;367:1108–1118. doi: 10.1056/NEJMoa1204942. [DOI] [PubMed] [Google Scholar]
- 68.Oddo M, Schmidt JM, Carrera E, Badjatia N, Connolly ES, Presciutti M, Ostapkovich ND, Levine JM, Le Roux P, Mayer SA. Impact of tight glycemic control on cerebral glucose metabolism after severe brain injury: a microdialysis study. Crit Care Med. 2008;36:3233–3238. doi: 10.1097/CCM.0b013e31818f4026. [DOI] [PubMed] [Google Scholar]
- 69.Egi M, Bellomo R, Stachowski E, French CJ, Hart G. Variability of blood glucose concentration and short-term mortality in critically ill patients. Anesthesiology. 2006;105:244–252. doi: 10.1097/00000542-200608000-00006. [DOI] [PubMed] [Google Scholar]
- 70.Krinsley JS. Glycemic variability: a strong independent predictor of mortality in critically ill patients. Crit Care Med. 2008;36:3008–3013. doi: 10.1097/CCM.0b013e31818b38d2. [DOI] [PubMed] [Google Scholar]
- 71.Smith BS, Yogaratnam D, Levasseur-Franklin KE, Forni A, Fong J. Introduction to drug pharmacokinetics in the critically ill patient. Chest. 2012;141:1327–1336. doi: 10.1378/chest.11-1396. [DOI] [PubMed] [Google Scholar]
- 72.Moghissi ES, Korytkowski MT, DiNardo M, Einhorn D, Hellman R, Hirsch IB, Inzucchi SE, Ismail-Beigi F, Kirkman MS, Umpierrez GE, American Association of Clinical Endocrinologists; American Diabetes Association American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Endocr Pract. 2009;15:353–369. doi: 10.4158/EP09102.RA. [DOI] [PubMed] [Google Scholar]
- 73.Pinelli NR, Jones MC, Monday LM, Smith Z, Rhoney DH. Exogenous glucagon-like peptide-1 for hyperglycemia in critically ill patients. Ann Pharmacother. 2012;46:124–129. doi: 10.1345/aph.1Q417. [DOI] [PubMed] [Google Scholar]
- 74.Zander M, Madsbad S, Madsen JL, Holst JJ. Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet. 2002;359:824–830. doi: 10.1016/S0140-6736(02)07952-7. [DOI] [PubMed] [Google Scholar]
- 75.Meier JJ, Weyhe D, Michaely M, Senkal M, Zumtobel V, Nauck MA, Holst JJ, Schmidt WE, Gallwitz B. Intravenous glucagon-like peptide 1 normalizes blood glucose after major surgery in patients with type 2 diabetes. Crit Care Med. 2004;32:848–851. doi: 10.1097/01.CCM.0000114811.60629.B5. [DOI] [PubMed] [Google Scholar]
- 76.Mussig K, Oncu A, Lindauer P, Heininger A, Aebert H, Unertl K, Ziemer G, Haring HU, Holst JJ, Gallwitz B. Effects of intravenous glucagon-like peptide-1 on glucose control and hemodynamics after coronary artery bypass surgery in patients with type 2 diabetes. Am J Cardiol. 2008;102:646–647. doi: 10.1016/j.amjcard.2008.06.029. [DOI] [PubMed] [Google Scholar]
- 77.Nikolaidis LA, Mankad S, Sokos GG, Miske G, Shah A, Elahi D, Shannon RP. Effects of glucagon-like peptide-1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation. 2004;109:962–965. doi: 10.1161/01.CIR.0000120505.91348.58. [DOI] [PubMed] [Google Scholar]
- 78.Deane AM, Chapman MJ, Fraser RJ, Burgstad CM, Besanko LK, Horowitz M. The effect of exogenous glucagon-like peptide-1 on the glycaemic response to small intestinal nutrient in the critically ill: a randomised double-blind placebo-controlled cross over study. Crit Care. 2009;13:R67. doi: 10.1186/cc7874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Deane AM, Chapman MJ, Fraser RJ, Summers MJ, Zaknic AV, Storey JP, Jones KL, Rayner CK, Horowitz M. Effects of exogenous glucagon-like peptide-1 on gastric emptying and glucose absorption in the critically ill: relationship to glycemia. Crit Care Med. 2010;38:1261–1269. doi: 10.1097/CCM.0b013e3181d9d87a. [DOI] [PubMed] [Google Scholar]
- 80.Deane AM, Summers MJ, Zaknic AV, Chapman MJ, Fraser RJ, Di Bartolomeo AE, Wishart JM, Horowitz M. Exogenous glucagon-like peptide-1 attenuates the glycaemic response to postpyloric nutrient infusion in critically ill patients with type-2 diabetes. Crit Care. 2011;15:R35. doi: 10.1186/cc9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Degn KB, Brock B, Juhl CB, Djurhuus CB, Grubert J, Kim D, Han J, Taylor K, Fineman M, Schmitz O. Effect of intravenous infusion of exenatide (synthetic exendin-4) on glucose-dependent insulin secretion and counterregulation during hypoglycemia. Diabetes. 2004;53:2397–2403. doi: 10.2337/diabetes.53.9.2397. [DOI] [PubMed] [Google Scholar]
- 82.Fehse F, Trautmann M, Holst JJ, Halseth AE, Nanayakkara N, Nielsen LL, Fineman MS, Kim DD, Nauck MA. Exenatide augments first- and second-phase insulin secretion in response to intravenous glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab. 2005;90:5991–5997. doi: 10.1210/jc.2005-1093. [DOI] [PubMed] [Google Scholar]
- 83.Abuannadi M, Kosiborod M, Riggs L, House JA, Hamburg MS, Kennedy KF, Marso SP. Management of hyperglycemia with the administration of intravenous exenatide to patients in the cardiac intensive care unit. Endocr Pract. 2013;19:81–90. doi: 10.4158/EP12196.OR. [DOI] [PubMed] [Google Scholar]
- 84.Schwartz S, Defronzo RA. Is incretin-based therapy ready for the care of hospitalized patients with type 2 diabetes? The time has come for GLP-1 receptor agonists! Diabetes Care. 2013;36:2107–2111. doi: 10.2337/dc12-2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hojberg PV, Vilsboll T, Rabol R, Knop FK, Bache M, Krarup T, Holst JJ, Madsbad S. Four weeks of near-normalisation of blood glucose improves the insulin response to glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes. Diabetologia. 2009;52:199–207. doi: 10.1007/s00125-008-1195-5. [DOI] [PubMed] [Google Scholar]
- 86.Lee MY, Fraser JD, Chapman MJ, Sundararajan K, Umapathysivam MM, Summers MJ, Zaknic AV, Rayner CK, Meier JJ, Horowitz M, Deane AM. The effect of exogenous glucose-dependent insulinotropic polypeptide in combination with glucagon-like peptide-1 on glycemia in the critically ill. Diabetes Care. 2013;36:3333–3336. doi: 10.2337/dc13-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Deane AM, Fraser RJ, Chapman MJ. Prokinetic drugs for feed intolerance in critical illness: current and potential therapies. Crit Care Resusc. 2009;11:132–143. [PubMed] [Google Scholar]
- 88.Masclee AA, Gielkens HA, Lam WF, de Boer SY, Lamers CB. Effects of parenteral nutrients on gastrointestinal motility and secretion. Scand J Gastroenterol Suppl. 1996;218:50–55. doi: 10.3109/00365529609094731. [DOI] [PubMed] [Google Scholar]