

Abstract
While the cure rates of childhood acute lymphoblastic leukemia (ALL) have improved dramatically in the past 40 years, not all children have benefited equally from this impressive progress. Racial and ethnic disparities in the incidence and treatment outcome of childhood ALL persist with Hispanic children having an elevated risk of developing ALL and one of the lowest survival rates after ALL therapy. A critical barrier to progress is that we lack the understanding of the causes of ALL disparities, particularly racial and ethnic differences in ALL biology. This review summarizes the current knowledge on population variation in childhood ALL incidence and treatment outcome, discusses the contributing genetic and non-genetic variables, and highlights possible therapeutic interventions to mitigate disparities in ALL.
Keywords: race, ethnicity, disparity, childhood, leukemia, genomics
Introduction
Acute lymphoblastic leukemia (ALL) is the most common cancer in children, with ∼3,500 new cases each year in the United States (U.S.).1 Cure rates have improved dramatically over the past 40 years, attributed largely to risk-adapted combination chemotherapy.2, 3 However, racial and ethnic disparities persist in both the incidence and treatment outcome of ALL. For example, Hispanic children have not only the highest incidence of ALL4, 5 but also one of the lowest survival rates in the U.S populations.6-8 African American children are least likely to develop ALL,5, 9 but they fare worse with ALL therapy than European Americans and Asians.6, 7, 10
While epidemiological studies have repeatedly documented racial and ethnic differences in ALL incidence and outcome, the underlying causes remain poorly understood. Recent high-throughput genomic profiling of ALL (e.g., genome-wide gene expression, DNA copy number, and single nucleotide polymorphism [SNP] genotype) has dramatically improved our understanding of the genetic landscape of this disease and has also led to the discovery of novel molecular markers for treatment individualization.1, 3, 11, 12 In this review, we will provide an overview of epidemiological findings of racial and ethnic disparities in childhood ALL and, more importantly, discuss the plausible genomic basis of these variations in ALL incidence and treatment outcome (Figure 1).
Figure 1. Genetic and non-genetic factors influencing racial and ethnic disparities in childhood ALL.

While there is a perception that race categorization is more based on physical appearance and ethnicity is defined more by cultural identity, the exact distinctions between these two terms are not well defined. For the purpose of this review, we will focus primarily on ALL disparities among the 4 population groups: European American (EA), African American (AA), Hispanic American (HA), Asian American (Asian or Pacific Islander), and Native American (American Indian/Alaska Native, NA) in the U.S. Self-reported race and ethnic categories are used throughout the article unless otherwise specified (e.g., genetic ancestry). There is also increasing evidence for critical roles of epigenetic regulation in ALL pathogenesis13-15, but its relevance in racial and ethnic disparities of ALL has yet to be characterized and thus not discussed here.
Race, Ethnicity and Genetic ancestry
The definition of race and ethnicity has a long and tumultuous history in medical research and a focal point of contention is whether or not there is a biologic basis of racial and ethnic classification16, 17. As humans evolved in the past 100,000 years, genetic variation inevitably arose across the genome as results of either random mutation or selection imposed by environmental factors, forming the basis of inter-individual variability in a wide range of phenotypic traits. During human diasporas before modern days, individuals were more likely to mate with one another if they lived in close proximity and this assortative mating pattern is likely to be the driving force of genetic differences among geographically divided populations (e.g., Africans, European, Asians, etc.)18, 19. However, we commonly divide individuals into groups (or self-identify) on the basis of their biological (e.g., physical appearance) or non-biological (e.g., language) features, without the appreciation of human genetics (i.e., genetic ancestry). Therefore, depending upon the criteria used, race and ethnicity can be completely genetics-based (genetic ancestry) or non-genetic at all (language). This introduces enormous heterogeneity within self-reported racial and ethnic groups: e.g., two individuals both self-identified as African American can have drastically different levels of African genetic ancestry. This is particularly problematic for Hispanics, arguably one of the most broadly defined ethnic groups in the US. With varying degrees of admixture among Europeans, Africans, and Native Americans, the genetic ancestry composition of Hispanics is extremely diverse20-22. Hispanics in Florida are more likely to be of Cuban origin and have much higher African genetic ancestry, compared to Hispanics in California of Mexican descent with high levels of Native American genetic ancestry. Conversely, replacing self-reported race or ethnicity by genetic ancestry can overlook potentially critical contributions of environmental or cultural factors. Therefore, it is prudent to recognize the limitations of using self-reported race and ethnicity as well as those associated with genetic ancestry. The discussion of racial and ethnic disparities in cancer would not be comprehensive without considering both genetic and non-genetic features as well as the interactions between the two.
Racial and ethnic Differences in Susceptibility to Childhood ALL
ALL incidence rate and molecular subtypes in different racial and ethnic groups
Based on the National Cancer Institute (NCI)'s Surveillance, Epidemiology and End Results (SEER) registries, the overall age-adjusted incidence of childhood ALL (diagnosed between 0-19 years) is 31.9 per million person-years,23, 24 accounting for approximately 27% of all pediatric cancers and disproportionally affecting children between the ages of 2 and 5 years. Although age patterns are consistent across racial or ethnic groups, the incidence of ALL differs markedly. Children of African descent have a significantly lower incidence of ALL compared to EAs (14.8 vs. 35.6 per million, respectively),4, 5 while childhood ALL is most common in HAs (40.9 per million person-years).4, 5 A higher incidence rate was also reported for NAs, although with a relatively small sample size.9
Although ALL is generally distinguished from other hematologic malignancies by over-representation of lymphoblast cells in the bone marrow, it can be further divided into subtypes with distinct immunophenotypes (B or T-ALL) and/or genetic abnormalities (chromosomal translocations: t(12;21) with ETV6-RUNX1 fusion, t(1;19) with TCF3-PBX1 fusion, t(9;22) with BCR-ABL1 fusion, MLL rearrangements at 11q23; numeric chromosomal gain [hyperdiploidy 47-50 or ≥51 chromosomes] or loss [hypodiploidy <45 chromosomes]). In a population-based survey using the SEER database of 4,952 cases of childhood ALL, Kadan-Lottick et al. observed a significant over-representation of T-cell ALL in AA (9%) compared to EA children (5%).7 Similarly, among 8,447 children with ALL treated on Children's Cancer Group (CCG) protocols from 1983 to 1995, the incidence of T-ALL was 1.7-fold higher in AAs than in EAs.6 In a study of 412 children with ALL consecutively treated at St. Jude Children's Research Hospital, higher prevalence of T-cell ALL was again evident in AAs.25 Similarly, TCF3-PBX1 fusion was also overrepresented in AAs with ALL, while ploidy abnormalities and other translocation events were not.25 In a small cohort of children with ALL in California (N=53), ETV6-RUNX1 translocation was more common in EAs than in HAs,26 but this was not validated in a larger national study of 2,534 children with ALL.27
Genetic basis for racial and ethnic differences in ALL incidence
The etiology of ALL is likely to be complex with genetic and environmental factors collectively contributing to leukemogenesis. Several congenital genetic abnormalities have been linked to predisposition to childhood ALL, lending themselves support to a genetic basis of ALL susceptibility. For example, children with Down Syndrome (constitutive chromosome 21 trisomy) are at a significantly elevated risk of developing acute leukemia,28 particularly ALL with somatic CRLF2 lesions.29 Inherited inter-individual genetic variations (e.g., differences in DNA sequence between individuals) are common across the human genome and are often related to geographic ancestry of racial or ethnic groups.19 Thus, genetic polymorphisms can contribute to racial and ethnic differences in ALL incidences if the frequency of a susceptibility variant differs by race or ethnicity, and/or when genetic variants are associated with ALL in a population specific manner.
The contribution of genetic variations in “candidate” pathways (e.g., carcinogen metabolism, folate metabolism, DNA repair) to ALL susceptibility has been extensively examined over the past two decades, with inconsistent results. A recent meta-analysis summarized 47 studies of 25 polymorphisms in 16 genes and observed statistically significant (P<0.05) albeit modest associations with ALL susceptibility for only 8 variants (e.g., GSTM1 deletion, SLC19A1 G80A), with an estimated false-positive probability of 20%.30 A similar pooled analysis of MTHFR polymorphisms in 12 studies observed a significant association for the C677T variant but not at the A1298C polymorphism.31 Germline SNPs in the IL12A and the HLA-DP genes were also linked to ALL risk in Hispanics,32, 33 suggesting that immune modulation plays a role in ALL etiology. However, a comprehensive analysis of the major histocompatibility complex region in 824 B-ALL cases and 4,737 controls of European genetic ancestry did not find statistically significant association between HLA variants and ALL susceptibility.34
Advances in high-throughput genotyping now allow genome-wide association studies (GWAS) to interrogate a large number of genetic variations across the entire human genome for associations with a variety of phenotypic traits. GWAS does not rely on prior knowledge of the disease biology, but instead systematically examines genetic variants in an agnostic fashion. To date, GWAS of childhood ALL susceptibility have thus far discovered 5 genomic loci at the genome-wide significance level (P<5×10-8)35-38: ARID5B (10q21.2), IKZF1 (7p12.2), CEBPE (14q11.2), CDKN2A (9p21.3), and BMI1-PIP4K2A (10p12.31-12.2). While these germline variants had never been associated with ALL prior to GWAS, there is compelling evidence implicating all 5 genes in the ALL pathogenesis. For example, germline variants in ARID5B have the strongest association with ALL susceptibility across the genome and the loss of Arid5b in mouse leads to significant defects in lymphoid cell development.39 IKZF1, an important transcription factor in all lymphoid lineages, is frequently targeted by copy number alterations in ALL blast cells (particularly in high-risk ALL), and IKZF1 deletion is associated with a poor prognosis.40 Loss of CDKN2A/CDKN2B occurs in up to 40% of B-precursor ALL and is likely to contribute to cell cycle deregulation in leukemia.41 CEBPE is related specifically to myeloid cell maturation and terminal differentiation,42, 43 but intrachromosomal translocations involving IGH and CEBPE have also been described in childhood ALL.44 The remarkable convergence of germline ALL susceptibility loci and somatic aberrations on genes involved in lymphoid cell development, cell cycle control, and tumor suppression reinforces the contribution of these key pathways to leukemogenesis and also points to the possibility that inherited and acquired genetic variations act synergistically in the development of childhood ALL. Importantly, unlike the candidate gene studies, these loci showing genome-wide significant association with the risk of ALL are repeatedly validated by subsequent reports,45-52 unequivocally establishing the importance of inherited genetic variations in ALL susceptibility.
Question naturally arises as to whether the racial and ethnic pattern of ALL incidence could be explained by population differences in the frequency of the ALL-predisposing genetic variations. In AAs, allele frequency at rs10821936 was significantly different between ALL cases and non-ALL controls, confirming the association of the ARID5B variant with ALL susceptibility.46 The risk variant was also substantially less common in subjects of African descent than in those with European ancestry, explaining an increase of 5.2 per million person-years in ALL incidence (e.g., 30% of the observed racial difference).46 Similarly, ARID5B SNP genotype was associated with ALL risk in HAs and the frequency of the risk allele was highest in HAs, consistent with the higher ALL incidence in this population.50 Additional genotyping at the ARID5B locus identified 5 and 3 susceptibility variants that were specific to EAs and HAs, respectively.50 At the novel susceptibility locus PIP4K2A, rs7088318 was associated with ALL risk across race and ethnicity, and the population differences in risk allele frequency at this SNP paralleled racial and ethnic differences in ALL incidence.37 In contrast, CEBPE SNPs were more strongly related to ALL risk in EAs, with variable effects in populations of non-European descent.37 IKZF1 SNPs were associated with ALL susceptibility across racial and ethnic groups, with comparable risk allele frequency and thus little relation to racial and ethnic differences in ALL incidence.37 Together, these findings consistently point to the ARID5B and PIP4K2A loci as important determinants of racial and ethnic differences in ALL susceptibility.
Non-genetic factors influencing racial and ethnic disparities in ALL incidence
In contrast to the remarkable advances in our understanding of the basic biology and treatment of childhood ALL, there has been little, if any, new information regarding an environmental etiology.53 In spite of large epidemiological studies of childhood leukemia and a substantial volume of reports of largely unconfirmed associations, we are still left with a limited list of potential environmental leukemogenic exposures, e.g., ionizing radiation, alkylating chemotherapeutic agents, and topoisomerase-II inhibitors.53 Based upon the distinct age-specific incidence pattern and international variations observed among populations of different social and economic development, a hypothesis was proposed for a role of infection in the etiology of childhood ALL.54 Subsequently, considerable interest and research has focused on infection with a proposed mechanism of proliferative stress resulting from delayed exposure to infectious agents during infancy (Greave hypothesis)55 or population mixing (Kinlen hypothesis).56
It is unlikely that any major environmental factors are driving racial and ethnic differences in the occurrence of ALL given the paucity of reproducible associations within the context of the substantial amount of epidemiological research.53, 57, 58 Conversely, possibility also exists that our ability to accurately document or quantify exposure remains inadequate. Given the genotypic and phenotypic heterogeneity of childhood ALL, focusing on distinct subsets of the disease may provide insights into environmental influences in ALL risk.59-61
Racial and ethnic Differences in ALL Treatment Outcome
Racial and ethnic differences in survival after childhood are well documented, with inferior treatment outcome in AAs and HAs compared to EAs.
In the Pediatric Oncology Group ALL trials between 1981 and 1994 (N=5,086), AAs and HAs had greater excess mortality than those of EAs.10 A retrospective study of 8,447 children with newly diagnosed ALL treated on CCG protocols during the same time period showed similar racial and ethnic differences in 5-year event-free survival (EFS): Asians, 75.1%±3.5%; EAs, 72.8%±0.6%; HAs, 65.9%±1.5%; and AAs, 61.5%±2.2%.6 These differences resulted mainly from differential risks of relapse, as nearly all children achieved clinical remission regardless of race/ethnicity. Racial and ethnic disparities in outcome remained significant even after adjusting for known clinical and molecular risk factors (e.g., age and leukocyte count at diagnosis, sex, ALL lineage and molecular subtypes), and persisted across the treatment eras (1983 to 1989 and 1989 to 1995). However, in a single-institution study of children with ALL treated at St. Jude Children's Research Hospital, there was a significant gap in survival between AAs and EAs during the early treatment era (1962 to 1983), which became nonsignificant in more recent treatment protocols (1984 to 1992 and 1991 to 1998).25, 62 The authors concluded that with equal access to contemporary protocol-based ALL therapy, AA children fare as well as EAs. This is also reflected in a recent meta-analysis of 21,626 children treated on the Children's Oncology Group (COG) frontline ALL trials between 1990 and 2005, where the absolute 5-year survival difference between AAs and EAs fell from 11.0% during 1990-1994, to 8.1% during 1995 to 1999, eventually then to 3.3% during 2000-2005.8 In contrast, even with risk-adapted treatment regimens, HA children with ALL continued to fare worse than patients of European descent across treatment eras. In contrast, a population-based study using the NCI SEER database (from 1988 to 2008) reported a continuing trend of worse treatment outcomes in AA, HA, and NA children, but also noted poorer survival in Asian children with ALL (particularly Vietnamese and Filipinos)63.
Genetic basis of racial and ethnic differences in ALL treatment outcome
Relapse is the primary cause of death in children with ALL, and racial and ethnic disparity in relapse can arise from differences in host disposition of and/or tumor response to anti-leukemic agents, which may be influenced by both inherited (germline) and acquired (somatic) genetic factors.
Genome-wide studies comparing germline SNPs in global populations revealed dramatic differences in the genetic make-up of racial and ethnic groups,19 raising the possibility that these ancestry-related genetic variations contribute to racial and ethnic disparities in ALL treatment response. To test this hypothesis, Yang et al. performed a near population-based GWAS that examined 444,044 genetic polymorphisms for their associations with ALL relapse in 2,534 children with newly-diagnosed disease from COG and St. Jude front-line protocols (Figure 2A).27 They first quantitatively assessed European, African, East Asian, and NA genetic ancestry in children with ALL, by genome-wide comparing SNP genotype against established reference subjects with clearly defined ancestry origin (e.g., indigenous Maya, Nahua, Aymara, Quechua populations as NA references20). Among 4 ancestries evaluated, only NA genetic ancestry were significantly associated with the cumulative incidence of ALL relapse (Figure 2B), independent of known prognostic factors (leukocyte count and age at diagnosis, ALL lineage and molecular subtypes). It is important to note that NA ancestry remained significantly associated with relapse even within self-declared EAs (for whom genetic ancestry was cryptic, Figure 2C), strongly arguing for a genetic basis for the elevated risk of leukemia relapse in HAs. Particularly of clinical relevance, NA genetic ancestry was predictive of relapse after adjusting for minimal residual disease, or even within the group of patients with no detectable residual leukemia at the end of induction therapy. A possible therapeutic intervention was also noted by the analysis of the COG P9904/9905 protocols in which children were randomized to receive or not to receive the delayed intensification therapy (e.g., an 8-week multi-agent treatment, Figures 2D and 2E). This additional phase of chemotherapy almost completely mitigated the poor prognosis attributed with NA ancestry. Although the exact benefit of delayed intensification treatment in the context of ancestry needs to be examined in future ALL trials with diverse treatment regimens, these results illustrate the importance and feasibility of individualizing therapy on the basis of race- and ethnicity-related genetic variation. Together, this study conveys two very important messages: 1) there is a biological basis for racial and ethnic disparities in ALL treatment outcome, and 2) race- and ethnicity-related relapse risk can be overcome by more individualized chemotherapy.
Figure 2. Genetic ancestry and risk of relapse in childhood ALL.
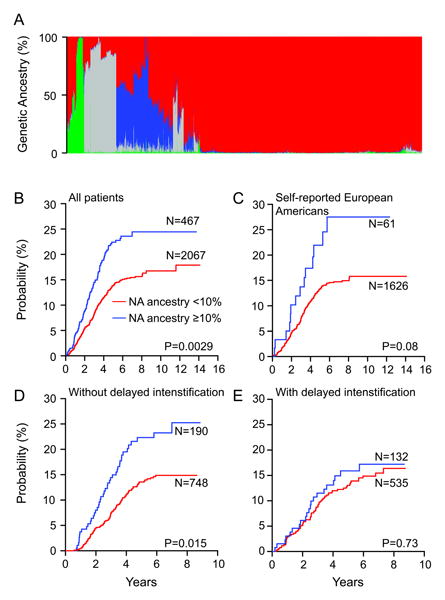
(a) Genetic ancestral composition of 2,534 children with ALL. Each patient's ancestry is shown as a column and the color represents the proportion of ancestry estimated for that patient (European, red; African, gray; Asian, green; Native American, blue). Genetic ancestry was estimated using STRUCTURE. Patients were clustered using the Ward clustering method based on dissimilarity in genetic ancestry measured by 1-minus pair-wise correlation. (b–e) Higher levels of Native American (NA) ancestry were linked to increased risk of relapse in all patients (b) and within the self-reported European American (c) and for those who did not receive delayed intensification (d) but not within those who did receive delayed intensification in the COG P9904/9905 trial (e). Although cumulative incidence of relapse is plotted separately for patients with <10% (red) versus ≥10% (blue) Native American ancestry, we estimated all P values using a Fine and Gray's cumulative incidence hazard regression model treating Native American ancestry as a continuous. (Reproduced with permission from Yang JJ, Cheng C, Devidas M et al., Nature Genetics, 43, 240, 2011)
As noted above, a spectrum of somatic chromosomal abnormalities have been described for ALL, some of which are highly prognostic and are used to assign patients to treatment intensification.3 There is little evidence that ETV6-RUNX1, TCF3-PBX1, BCR-ABL1, or MLL rearranged ALL is disproportionally distributed between HA and EA children with ALL, and multivariate analyses indicate racial and ethnic differences in relapse remain significant after adjusting for leukocyte count and age, ALL lineage and molecular subtypes, and/or minimal residual disease.6, 27 More recently, global gene expression profiling of leukemia blasts identified up to 15% of childhood ALL with a transcription signature similar to that of Philadelphia chromosome-positive ALL, and this novel subtype is thus termed Ph-like ALL.40, 64-66 Up to 50% of Ph-like ALL exhibit overexpression of CRLF2 as a result of balanced translocation or gene fusion and is associated with a very poor prognosis.66 Interestingly, Ph-like ALL with CRLF2 lesions is significantly over-represented in self-reported Hispanics (35.3%) compared to non-Hispanic children (7.1%)66, plausibly contributing to the gap in treatment outcome between the two groups. In fact, our group recently identified germline variants in the GATA3 gene that predispose children to Ph-like ALL and were associated with ALL relapse67. These 2 variants are strikingly over-represented in patients with high levels of NA genetic ancestry, consistent with the inferior treatment outcome of Hispanic children with ALL.
Non-genetic factors influencing racial and ethnic disparities in ALL treatment outcomes
While the discovery of genetic ancestry as a prognostic factor in ALL indicates a biological basis of racial and ethnic disparities, it by no means precludes the contribution of non-genetic factors, e.g., the timing of diagnosis, access to quality health care, and adherence to treatment.68 In fact, inclusion of socio-economic status in the multivariate model substantially reduced the statistical significance of the difference in relapse between HA and EA children with ALL6.
Poor adherence to medication can negatively influence cancer treatment outcome and is of particular importance in childhood ALL therapy which requires prolonged daily oral administration of antimetabolites (6-mercaptopurine [6MP] for up to 2 years). In a recent study, it was observed that the mean oral 6MP adherence rate was significantly lower among relapse patients as compared with those in continuous complete remission (88.2% and 96.2% respectively),69 with a progressive increase in the risk of relapse with decreasing levels of adherence. Adherence was significantly lower among self-reported HAs for whom there was also a higher rate of relapse compared to EAs. In a multivariate model, the prognostic value of HA ethnicity became nonsignificant after adjusting for 6MP adherence and socioeconomic status. However, when the analysis was restricted to patients with high medication compliance (≥90% adherence rate), relapse was still more common in HAs than in EAs, highlighting the contribution of genetic and biological factors to the racial and ethnic gap in ALL treatment outcome. A comprehensive evaluation of both genetic and non-genetic factors is warranted in future studies to accurately ascertain the causes underlying the racial and ethnic disparities in ALL survival.
Conclusion
It is hard to over-emphasize the need to address racial and ethnic gaps in the incidence and outcome of childhood ALL, the most common pediatric cancer. Differences in ALL biology by race and ethnicity are poorly characterized, contributing to the persisting disparity in spite of overall improvement in cure rates. Recent genomic profiling of ALL described the genetic landscape of this disease with an unprecedented resolution, discovering novel molecular markers associated with disease biology and prognosis. In particular, inherited genetic variants associated with NA ancestry were linked to the susceptibility and treatment outcome of childhood ALL, partly explaining higher ALL incidence and poorer survival in HAs. With these advances in cancer genomics and novel approaches to investigate non-genetic variables, the timing is opportune for comprehensive research of ALL disparity to finally close the racial and ethnic gaps in this catastrophic disease in children.
Acknowledgments
This work was supported by the National Institutes of Health grant U01GM92666 and by the American Lebanese Syrian Associated Charities (ALSAC). J.J. Yang is an American Society of Hematology Scholar.
We thank Sharon Naron for her editorial assistance.
Footnotes
Conflict of Interest/Disclosures: The authors declare no competing financial interests.
References
- 1.Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet. 2013;381(9881):1943–55. doi: 10.1016/S0140-6736(12)62187-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pui CH, Campana D, Pei D, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med. 2009;360(26):2730–41. doi: 10.1056/NEJMoa0900386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pui CH, Mullighan CG, Evans WE, Relling MV. Pediatric acute lymphoblastic leukemia: where are we going and how do we get there? Blood. 2012;120(6):1165–74. doi: 10.1182/blood-2012-05-378943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dores GM, Devesa SS, Curtis RE, Linet MS, Morton LM. Acute leukemia incidence and patient survival among children and adults in the United States, 2001-2007. Blood. 2012;119(1):34–43. doi: 10.1182/blood-2011-04-347872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Linabery AM, Ross JA. Trends in childhood cancer incidence in the U.S. (1992-2004) Cancer. 2008;112(2):416–32. doi: 10.1002/cncr.23169. [DOI] [PubMed] [Google Scholar]
- 6.Bhatia S, Sather HN, Heerema NA, Trigg ME, Gaynon PS, Robison LL. Racial and ethnic differences in survival of children with acute lymphoblastic leukemia. Blood. 2002;100(6):1957–64. doi: 10.1182/blood-2002-02-0395. [DOI] [PubMed] [Google Scholar]
- 7.Kadan-Lottick NS, Ness KK, Bhatia S, Gurney JG. Survival variability by race and ethnicity in childhood acute lymphoblastic leukemia. JAMA. 2003;290(15):2008–14. doi: 10.1001/jama.290.15.2008. [DOI] [PubMed] [Google Scholar]
- 8.Hunger SP, Lu X, Devidas M, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: A report from the Children's Oncology Group. J Clin Oncol. 2012;30(14):1663–69. doi: 10.1200/JCO.2011.37.8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chow EJ, Puumala SE, Mueller BA, et al. Childhood cancer in relation to parental race and ethnicity: a 5-state pooled analysis. Cancer. 2010;116(12):3045–53. doi: 10.1002/cncr.25099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pollock BH, DeBaun MR, Camitta BM, et al. Racial differences in the survival of childhood B-precursor acute lymphoblastic leukemia: a Pediatric Oncology Group Study. J Clin Oncol. 2000;18(4):813–23. doi: 10.1200/JCO.2000.18.4.813. [DOI] [PubMed] [Google Scholar]
- 11.Mullighan CG. Molecular genetics of B-precursor acute lymphoblastic leukemia. J Clin Invest. 2012;122(10):3407–15. doi: 10.1172/JCI61203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pui CH, Carroll WL, Meshinchi S, Arceci RJ. Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol. 2011;29(5):551–65. doi: 10.1200/JCO.2010.30.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Figueroa ME, Chen SC, Andersson AK, et al. Integrated genetic and epigenetic analysis of childhood acute lymphoblastic leukemia. J Clin Invest. 2013;123(7):3099–111. doi: 10.1172/JCI66203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor KH, Kramer RS, Davis JW, et al. Ultradeep bisulfite sequencing analysis of DNA methylation patterns in multiple gene promoters by 454 sequencing. Cancer Res. 2007;67(18):8511–8. doi: 10.1158/0008-5472.CAN-07-1016. [DOI] [PubMed] [Google Scholar]
- 15.Schafer E, Irizarry R, Negi S, et al. Promoter hypermethylation in MLL-r infant acute lymphoblastic leukemia: biology and therapeutic targeting. Blood. 2010;115(23):4798–809. doi: 10.1182/blood-2009-09-243634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bamshad M, Wooding S, Salisbury BA, Stephens JC. Deconstructing the relationship between genetics and race. Nat Rev Genet. 2004;5(8):598–609. doi: 10.1038/nrg1401. [DOI] [PubMed] [Google Scholar]
- 17.Bloche MG. Health care disparities--science, politics, and race. N Engl J Med. 2004;350(15):1568–70. doi: 10.1056/NEJMsb045005. [DOI] [PubMed] [Google Scholar]
- 18.Ramachandran S, Deshpande O, Roseman CC, Rosenberg NA, Feldman MW, Cavalli-Sforza LL. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc Natl Acad Sci U S A. 2005;102(44):15942–7. doi: 10.1073/pnas.0507611102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li JZ, Absher DM, Tang H, et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319(5866):1100–4. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 20.Mao X, Bigham AW, Mei R, et al. A genomewide admixture mapping panel for Hispanic/Latino populations. Am J Hum Genet. 2007;80(6):1171–8. doi: 10.1086/518564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Price AL, Patterson N, Yu F, et al. A genomewide admixture map for Latino populations. Am J Hum Genet. 2007;80(6):1024–36. doi: 10.1086/518313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang S, Lewis CM, Jakobsson M, et al. Genetic variation and population structure in native Americans. PLoS Genet. 2007;3(11):e185. doi: 10.1371/journal.pgen.0030185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.National Cancer Institute. [accessed 22 Jul 2013];SEER Cancer Statistics Review, 1975-2006. 2009 Available from URL: http://seer.cancer.gov/csr/1975_2006/
- 24.Ferlay J, Shin H, Bray F, Forman D, Mathers C, Parkin D. [accessed 6 Mar 2012];GLOBOCAN 2000, Cancer Incidence and Mortality Worldwide. 2001 Available from URL: http://www-dep.iarc.fr/globocan/globocan.htm.
- 25.Pui CH, Sandlund JT, Pei D, et al. Results of therapy for acute lymphoblastic leukemia in black and white children. JAMA. 2003;290(15):2001–7. doi: 10.1001/jama.290.15.2001. [DOI] [PubMed] [Google Scholar]
- 26.Aldrich MC, Zhang L, Wiemels JL, et al. Cytogenetics of Hispanic and White children with acute lymphoblastic leukemia in California. Cancer Epidemiol Biomarkers Prev. 2006;15(3):578–81. doi: 10.1158/1055-9965.EPI-05-0833. [DOI] [PubMed] [Google Scholar]
- 27.Yang JJ, Cheng C, Devidas M, et al. Ancestry and pharmacogenomics of relapse in acute lymphoblastic leukemia. Nat Genet. 2011;43(3):237–41. doi: 10.1038/ng.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hasle H, Clemmensen IH, Mikkelsen M. Risks of leukaemia and solid tumours in individuals with Down's syndrome. Lancet. 2000;355(9199):165–9. doi: 10.1016/S0140-6736(99)05264-2. [DOI] [PubMed] [Google Scholar]
- 29.Mullighan CG, Collins-Underwood JR, Phillips LA, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009;41(11):1243–6. doi: 10.1038/ng.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vijayakrishnan J, Houlston RS. Candidate gene association studies and risk of childhood acute lymphoblastic leukemia: a systematic review and meta-analysis. Haematologica. 2010;95(8):1405–14. doi: 10.3324/haematol.2010.022095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koppen IJ, Hermans FJ, Kaspers GJ. Folate related gene polymorphisms and susceptibility to develop childhood acute lymphoblastic leukaemia. Br J Haematol. 2010;148(1):3–14. doi: 10.1111/j.1365-2141.2009.07898.x. [DOI] [PubMed] [Google Scholar]
- 32.Urayama KY, Chokkalingam AP, Metayer C, et al. HLA-DP genetic variation, proxies for early life immune modulation and childhood acute lymphoblastic leukemia risk. Blood. 2012;120(15):3039–47. doi: 10.1182/blood-2012-01-404723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang JS, Wiemels JL, Chokkalingam AP, et al. Genetic polymorphisms in adaptive immunity genes and childhood acute lymphoblastic leukemia. Cancer Epidemiol Biomarkers Prev. 2010;19(9):2152–63. doi: 10.1158/1055-9965.EPI-10-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hosking FJ, Leslie S, Dilthey A, et al. MHC variation and risk of childhood B-cell precursor acute lymphoblastic leukemia. Blood. 2011;117(5):1633–40. doi: 10.1182/blood-2010-08-301598. [DOI] [PubMed] [Google Scholar]
- 35.Trevino LR, Yang W, French D, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet. 2009;41(9):1001–5. doi: 10.1038/ng.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Papaemmanuil E, Hosking FJ, Vijayakrishnan J, et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet. 2009;41(9):1006–10. doi: 10.1038/ng.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu H, Yang W, Perez-Andreu V, et al. Novel susceptibility variants at 10p12.31-12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J Natl Cancer Inst. 2013;105(10):733–42. doi: 10.1093/jnci/djt042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sherborne AL, Hosking FJ, Prasad RB, et al. Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nat Genet. 2010;42(6):492–4. doi: 10.1038/ng.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lahoud MH, Ristevski S, Venter DJ, et al. Gene targeting of Desrt, a novel ARID class DNA-binding protein, causes growth retardation and abnormal development of reproductive organs. Genome Res. 2001;11(8):1327–34. doi: 10.1101/gr.168801. [DOI] [PubMed] [Google Scholar]
- 40.Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360(5):470–80. doi: 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mullighan CG, Goorha S, Radtke I, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446(7137):758–64. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 42.Yamanaka R, Kim GD, Radomska HS, et al. CCAAT/enhancer binding protein epsilon is preferentially up-regulated during granulocytic differentiation and its functional versatility is determined by alternative use of promoters and differential splicing. Proc Natl Acad Sci U S A. 1997;94(12):6462–7. doi: 10.1073/pnas.94.12.6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakajima H, Watanabe N, Shibata F, Kitamura T, Ikeda Y, Handa M. N-terminal region of CCAAT/enhancer-binding protein epsilon is critical for cell cycle arrest, apoptosis, and functional maturation during myeloid differentiation. J Biol Chem. 2006;281(20):14494–502. doi: 10.1074/jbc.M600575200. [DOI] [PubMed] [Google Scholar]
- 44.Akasaka T, Balasas T, Russell LJ, et al. Five members of the CEBP transcription factor family are targeted by recurrent IGH translocations in B-cell precursor acute lymphoblastic leukemia (BCP-ALL) Blood. 2007;109(8):3451–61. doi: 10.1182/blood-2006-08-041012. [DOI] [PubMed] [Google Scholar]
- 45.Prasad RB, Hosking FJ, Vijayakrishnan J, et al. Verification of the susceptibility loci on 7p12.2, 10q21.2, and 14q11.2 in precursor B-cell acute lymphoblastic leukemia of childhood. Blood. 2010;115(9):1765–7. doi: 10.1182/blood-2009-09-241513. [DOI] [PubMed] [Google Scholar]
- 46.Yang W, Trevino LR, Yang JJ, et al. ARID5B SNP rs10821936 is associated with risk of childhood acute lymphoblastic leukemia in blacks and contributes to racial differences in leukemia incidence. Leukemia. 2010;24(4):894–6. doi: 10.1038/leu.2009.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Healy J, Richer C, Bourgey M, Kritikou EA, Sinnett D. Replication analysis confirms the association of ARID5B with childhood B-cell acute lymphoblastic leukemia. Haematologica. 2010;95(9):1608–11. doi: 10.3324/haematol.2010.022459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pastorczak A, Gorniak P, Sherborne A, et al. Role of 657del5 NBN mutation and 7p12.2 (IKZF1), 9p21 (CDKN2A), 10q21.2 (ARID5B) and 14q11.2 (CEBPE) variation and risk of childhood ALL in the Polish population. Leukemia Res. 2011;35(11):1534–6. doi: 10.1016/j.leukres.2011.07.034. [DOI] [PubMed] [Google Scholar]
- 49.Walsh KM, de Smith AJ, Chokkalingam AP, et al. Novel childhood ALL susceptibility locus BMI1-PIP4K2A is specifically associated with the hyperdiploid subtype. Blood. 2013;121(23):4808–9. doi: 10.1182/blood-2013-04-495390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu H, Cheng C, Devidas M, et al. ARID5B genetic polymorphisms contribute to racial disparities in the incidence and treatment outcome of childhood acute lymphoblastic leukemia. J Clin Oncol. 2012;30(7):751–57. doi: 10.1200/JCO.2011.38.0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ellinghaus E, Stanulla M, Richter G, et al. Identification of germline susceptibility loci in ETV6-RUNX1-rearranged childhood acute lymphoblastic leukemia. Leukemia. 2012;26(5):902–9. doi: 10.1038/leu.2011.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Orsi L, Rudant J, Bonaventure A, et al. Genetic polymorphisms and childhood acute lymphoblastic leukemia: GWAS of the ESCALE study (SFCE) Leukemia. 2012;26(12):2561–4. doi: 10.1038/leu.2012.148. [DOI] [PubMed] [Google Scholar]
- 53.Eden T. Aetiology of childhood leukaemia. Cancer Treat Rev. 2010;36(4):286–97. doi: 10.1016/j.ctrv.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 54.Kinlen L. Infective cause of childhood leukaemia. Lancet. 1989;1(8629):94–5. [PubMed] [Google Scholar]
- 55.Greaves M. Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev. 2006;6(3):193–203. doi: 10.1038/nrc1816. [DOI] [PubMed] [Google Scholar]
- 56.Kinlen L. Childhood leukaemia, nuclear sites, and population mixing. Br J Cancer. 2011;104(1):12–8. doi: 10.1038/sj.bjc.6605982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Buffler PA, Kwan ML, Reynolds P, Urayama KY. Environmental and genetic risk factors for childhood leukemia: appraising the evidence. Cancer Invest. 2005;23(1):60–75. [PubMed] [Google Scholar]
- 58.Spector LG, Ross JA, Robison LL, Bhatia S. Epidemiology and etiology. In: Pui CH, editor. Childhood Leukemias. 3rd. New York: Cambridge University Press; 2012. [Google Scholar]
- 59.Shu XO, Ross JA, Pendergrass TW, Reaman GH, Lampkin B, Robison LL. Parental alcohol consumption, cigarette smoking, and risk of infant leukemia: a Childrens Cancer Group study. Journal of the National Cancer Institute. 1996;88(1):24–31. doi: 10.1093/jnci/88.1.24. [DOI] [PubMed] [Google Scholar]
- 60.Linabery AM, Olshan AF, Gamis AS, et al. Exposure to medical test irradiation and acute leukemia among children with Down syndrome: a report from the Children's Oncology Group. Pediatrics. 2006;118(5):e1499–508. doi: 10.1542/peds.2006-0644. [DOI] [PubMed] [Google Scholar]
- 61.Slater ME, Linabery AM, Spector LG, et al. Maternal exposure to household chemicals and risk of infant leukemia: a report from the Children's Oncology Group. Cancer Causes Control. 2011;22(8):1197–204. doi: 10.1007/s10552-011-9798-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pui CH, Boyett JM, Hancock ML, Pratt CB, Meyer WH, Crist WM. Outcome of treatment for childhood cancer in black as compared with white children. The St Jude Children's Research Hospital experience, 1962 through 1992. JAMA. 1995;273(8):633–7. [PubMed] [Google Scholar]
- 63.Goggins WB, Lo FF. Racial and ethnic disparities in survival of US children with acute lymphoblastic leukemia: evidence from the SEER database 1988-2008. Cancer Causes Control. 2012;23(5):737–43. doi: 10.1007/s10552-012-9943-8. [DOI] [PubMed] [Google Scholar]
- 64.Roberts KG, Morin RD, Zhang J, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22(2):153–66. doi: 10.1016/j.ccr.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10(2):125–34. doi: 10.1016/S1470-2045(08)70339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harvey RC, Mullighan CG, Chen IM, et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood. 2010;115(26):5312–21. doi: 10.1182/blood-2009-09-245944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Perez-Andreu V, Roberts KG, Harvey RC, et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet. 2013 doi: 10.1038/ng.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pappas G, Queen S, Hadden W, Fisher G. The increasing disparity in mortality between socioeconomic groups in the United States, 1960 and 1986. N Engl J Med. 1993;329(2):103–9. doi: 10.1056/NEJM199307083290207. [DOI] [PubMed] [Google Scholar]
- 69.Bhatia S, Landier W, Shangguan M, et al. Nonadherence to oral mercaptopurine and risk of relapse in Hispanic and non-Hispanic white children with acute lymphoblastic leukemia: a report from the children's oncology group. J Clin Oncol. 2012;30(17):2094–101. doi: 10.1200/JCO.2011.38.9924. [DOI] [PMC free article] [PubMed] [Google Scholar]